



Comprehensive Integrated Analysis Reveals the Spatiotemporal Microevolution of Cancer Cells in Patients with Bone-Metastatic Prostate Cancer
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Result
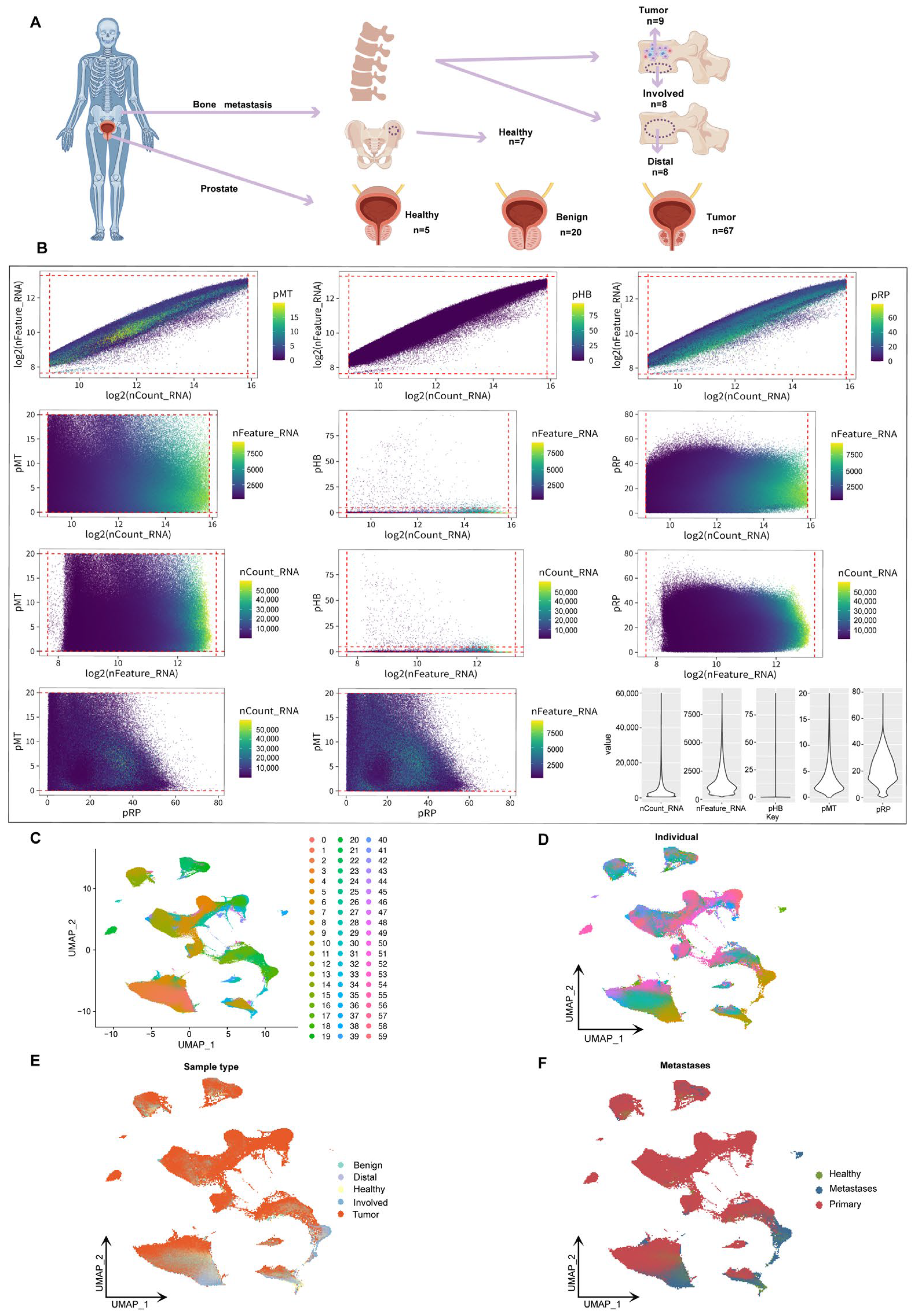
2.1. Sample Distribution and Data Integration in the Spatiotemporal Landscape of Prostate Cancer Bone Metastasis
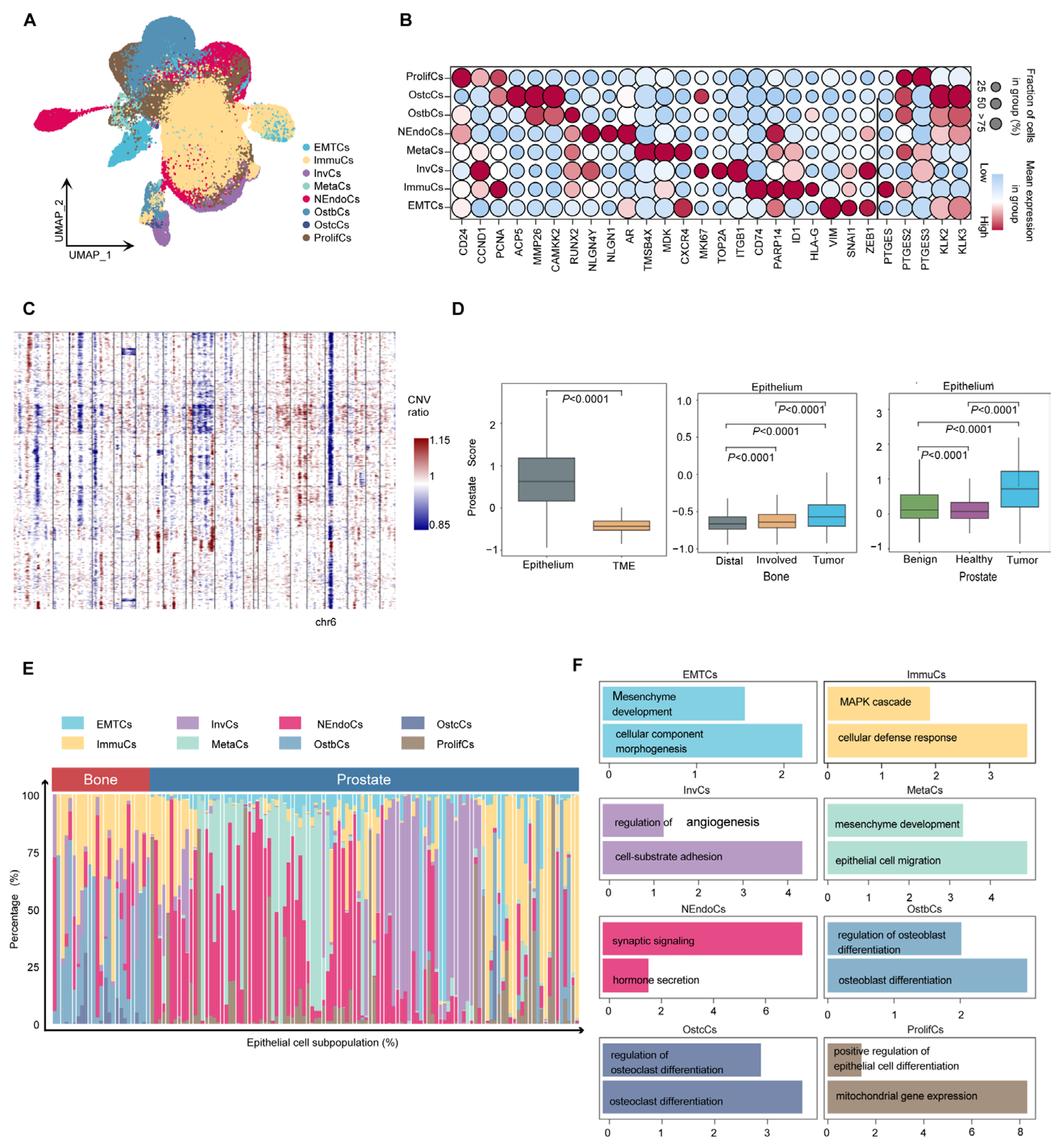
2.2. Single-Cell Landscape of Human Metastatic Prostate Cancer
2.3. Heterogeneity and Biological Characteristics of Epithelial Cells in Prostate Cancer
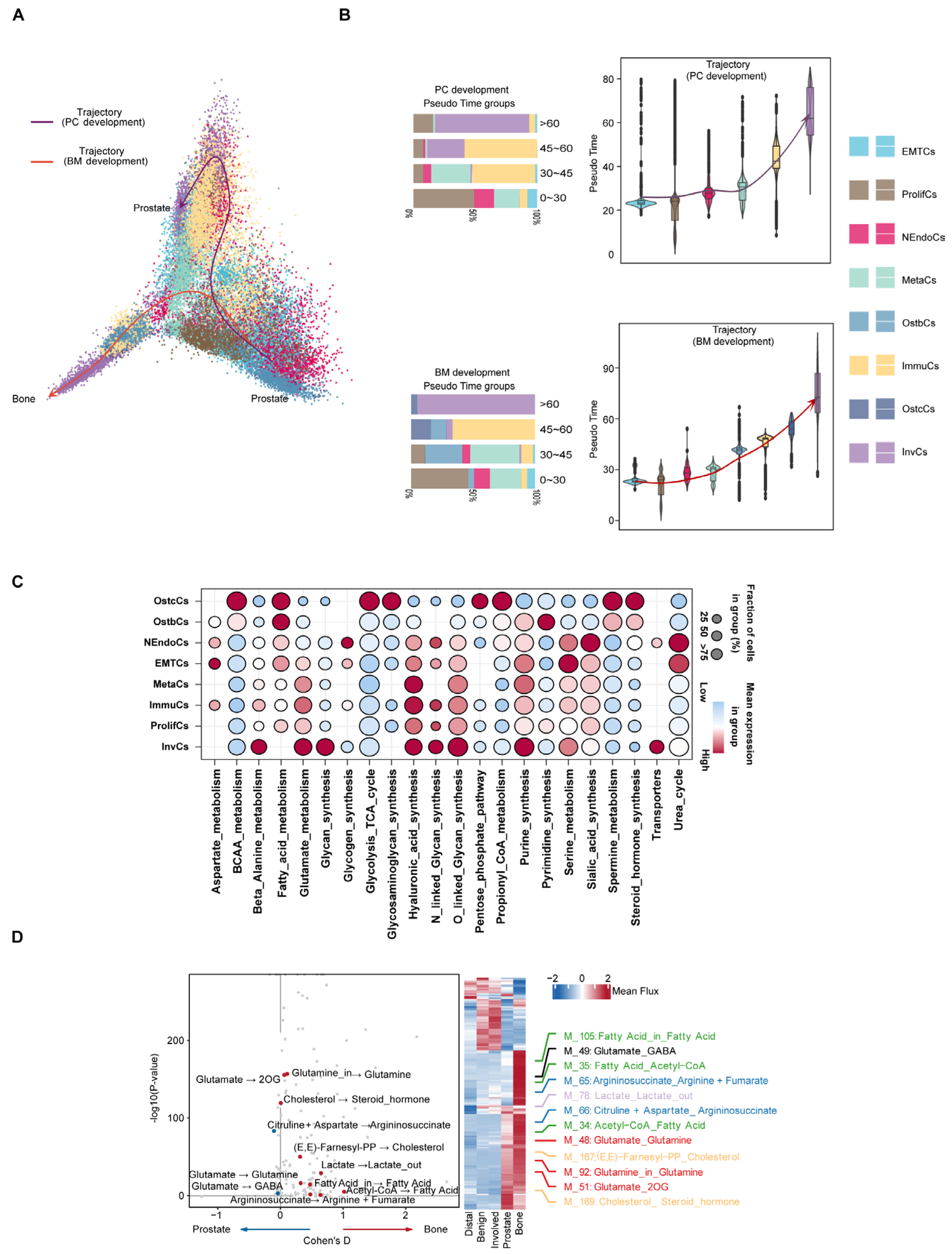
2.4. Inferring Metabolic Heterogeneity of Cancer Cells from scRNA-seq Data
2.5. Single-Cell Mutation Analysis Reveals the Microevolutionary Process of Cancer Cells
2.6. Cell Mutation Analysis Reveals the Mutation Landscape of Cancer Cells
3. Discussion
4. Methods
4.1. Patient Sample Collection
4.2. Single-Cell RNA Sequencing
4.3. Cell Type Annotation
4.4. Copy Number Variation Analysis
4.5. Calculation of Gene Signature Scores
4.6. Pathway Enrichment Analyses
4.7. Single-Cell Level Metabolic Analysis
4.8. De Novo Detection of Somatic Mutations
4.9. Pseudo Trajectory Inference Analyses
4.10. Pseudo-Evolution Tree
4.11. Pseudo-Bulk Differential Expression Analysis
4.12. TCGA Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zi, H.; Liu, M.-Y.; Luo, L.-S.; Huang, Q.; Luo, P.-C.; Luan, H.-H.; Huang, J.; Wang, D.-Q.; Wang, Y.-B.; Zhang, Y.-Y.; et al. Global burden of benign prostatic hyperplasia, urinary tract infections, urolithiasis, bladder cancer, kidney cancer, and prostate cancer from 1990 to 2021. Mil. Med. Res. 2024, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.J.; Laversanne, M.; Sung, H.; Soerjomataram, I.; Briganti, A.; Dahut, W.; Bray, F.; Jemal, A. Recent Patterns and Trends in Global Prostate Cancer Incidence and Mortality: An Update. Eur. Urol. 2024, 87, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Al Hussein Al Awamlh, B.; Wallis, C.J.D.; Penson, D.F.; Huang, L.-C.; Zhao, Z.; Conwill, R.; Talwar, R.; Morgans, A.K.; Goodman, M.; Hamilton, A.S.; et al. Functional Outcomes After Localized Prostate Cancer Treatment. JAMA 2024, 331, 302–317. [Google Scholar] [CrossRef]
- Berish, R.B.; Ali, A.N.; Telmer, P.G.; Ronald, J.A.; Leong, H.S. Translational models of prostate cancer bone metastasis. Nat. Rev. Urol. 2018, 15, 403–421. [Google Scholar] [CrossRef]
- Kang, J.; La Manna, F.; Bonollo, F.; Sampson, N.; Alberts, I.L.; Mingels, C.; Afshar-Oromieh, A.; Thalmann, G.N.; Karkampouna, S. Tumor microenvironment mechanisms and bone metastatic disease progression of prostate cancer. Cancer Lett. 2022, 530, 156–169. [Google Scholar] [CrossRef]
- Gebrael, G.; Jo, Y.; Swami, U.; Plets, M.; Hage Chehade, C.; Narang, A.; Gupta, S.; Myint, Z.W.; Sayegh, N.; Tangen, C.M.; et al. Bone Pain and Survival Among Patients with Metastatic, Hormone-Sensitive Prostate Cancer: A Secondary Analysis of the SWOG-1216 Trial. JAMA Netw. Open 2024, 7, e2419966. [Google Scholar] [CrossRef]
- Jakob, A.; Zahn, M.-O.; Nusch, A.; Werner, T.; Schnell, R.; Frank, M.; Hamm, N.; Däßler, K.-U.; Losem, C.; Welslau, M.; et al. Real-world patient-reported outcomes of breast cancer or prostate cancer patients receiving antiresorptive therapy for bone metastases: Final results of the PROBone registry study. J. Bone Oncol. 2022, 33, 100420. [Google Scholar] [CrossRef]
- Wilkinson, E.J.; Woodworth, A.M.; Parker, M.; Phillips, J.L.; Malley, R.C.; Dickinson, J.L.; Holloway, A.F. Epigenetic regulation of the ITGB4 gene in prostate cancer. Exp. Cell Res. 2020, 392, 112055. [Google Scholar] [CrossRef]
- Ren, S.; Li, J.; Dorado, J.; Sierra, A.; González-Díaz, H.; Duardo, A.; Shen, B. From molecular mechanisms of prostate cancer to translational applications: Based on multi-omics fusion analysis and intelligent medicine. Health Inf. Sci. Syst. 2024, 12, 6. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Chu, H.; Jin, Z.; Long, H.; Zhu, B. High-throughput single-cell sequencing in cancer research. Signal Transduct. Target Ther. 2022, 7, 145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, X.; Teng, F.; Yang, Q.; Wang, J.; Sun, B.; Liu, J.; Zhang, J.; Sun, X.; Zhao, H.; et al. Research progress and the prospect of using single-cell sequencing technology to explore the characteristics of the tumor microenvironment. Genes Dis. 2025, 12, 101239. [Google Scholar] [CrossRef]
- Fan, J.; Slowikowski, K.; Zhang, F. Single-cell transcriptomics in cancer: Computational challenges and opportunities. Exp. Mol. Med. 2020, 52, 1452–1465. [Google Scholar] [CrossRef]
- Lusby, R.; Demirdizen, E.; Inayatullah, M.; Kundu, P.; Maiques, O.; Zhang, Z.; Terp, M.G.; Sanz-Moreno, V.; Tiwari, V.K. Pan-cancer drivers of metastasis. Mol. Cancer 2025, 24, 2. [Google Scholar] [CrossRef]
- Bian, X.; Wang, W.; Abudurexiti, M.; Zhang, X.; Ma, W.; Shi, G.; Du, L.; Xu, M.; Wang, X.; Tan, C.; et al. Integration Analysis of Single-Cell Multi-Omics Reveals Prostate Cancer Heterogeneity. Adv. Sci. 2024, 11, e2305724. [Google Scholar] [CrossRef]
- Yu, W.; Wang, C.; Shang, Z.; Tian, J. Unveiling novel insights in prostate cancer through single-cell RNA sequencing. Front. Oncol. 2023, 13, 1224913. [Google Scholar] [CrossRef]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2024, 42, 293–304. [Google Scholar] [CrossRef]
- Ihle, C.L.; Provera, M.D.; Straign, D.M.; Smith, E.E.; Edgerton, S.M.; Van Bokhoven, A.; Lucia, M.S.; Owens, P. Distinct tumor microenvironments of lytic and blastic bone metastases in prostate cancer patients. J. Immunother. Cancer 2019, 7, 293. [Google Scholar] [CrossRef]
- Rucci, N.; Teti, A. Osteomimicry: How the Seed Grows in the Soil. Calcif. Tissue Int. 2018, 102, 131–140. [Google Scholar] [CrossRef]
- Mandelin, J.; Cardó-Vila, M.; Driessen, W.H.P.; Mathew, P.; Navone, N.M.; Lin, S.-H.; Logothetis, C.J.; Rietz, A.C.; Dobroff, A.S.; Proneth, B.; et al. Selection and identification of ligand peptides targeting a model of castrate-resistant osteogenic prostate cancer and their receptors. Proc. Natl. Acad. Sci. USA 2015, 112, 3776–3781. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-A.; Li, X.-L.; Mo, Y.-Z.; Fan, C.-M.; Tang, L.; Xiong, F.; Guo, C.; Xiang, B.; Zhou, M.; Ma, J.; et al. Effects of tumor metabolic microenvironment on regulatory T cells. Mol. Cancer 2018, 17, 168. [Google Scholar] [CrossRef]
- Ren, S.; Peng, Z.; Mao, J.-H.; Yu, Y.; Yin, C.; Gao, X.; Cui, Z.; Zhang, J.; Yi, K.; Xu, W.; et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res. 2012, 22, 806–821. [Google Scholar] [CrossRef]
- Lacher, S.B.; Dörr, J.; de Almeida, G.P.; Hönninger, J.; Bayerl, F.; Hirschberger, A.; Pedde, A.-M.; Meiser, P.; Ramsauer, L.; Rudolph, T.J.; et al. PGE2 limits effector expansion of tumour-infiltrating stem-like CD8+ T cells. Nature 2024, 629, 417–425. [Google Scholar] [CrossRef]
- Morotti, M.; Grimm, A.J.; Hope, H.C.; Arnaud, M.; Desbuisson, M.; Rayroux, N.; Barras, D.; Masid, M.; Murgues, B.; Chap, B.S.; et al. PGE2 inhibits TIL expansion by disrupting IL-2 signalling and mitochondrial function. Nature 2024, 629, 426–434. [Google Scholar] [CrossRef]
- Cao, T.; Zhang, W.; Wang, Q.; Wang, C.; Ma, W.; Zhang, C.; Ge, M.; Tian, M.; Yu, J.; Jiao, A.; et al. Cancer SLC6A6-mediated taurine uptake transactivates immune checkpoint genes and induces exhaustion in CD8+ T cells. Cell 2024, 187, 2288–2304.e27. [Google Scholar] [CrossRef]
- Luk, I.S.U.; Shrestha, R.; Xue, H.; Wang, Y.; Zhang, F.; Lin, D.; Haegert, A.; Wu, R.; Dong, X.; Collins, C.C.; et al. BIRC6 Targeting as Potential Therapy for Advanced, Enzalutamide-Resistant Prostate Cancer. Clin. Cancer Res. 2017, 23, 1542–1551. [Google Scholar] [CrossRef]
- Alghamdi, N.; Chang, W.; Dang, P.; Lu, X.; Wan, C.; Gampala, S.; Huang, Z.; Wang, J.; Ma, Q.; Zang, Y.; et al. A graph neural network model to estimate cell-wise metabolic flux using single-cell RNA-seq data. Genome Res. 2021, 31, 1867–1884. [Google Scholar] [CrossRef]
- Xue, R.; Zhang, Q.; Cao, Q.; Kong, R.; Xiang, X.; Liu, H.; Feng, M.; Wang, F.; Cheng, J.; Li, Z.; et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature 2022, 612, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Bhyravbhatla, N.; Kisling, A.; Li, X.; Batra, S.K.; Kumar, S. Cytokines chattering in pancreatic ductal adenocarcinoma tumor microenvironment. Semin. Cancer Biol. 2022, 86, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Lanzetti, L. Oncometabolites at the crossroads of genetic, epigenetic and ecological alterations in cancer. Cell Death Differ. 2024, 31, 1582–1594. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.-K.; Liu, J.; Chen, S.; Storrs, E.; Targino da Costa, A.L.N.; Houston, A.; Wendl, M.C.; Jayasinghe, R.G.; Iglesia, M.D.; Ma, C.; et al. Tumour evolution and microenvironment interactions in 2D and 3D space. Nature 2024, 634, 1178–1186. [Google Scholar] [CrossRef]
- Muyas, F.; Sauer, C.M.; Valle-Inclán, J.E.; Li, R.; Rahbari, R.; Mitchell, T.J.; Hormoz, S.; Cortés-Ciriano, I. De novo detection of somatic mutations in high-throughput single-cell profiling data sets. Nat. Biotechnol. 2024, 42, 758–767. [Google Scholar] [CrossRef]
- Li, C.; Wang, F.; Cui, L.; Li, S.; Zhao, J.; Liao, L. Association between abnormal lipid metabolism and tumor. Front. Endocrinol. 2023, 14, 1134154. [Google Scholar] [CrossRef]
- Liu, C.; Chikina, M.; Deshpande, R.; Menk, A.V.; Wang, T.; Tabib, T.; Brunazzi, E.A.; Vignali, K.M.; Sun, M.; Stolz, D.B.; et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8+ T Cell-Derived Interferon-γ. Immunity 2019, 51, 381–397.e6. [Google Scholar] [CrossRef]
- Verbruggen, S.W.; Nolan, J.; Duffy, M.P.; Pearce, O.M.T.; Jacobs, C.R.; Knight, M.M. A Novel Primary Cilium-Mediated Mechanism Through which Osteocytes Regulate Metastatic Behavior of Both Breast and Prostate Cancer Cells. Adv. Sci. 2024, 11, e2305842. [Google Scholar] [CrossRef]
- Wang, J.; He, Y.; Hu, F.; Hu, C.; Sun, Y.; Yang, K.; Yang, S. Metabolic Reprogramming of Immune Cells in the Tumor Microenvironment. Int. J. Mol. Sci. 2024, 25, 12223. [Google Scholar] [CrossRef]
- Whitburn, J.; Edwards, C.M. Metabolism in the Tumour-Bone Microenvironment. Curr. Osteoporos. Rep. 2021, 19, 494–499. [Google Scholar] [CrossRef]
- Ma, X.; Yu, J. Role of the bone microenvironment in bone metastasis of malignant tumors—Therapeutic implications. Cell. Oncol. 2020, 43, 751–761. [Google Scholar] [CrossRef]
- Greenfield, A.; Scott, D.; Pennisi, D.; Ehrmann, I.; Ellis, P.; Cooper, L.; Simpson, E.; Koopman, P. An H-YDb epitope is encoded by a novel mouse Y chromosome gene. Nat. Genet. 1996, 14, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.H.; Furlong, R.A.; Burkin, H.; Chalmers, I.J.; Brown, G.M.; Khwaja, O.; Affara, N.A. The Drosophila developmental gene fat facets has a human homologue in Xp11.4 which escapes X-inactivation and has related sequences on Yq11.2. Hum. Mol. Genet. 1996, 5, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Walport, L.J.; Hopkinson, R.J.; Vollmar, M.; Madden, S.K.; Gileadi, C.; Oppermann, U.; Schofield, C.J.; Johansson, C. Human UTY(KDM6C) is a male-specific Nϵ-methyl lysyl demethylase. J. Biol. Chem. 2014, 289, 18302–18313. [Google Scholar] [CrossRef]
- Lee, K.H.; Song, G.J.; Kang, I.S.; Kim, S.W.; Paick, J.-S.; Chung, C.H.; Rhee, K. Ubiquitin-specific protease activity of USP9Y, a male infertility gene on the Y chromosome. Reprod. Fertil. Dev. 2003, 15, 129–133. [Google Scholar] [CrossRef]
- Shpargel, K.B.; Sengoku, T.; Yokoyama, S.; Magnuson, T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012, 8, e1002964. [Google Scholar] [CrossRef]
- Luddi, A.; Margollicci, M.; Gambera, L.; Serafini, F.; Cioni, M.; De Leo, V.; Balestri, P.; Piomboni, P. Spermatogenesis in a man with complete deletion of USP9Y. N. Engl. J. Med. 2009, 360, 881–885. [Google Scholar] [CrossRef]
- Tang, F.; Li, J.; Qi, L.; Liu, D.; Bo, Y.; Qin, S.; Miao, Y.; Yu, K.; Hou, W.; Li, J.; et al. A pan-cancer single-cell panorama of human natural killer cells. Cell 2023, 186, 4235–4251.e20. [Google Scholar] [CrossRef]
- Zheng, L.; Qin, S.; Si, W.; Wang, A.; Xing, B.; Gao, R.; Ren, X.; Wang, L.; Wu, X.; Zhang, J.; et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021, 374, abe6474. [Google Scholar] [CrossRef]
- Glass, D.R.; Tsai, A.G.; Oliveria, J.P.; Hartmann, F.J.; Kimmey, S.C.; Calderon, A.A.; Borges, L.; Glass, M.C.; Wagar, L.E.; Davis, M.M.; et al. An Integrated Multi-omic Single-Cell Atlas of Human B Cell Identity. Immunity 2020, 53, 217–232.e5. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Q.; Xing, B.; Luo, N.; Gao, R.; Yu, K.; Hu, X.; Bu, Z.; Peng, J.; Ren, X.; et al. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell 2022, 40, 424–437.e5. [Google Scholar] [CrossRef] [PubMed]
- St Croix, B.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montgomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Vogelstein, B.; et al. Genes expressed in human tumor endothelium. Science 2000, 289, 1197–1202. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovations 2021, 2, 100141. [Google Scholar] [CrossRef]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef]
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef]
- Wolf, F.A.; Hamey, F.K.; Plass, M.; Solana, J.; Dahlin, J.S.; Göttgens, B.; Rajewsky, N.; Simon, L.; Theis, F.J. PAGA: Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019, 20, 59. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Y.; Zhang, X.; Wang, G.; Yang, F.; Li, R.; Yin, L.; Chen, D.; Wang, W.; Wang, M.; Hu, Z.; et al. Comprehensive Integrated Analysis Reveals the Spatiotemporal Microevolution of Cancer Cells in Patients with Bone-Metastatic Prostate Cancer. Biomedicines 2025, 13, 909. https://doi.org/10.3390/biomedicines13040909
Feng Y, Zhang X, Wang G, Yang F, Li R, Yin L, Chen D, Wang W, Wang M, Hu Z, et al. Comprehensive Integrated Analysis Reveals the Spatiotemporal Microevolution of Cancer Cells in Patients with Bone-Metastatic Prostate Cancer. Biomedicines. 2025; 13(4):909. https://doi.org/10.3390/biomedicines13040909
Chicago/Turabian StyleFeng, Yinghua, Xiuli Zhang, Guangpeng Wang, Feiya Yang, Ruifang Li, Lu Yin, Dong Chen, Wenkuan Wang, Mingshuai Wang, Zhiyuan Hu, and et al. 2025. "Comprehensive Integrated Analysis Reveals the Spatiotemporal Microevolution of Cancer Cells in Patients with Bone-Metastatic Prostate Cancer" Biomedicines 13, no. 4: 909. https://doi.org/10.3390/biomedicines13040909
APA StyleFeng, Y., Zhang, X., Wang, G., Yang, F., Li, R., Yin, L., Chen, D., Wang, W., Wang, M., Hu, Z., Sh, Y., & Xing, N. (2025). Comprehensive Integrated Analysis Reveals the Spatiotemporal Microevolution of Cancer Cells in Patients with Bone-Metastatic Prostate Cancer. Biomedicines, 13(4), 909. https://doi.org/10.3390/biomedicines13040909