

The Role of mTOR in Amyotrophic Lateral Sclerosis

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
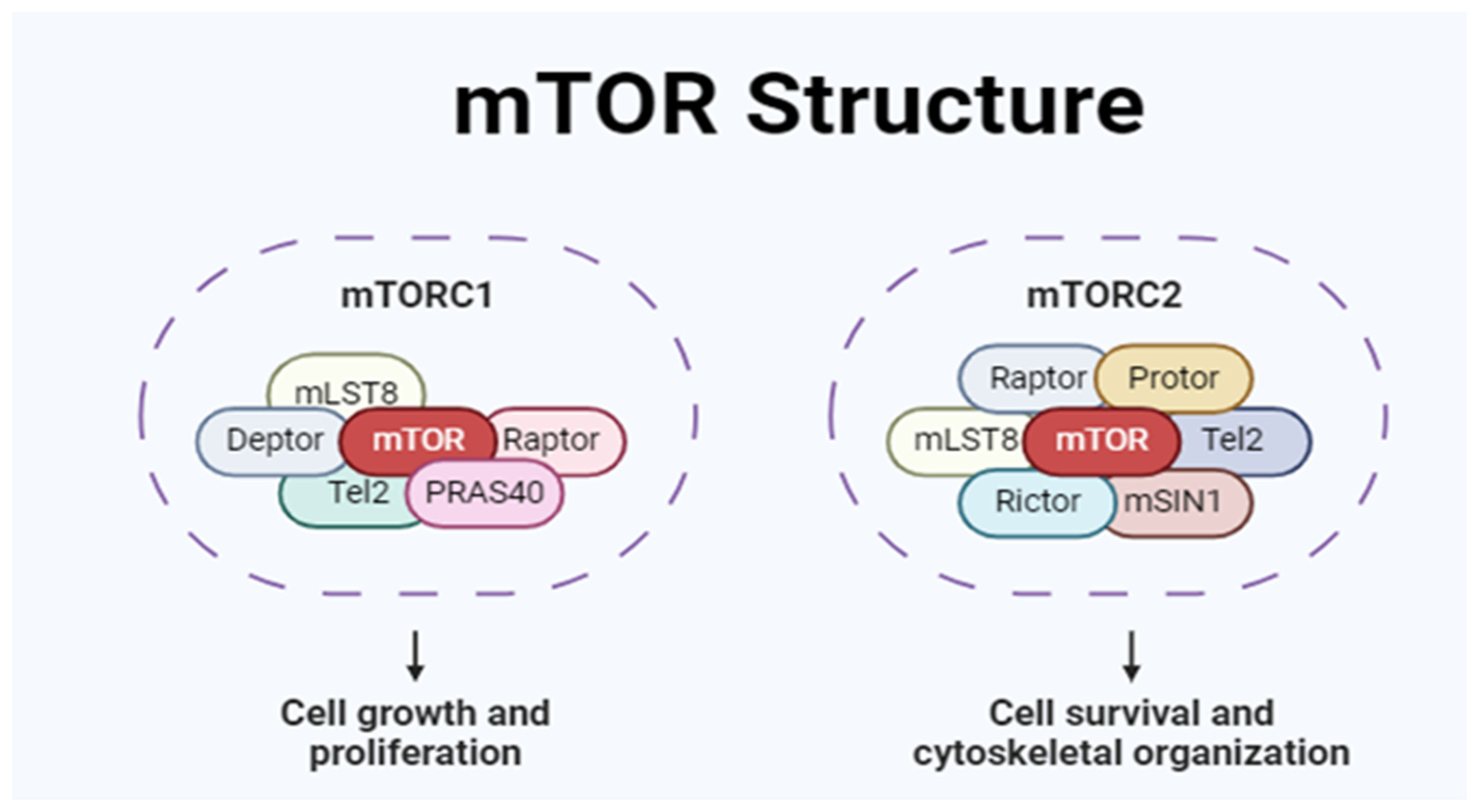
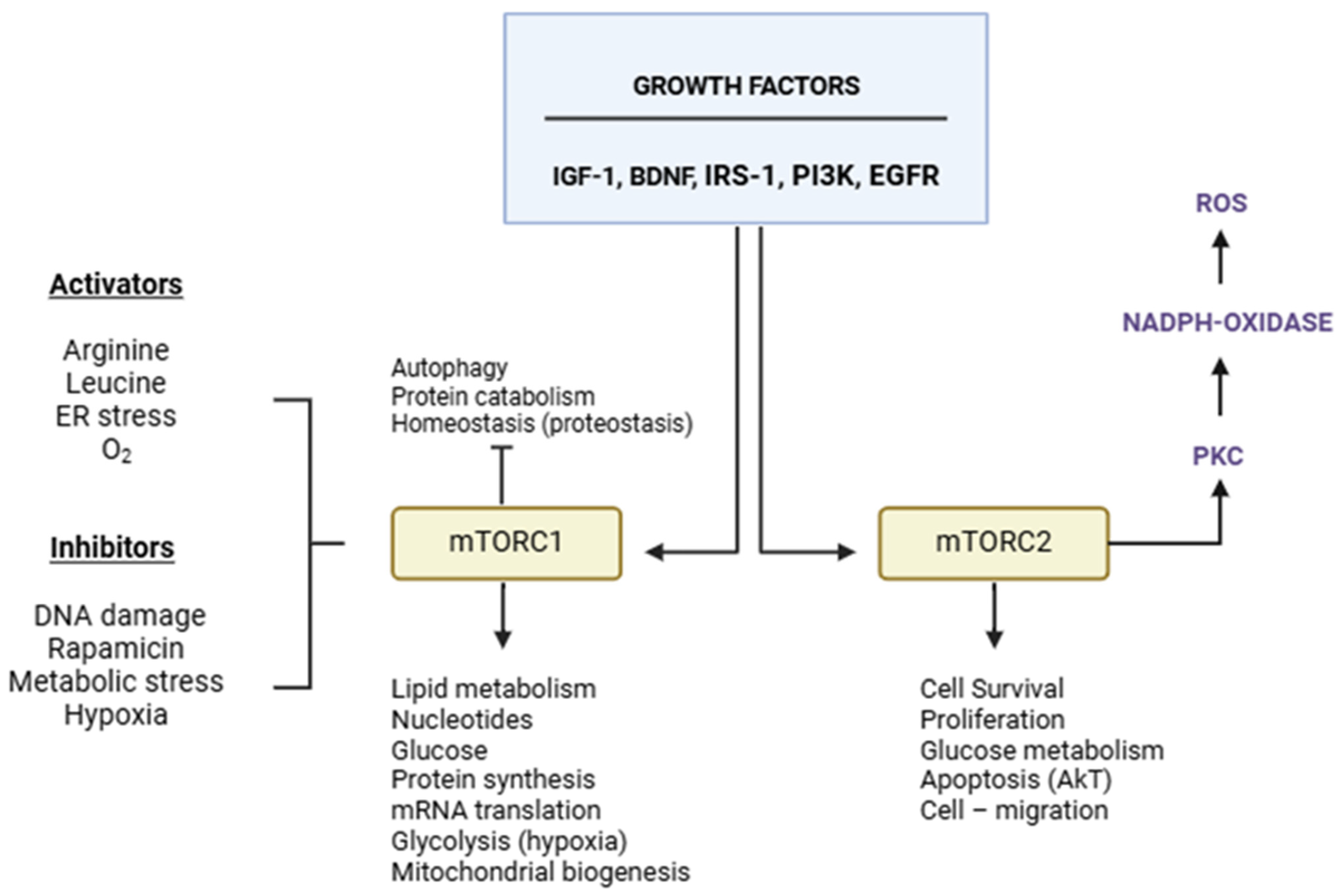
2. mTOR Structure and Function
3. mTOR and ALS
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. New Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D. Current Hypotheses for the Underlying Biology of Amyotrophic Lateral Sclerosis. Ann. Neurol. 2009, 65 (Suppl. S1), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- De Souza, P.V.S.; de Pinto, W.B.V.R.; Chieia, M.A.T.; Oliveira, A.S.B. Clinical and Genetic Basis of Familial Amyotrophic Lateral Sclerosis. Arq. De Neuro-Psiquiatr. 2015, 73, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Shandilya, A.; Mehan, S. Dysregulation of IGF-1/GLP-1 Signaling in the Progression of ALS: Potential Target Activators and Influences on Neurological Dysfunctions. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2021, 42, 3145–3166. [Google Scholar] [CrossRef]
- Tang, S.J.; Reis, G.; Kang, H.; Gingras, A.-C.; Sonenberg, N.; Schuman, E.M. A Rapamycin-Sensitive Signaling Pathway Contributes to Long-Term Synaptic Plasticity in the Hippocampus. Proc. Natl. Acad. Sci. USA 2002, 99, 467–472. [Google Scholar] [CrossRef]
- Hoeffer, C.A.; Klann, E. MTOR Signaling: At the Crossroads of Plasticity, Memory and Disease. Trends Neurosci. 2010, 33, 67–75. [Google Scholar] [CrossRef]
- Stoica, L.; Zhu, P.J.; Huang, W.; Zhou, H.; Kozma, S.C.; Costa-Mattioli, M. Selective Pharmacogenetic Inhibition of Mammalian Target of Rapamycin Complex I (MTORC1) Blocks Long-Term Synaptic Plasticity and Memory Storage. Proc. Natl. Acad. Sci. USA 2011, 108, 3791–3796. [Google Scholar] [CrossRef]
- Schieke, S.M.; Phillips, D.; McCoy, J.P.J.; Aponte, A.M.; Shen, R.-F.; Balaban, R.S.; Finkel, T. The Mammalian Target of Rapamycin (mTOR) Pathway Regulates Mitochondrial Oxygen Consumption and Oxidative Capacity. J. Biol. Chem. 2006, 281, 27643–27652. [Google Scholar] [CrossRef]
- Ramanathan, A.; Schreiber, S.L. Direct Control of Mitochondrial Function by MTOR. Proc. Natl. Acad. Sci. USA 2009, 106, 22229–22232. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP Activity Is Regulated by MTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de Novo Pyrimidine Synthesis by Growth Signaling through MTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. MTORC1 Induces Purine Synthesis through Control of the Mitochondrial Tetrahydrofolate Cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef]
- Brunn, G.J.; Hudson, C.C.; Sekulić, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Lawrence, J.C.J.; Abraham, R.T. Phosphorylation of the Translational Repressor PHAS-I by the Mammalian Target of Rapamycin. Science 1997, 277, 99–101. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 Functions as a Transcriptional Regulator of Autophagy by Preventing Nuclear Transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The Transcription Factor TFEB Links MTORC1 Signaling to Transcriptional Control of Lysosome Homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A Lysosome-to-Nucleus Signalling Mechanism Senses and Regulates the Lysosome via MTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef]
- Zhang, Y.; Manning, B.D. MTORC1 Signaling Activates NRF1 to Increase Cellular Proteasome Levels. Cell Cycle 2015, 14, 2011–2017. [Google Scholar] [CrossRef]
- Dibble, C.C.; Manning, B.D. Signal Integration by MTORC1 Coordinates Nutrient Input with Biosynthetic Output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/MTOR Pathway Is a Crucial Regulator of Skeletal Muscle Hypertrophy and Can Prevent Muscle Atrophy in Vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.H.; Widenmaier, S.B.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated Regulation of Protein Synthesis and Degradation by MTORC1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-Dependent MTORC1 Association with the ULK1-Atg13-FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate MTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Anthony, J.C.; Yoshizawa, F.; Anthony, T.G.; Vary, T.C.; Jefferson, L.S.; Kimball, S.R. Leucine Stimulates Translation Initiation in Skeletal Muscle of Postabsorptive Rats via a Rapamycin-Sensitive Pathway. J. Nutr. 2000, 130, 2413–2419. [Google Scholar] [CrossRef]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-Induced Skeletal Myotube Hypertrophy by PI(3)K/Akt/MTOR and PI(3)K/Akt/GSK3 Pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546.e5. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Romanino, K.; Cloëtta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M.; et al. Skeletal Muscle-Specific Ablation of Raptor, but Not of Rictor, Causes Metabolic Changes and Results in Muscle Dystrophy. Cell Metab. 2008, 8, 411–424. [Google Scholar] [CrossRef]
- Gomes, N.A.; das Chagas Lima, E.; Silva, F.; de Oliveira Volpe, C.M.; Villar-Delfino, P.H.; de Sousa, C.F.; Rocha-Silva, F.; Nogueira-Machado, J.A. Overexpression of MTOR in Leukocytes from ALS8 Patients. Curr. Neuropharmacol. 2023, 21, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.; Chen, S.; Yang, D.; Wang, Y.; Zhang, X.; Wang, Z.; Le, W. Rapamycin Treatment Augments Motor Neuron Degeneration in SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Autophagy 2011, 7, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Roselli, F.; Singh, K.; Leptien, K.; Julien, J.-P.; Gros-Louis, F.; Caroni, P. Neuroprotection through Excitability and MTOR Required in ALS Motoneurons to Delay Disease and Extend Survival. Neuron 2013, 80, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-F.; Tsai, K.-J.; Shen, C.-K.J. Autophagy Activation Ameliorates Neuronal Pathogenesis of FTLD-U Mice: A New Light for Treatment of TARDBP/TDP-43 Proteinopathies. Autophagy 2013, 9, 239–240. [Google Scholar] [CrossRef]
- Ching, J.K.; Weihl, C.C. Rapamycin-Induced Autophagy Aggravates Pathology and Weakness in a Mouse Model of VCP-Associated Myopathy. Autophagy 2013, 9, 799–800. [Google Scholar] [CrossRef]
- Granatiero, V.; Sayles, N.M.; Savino, A.M.; Konrad, C.; Kharas, M.G.; Kawamata, H.; Manfredi, G. Modulation of the IGF1R-MTOR Pathway Attenuates Motor Neuron Toxicity of Human ALS SOD1G93A Astrocytes. Autophagy 2021, 17, 4029–4042. [Google Scholar] [CrossRef]
- Glass, D.J. Molecular Mechanisms Modulating Muscle Mass. Trends Mol. Med. 2003, 9, 344–350. [Google Scholar] [CrossRef]
- Yoon, M.-S. MTOR as a Key Regulator in Maintaining Skeletal Muscle Mass. Front. Physiol. 2017, 8, 788. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 Regulates Its MRNA Levels through a Negative Feedback Loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How Neuroinflammation Contributes to Neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Pacheco, A.; Franco, J.M.; Lopez, S.; Gomez-Zumaquero, J.M.; Magdalena Leal-Lasarte, M.; Caballero-Hernandez, D.E.; Cejudo-Guillén, M.; Pozo, D. Epigenetic Mechanisms of Gene Regulation in Amyotrophic Lateral Sclerosis. Adv. Exp. Med. Biol. 2017, 978, 255–275. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Neukomm, L.J.; Brown, R.H.J.; Freeman, M.R. Age-Dependent TDP-43-Mediated Motor Neuron Degeneration Requires GSK3, Hat-Trick, and Xmas-2. Curr. Biol. 2015, 25, 2130–2136. [Google Scholar] [CrossRef]
- Mitchell, J.C.; Constable, R.; So, E.; Vance, C.; Scotter, E.; Glover, L.; Hortobagyi, T.; Arnold, E.S.; Ling, S.-C.; McAlonis, M.; et al. Wild Type Human TDP-43 Potentiates ALS-Linked Mutant TDP-43 Driven Progressive Motor and Cortical Neuron Degeneration with Pathological Features of ALS. Acta Neuropathol. Commun. 2015, 3, 36. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Majumder, S.; Deng, J.J.; Bai, Y.; Thornton, F.B.; Oddo, S. Rapamycin Rescues TDP-43 Mislocalization and the Associated Low Molecular Mass Neurofilament Instability. J. Biol. Chem. 2009, 284, 27416–27424. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-Prone Proteins with Polyglutamine and Polyalanine Expansions Are Degraded by Autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef]
- Ravikumar, B.; Berger, Z.; Vacher, C.; O’Kane, C.J.; Rubinsztein, D.C. Rapamycin Pre-Treatment Protects against Apoptosis. Hum. Mol. Genet. 2006, 15, 1209–1216. [Google Scholar] [CrossRef]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin Alleviates Toxicity of Different Aggregate-Prone Proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. MTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex That Signals to the Cell Growth Machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits MTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Yang, Y.-P.; Liang, Z.-Q.; Gu, Z.-L.; Qin, Z.-H. Molecular Mechanism and Regulation of Autophagy. Acta Pharmacol. Sin. 2005, 26, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D.C. Chemical Modulators of Autophagy as Biological Probes and Potential Therapeutics. Nat. Chem. Biol. 2011, 7, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The Role of PI3K/AKT/MTOR Pathway in the Modulation of Autophagy and the Clearance of Protein Aggregates in Neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T.; Cozzolino, M. SOD1 and Mitochondria in ALS: A Dangerous Liaison. J. Bioenerg. Biomembr. 2011, 43, 593–599. [Google Scholar] [CrossRef]
- Heath-Engel, H.M.; Chang, N.C.; Shore, G.C. The Endoplasmic Reticulum in Apoptosis and Autophagy: Role of the BCL-2 Protein Family. Oncogene 2008, 27, 6419–6433. [Google Scholar] [CrossRef]
- Hellwig, C.T.; Passante, E.; Rehm, M. The Molecular Machinery Regulating Apoptosis Signal Transduction and Its Implication in Human Physiology and Pathophysiologies. Curr. Mol. Med. 2011, 11, 31–47. [Google Scholar] [CrossRef]
- Hetz, C.; Thielen, P.; Fisher, J.; Pasinelli, P.; Brown, R.H.; Korsmeyer, S.; Glimcher, L. The Proapoptotic BCL-2 Family Member BIM Mediates Motoneuron Loss in a Model of Amyotrophic Lateral Sclerosis. Cell Death Differ. 2007, 14, 1386–1389. [Google Scholar] [CrossRef]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 Deficiency in the Nervous System Protects against Amyotrophic Lateral Sclerosis by Increasing Autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef]
- Hetz, C.A. ER Stress Signaling and the BCL-2 Family of Proteins: From Adaptation to Irreversible Cellular Damage. Antioxid. Redox Signal. 2007, 9, 2345–2355. [Google Scholar] [CrossRef]
- Kye, M.J.; Niederst, E.D.; Wertz, M.H.; Gonçalves, I.D.C.G.; Akten, B.; Dover, K.Z.; Peters, M.; Riessland, M.; Neveu, P.; Wirth, B.; et al. SMN Regulates Axonal Local Translation via MiR-183/MTOR Pathway. Hum. Mol. Genet. 2014, 23, 6318–6331. [Google Scholar] [CrossRef]
- Biondi, O.; Branchu, J.; Ben Salah, A.; Houdebine, L.; Bertin, L.; Chali, F.; Desseille, C.; Weill, L.; Sanchez, G.; Lancelin, C.; et al. IGF-1R Reduction Triggers Neuroprotective Signaling Pathways in Spinal Muscular Atrophy Mice. J. Neurosci. Off. J. Soc. For Neurosci. 2015, 35, 12063–12079. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Shen, H.-M.; Gopalakrishnan, V.; Gordon, N. Epigenetic Regulation of Autophagy Beyond the Cytoplasm: A Review. Front. Cell Dev. Biol. 2021, 9, 675599. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Liu, L.; Gan, W. The Roles of Post-Translational Modifications on MTOR Signaling. Int. J. Mol. Sci. 2021, 22, 1784. [Google Scholar] [CrossRef]
- Chen, J.; Bassot, A.; Giuliani, F.; Simmen, T. Amyotrophic Lateral Sclerosis (ALS): Stressed by Dysfunctional Mitochondria-Endoplasmic Reticulum Contacts (MERCs). Cells 2021, 10, 71789. [Google Scholar] [CrossRef]
- Ramesh, N.; Pandey, U.B. Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Front. Mol. Neurosci. 2017, 10, 263. [Google Scholar] [CrossRef]
- Rehorst, W.A.; Thelen, M.P.; Nolte, H.; Türk, C.; Cirak, S.; Peterson, J.M.; Wong, G.W.; Wirth, B.; Krüger, M.; Winter, D.; et al. Muscle Regulates MTOR Dependent Axonal Local Translation in Motor Neurons via CTRP3 Secretion: Implications for a Neuromuscular Disorder, Spinal Muscular Atrophy. Acta Neuropathol. Commun. 2019, 7, 154. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Sossin, W.S.; Klann, E.; Sonenberg, N. Translational control of long-lasting synaptic plasticity and memory. Neuron 2009, 61, 10–26. [Google Scholar] [CrossRef]
- Crayle, J.I.; Rampersaud, E.; Myers, J.R.; Wuu, J.; Taylor, J.P.; Wu, G.; Benatar, M.; Bedlack, R.S. Genetic Associations with an Amyotrophic Lateral Sclerosis Reversal Phenotype. Neurology 2024, 103, e209696. [Google Scholar] [CrossRef]
- Gao, J.; Sterling, E.; Hankin, R.; Sikal, A.; Yao, Y. Therapeutics Targeting Skeletal Muscle in Amyotrophic Lateral Sclerosis. Biomolecules 2024, 14, 878. [Google Scholar] [CrossRef]
- Kubinski, S.; Claus, P. Protein network analysis reveals a functional connectivity of dysregulated processes in ALS and SMA. Neurosci. Insights 2022, 17. [Google Scholar] [CrossRef]
 ) IRS = insulin receptor substrate; PI3K = phosphatidylinositol (PI) 3-kinase; Akt = serine/th reonine kinase Akt, also known as protein kinase B (PKB); PIP2 = phosphatidylinositol biphosphate; PIP3 = phosphatidylinositol triphosphate.
) IRS = insulin receptor substrate; PI3K = phosphatidylinositol (PI) 3-kinase; Akt = serine/th reonine kinase Akt, also known as protein kinase B (PKB); PIP2 = phosphatidylinositol biphosphate; PIP3 = phosphatidylinositol triphosphate.
) IRS = insulin receptor substrate; PI3K = phosphatidylinositol (PI) 3-kinase; Akt = serine/th reonine kinase Akt, also known as protein kinase B (PKB); PIP2 = phosphatidylinositol biphosphate; PIP3 = phosphatidylinositol triphosphate.
) IRS = insulin receptor substrate; PI3K = phosphatidylinositol (PI) 3-kinase; Akt = serine/th reonine kinase Akt, also known as protein kinase B (PKB); PIP2 = phosphatidylinositol biphosphate; PIP3 = phosphatidylinositol triphosphate.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogueira-Machado, J.A.; Rocha-Silva, F.; Gomes, N.A. The Role of mTOR in Amyotrophic Lateral Sclerosis. Biomedicines 2025, 13, 952. https://doi.org/10.3390/biomedicines13040952
Nogueira-Machado JA, Rocha-Silva F, Gomes NA. The Role of mTOR in Amyotrophic Lateral Sclerosis. Biomedicines. 2025; 13(4):952. https://doi.org/10.3390/biomedicines13040952
Chicago/Turabian StyleNogueira-Machado, José Augusto, Fabiana Rocha-Silva, and Nathalia Augusta Gomes. 2025. "The Role of mTOR in Amyotrophic Lateral Sclerosis" Biomedicines 13, no. 4: 952. https://doi.org/10.3390/biomedicines13040952
APA StyleNogueira-Machado, J. A., Rocha-Silva, F., & Gomes, N. A. (2025). The Role of mTOR in Amyotrophic Lateral Sclerosis. Biomedicines, 13(4), 952. https://doi.org/10.3390/biomedicines13040952