

Integrative Bioinformatic Analysis of Cellular Senescence Genes in Ovarian Cancer: Molecular Subtyping, Prognostic Risk Stratification, and Chemoresistance Prediction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Preprocessing
2.2. Differential Expression Profiling
2.3. Molecular Subtyping Driven by Senescence Signatures
2.4. Prognostic Model Construction
2.5. Hub Genes Identification and Nomogram Construction
2.6. Functional and Genomic Characterization of Risk Groups
2.7. Immune Infiltration
2.8. Drug Sensitivity Analysis
2.9. Statistical Methods
3. Results
3.1. Transcriptomic Landscape of DE-CSGs
3.2. CNV Analysis of DE-CSGs
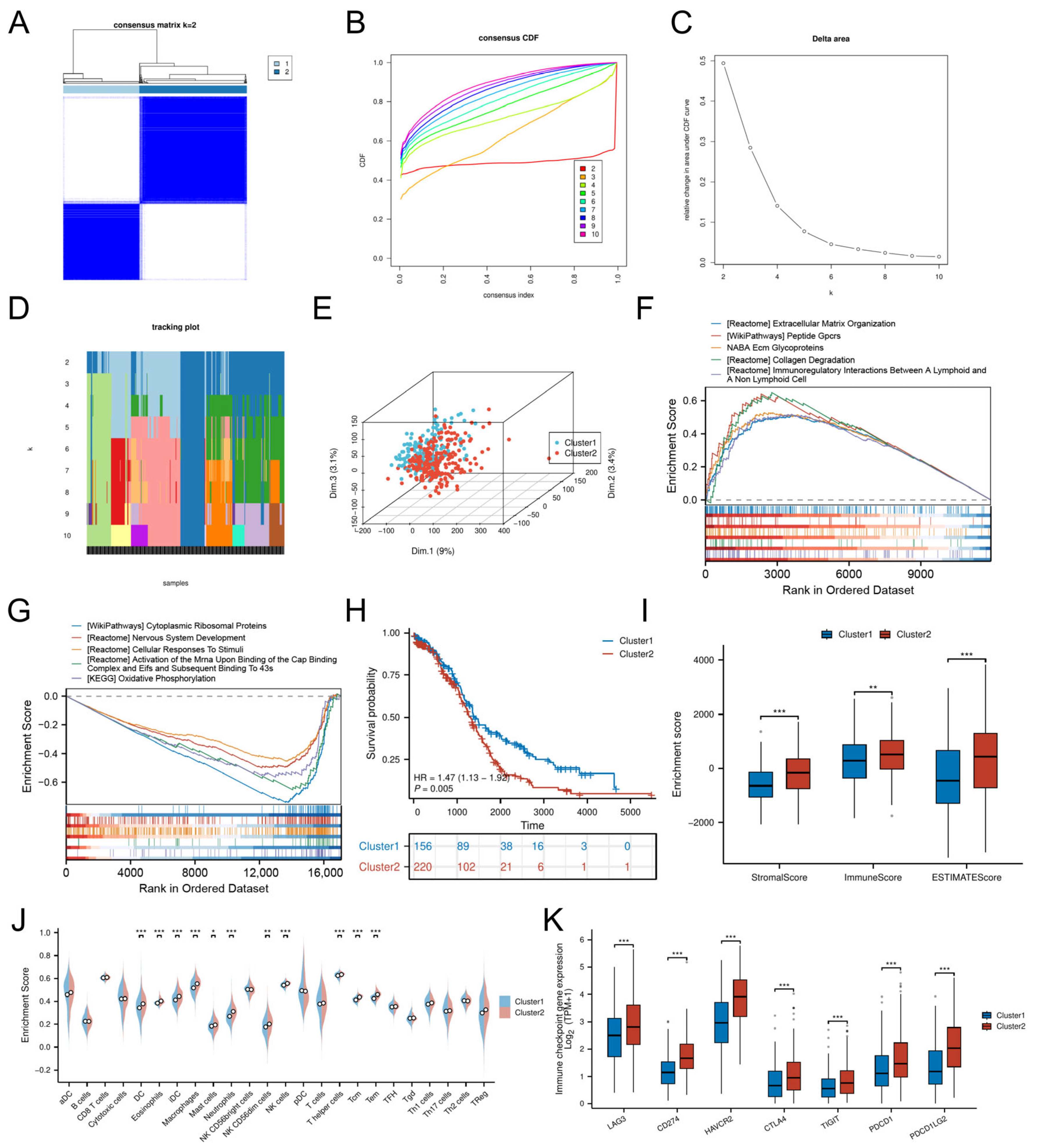
3.3. Cellular Senescence-Driven Molecular Subtyping
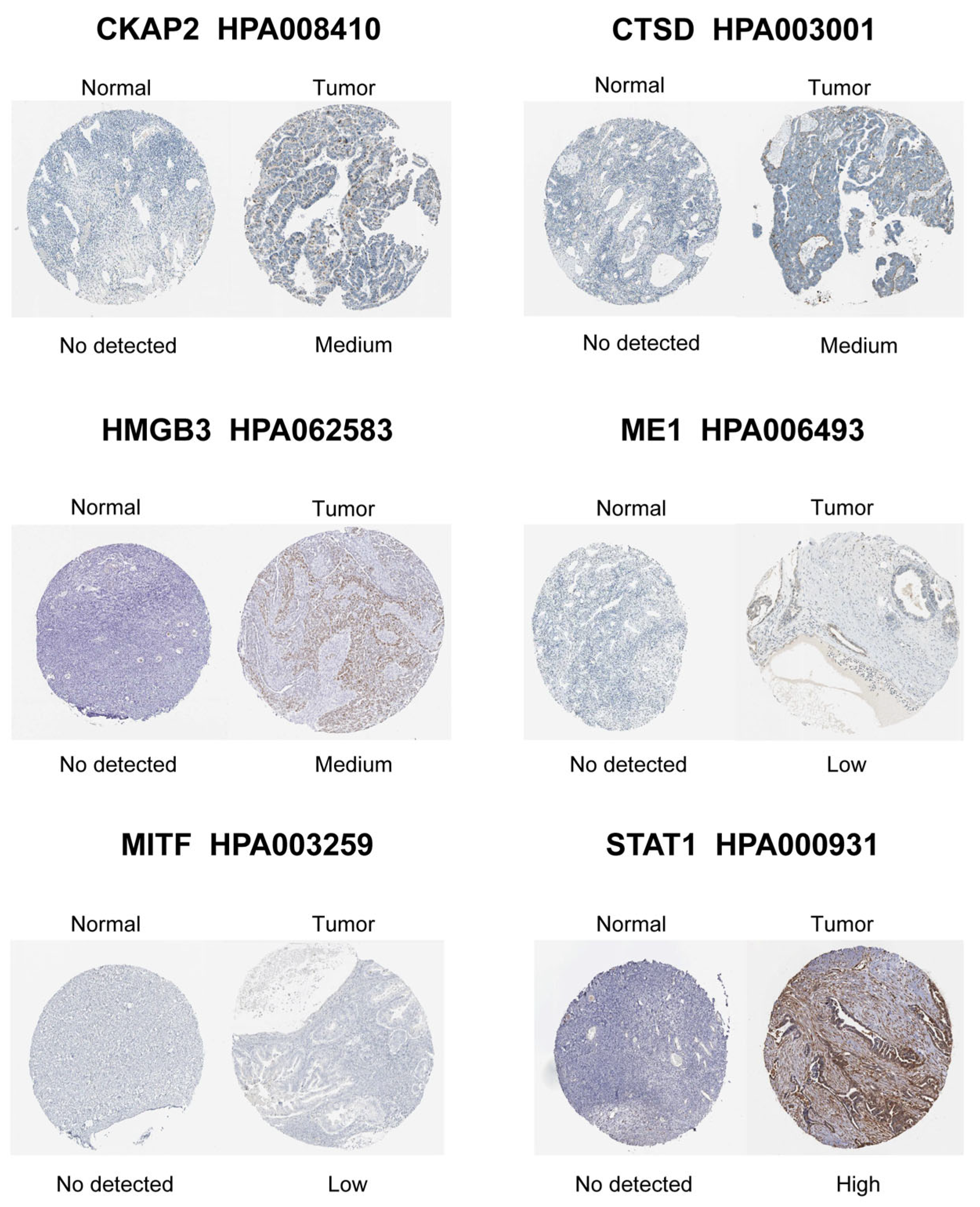
3.4. Identification and Validation of Hub CSGs
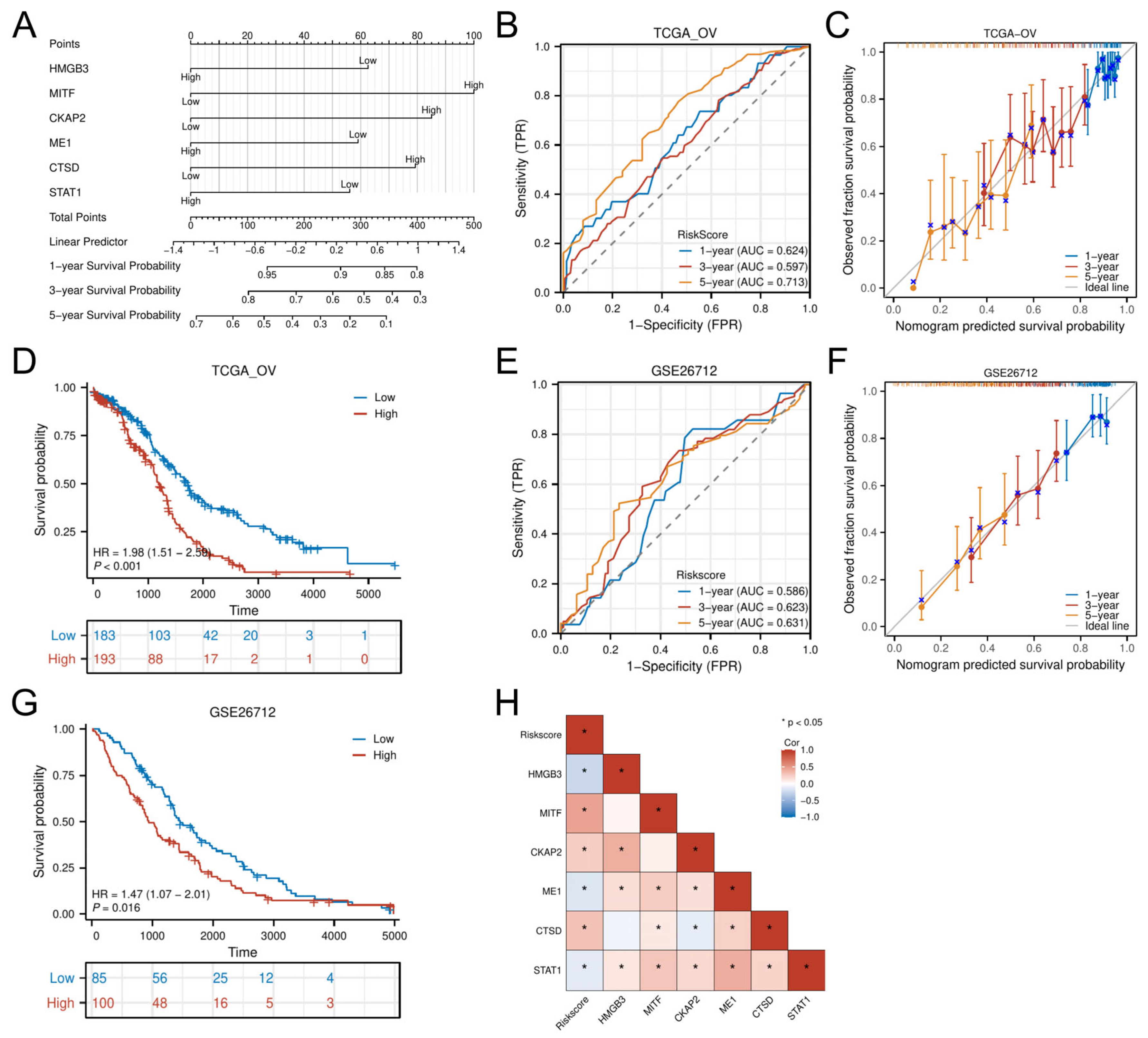
3.5. Nomogram Development and External Validation
3.6. Mutational Landscape and TMB-Driven Prognostic Stratification
3.7. Functional Pathway Enrichment in Risk Groups
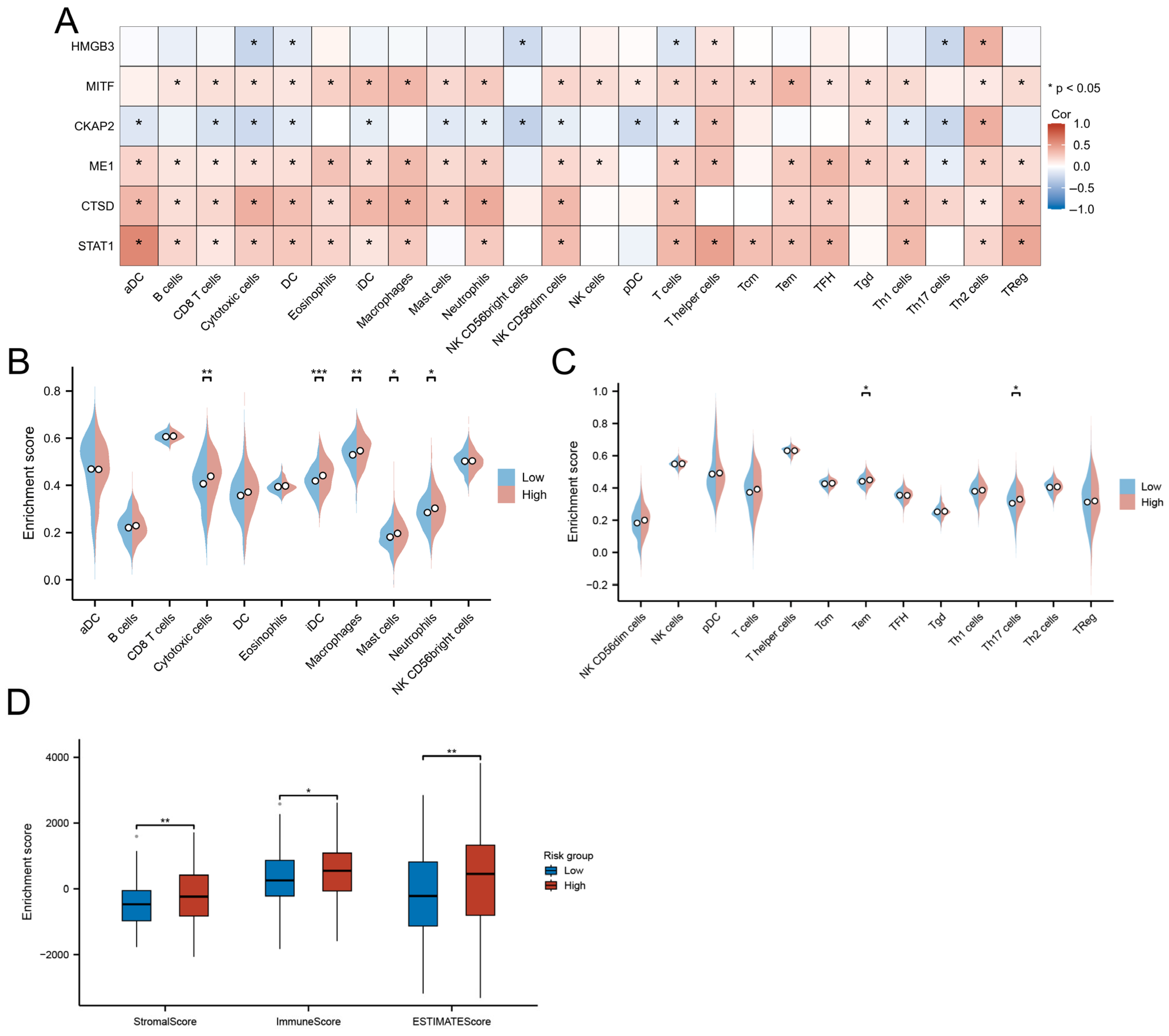
3.8. Immune Landscape and TME Characteristics
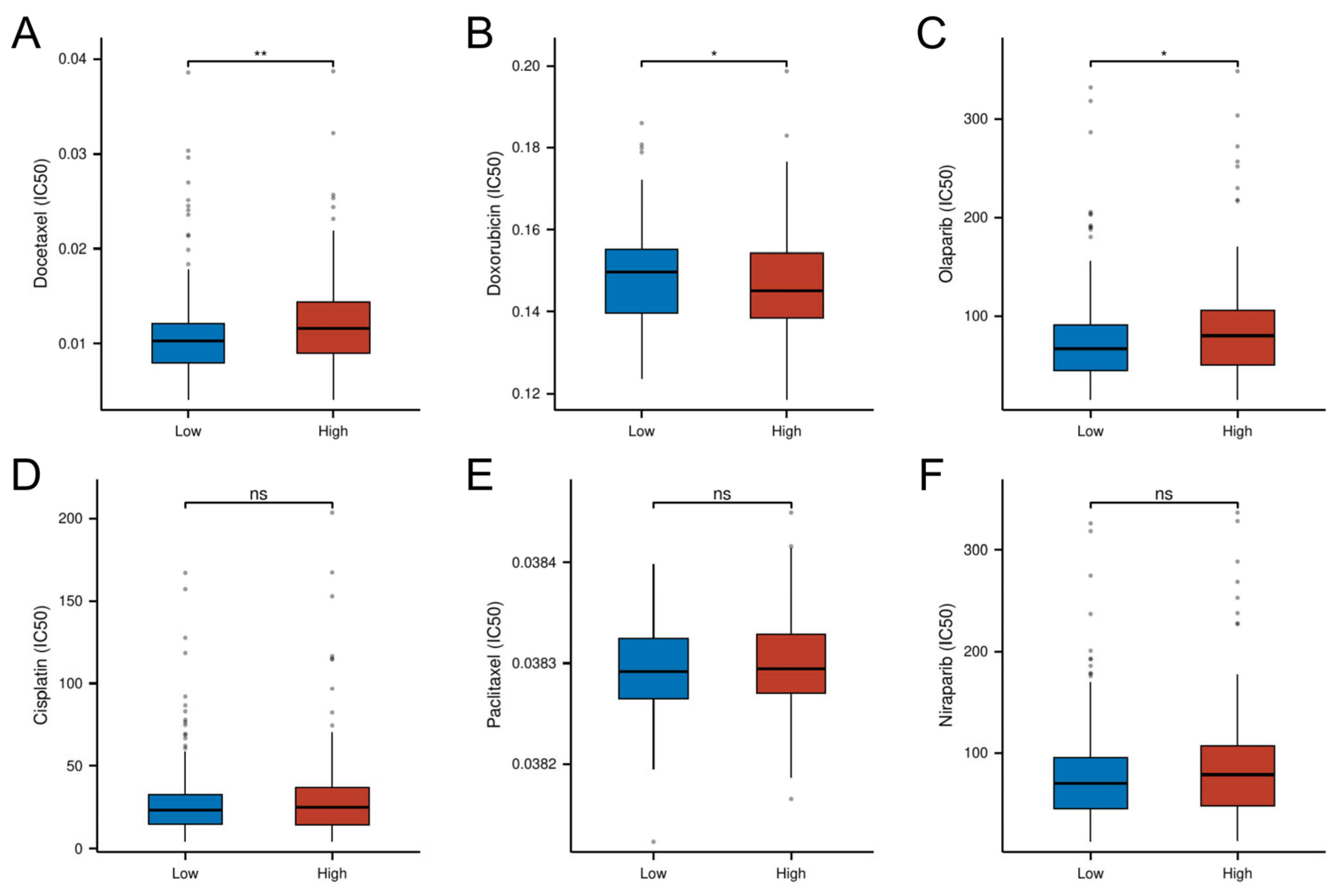
3.9. Risk-Stratified Drug Sensitivity Patterns
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OC | Ovarian cancer |
| SASP | Senescence-associated secretory phenotype |
| TME | Tumor microenvironment |
| CSGs | Cellular senescence-related genes |
| CNV | Copy number variation |
| FC | FoldChange |
| DEGs | Differentially expressed genes |
| OS | Overall survival |
| GO | Gene Ontology (GO) |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DE-CSGs | Differentially expressed cellular senescence-related genes |
| PCA | Principal component analysis |
| GSEA | Gene Set Enrichment Analysis |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| K–M | Kaplan–Meier |
| TMB | Tumor mutation burden |
| IC50 | Half-maximal inhibitory concentration |
| ECM | Extracellular matrix |
| DC | Dendritic cell |
| iDC | Immature dendritic cell |
| AUC | Area under curve |
| GPCRs | G-protein-coupled receptors |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, S.; Ikeda, K.; Horie-Inoue, K.; Sato, S.; Itakura, A.; Takeda, S.; Hasegawa, K.; Inoue, S. Systematic Identification of Characteristic Genes of Ovarian Clear Cell Carcinoma Compared with High-Grade Serous Carcinoma Based on RNA-Sequencing. Int. J. Mol. Sci. 2019, 20, 4330. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Laberge, R.M.; Awad, P.; Campisi, J.; Desprez, P.Y. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2012, 5, 39–44. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Wang, T.; Notta, F.; Navab, R.; Joseph, J.; Ibrahimov, E.; Xu, J.; Zhu, C.Q.; Borgida, A.; Gallinger, S.; Tsao, M.S. Senescent Carcinoma-Associated Fibroblasts Upregulate IL8 to Enhance Prometastatic Phenotypes. Mol. Cancer Res. MCR 2017, 15, 3–14. [Google Scholar] [CrossRef]
- Capparelli, C.; Guido, C.; Whitaker-Menezes, D.; Bonuccelli, G.; Balliet, R.; Pestell, T.G.; Goldberg, A.F.; Pestell, R.G.; Howell, A.; Sneddon, S.; et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle 2012, 11, 2285–2302. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Jana, S.; Chatterjee, S.; Wastall, L.M.; Mandal, G.; Nargis, N.; Roy, H.; Hughes, T.A.; Bhattacharyya, A. MicroRNA-222 reprogrammed cancer-associated fibroblasts enhance growth and metastasis of breast cancer. Br. J. Cancer 2019, 121, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Avelar, R.A.; Ortega, J.G.; Tacutu, R.; Tyler, E.J.; Bennett, D.; Binetti, P.; Budovsky, A.; Chatsirisupachai, K.; Johnson, E.; Murray, A.; et al. A multidimensional systems biology analysis of cellular senescence in aging and disease. Genome Biol. 2020, 21, 91. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Gundem, G.; Lopez-Bigas, N. Sample-level enrichment analysis unravels shared stress phenotypes among multiple cancer types. Genome Med. 2012, 4, 28. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.H.; Park, T.J. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 2017, 8, 15208. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, T.; Zhang, S.; Yang, T.; Gong, Y.; Deng, H.W.; Bai, D.; Tian, W.; Chen, Y. Discovery and functional assessment of a novel adipocyte population driven by intracellular Wnt/β-catenin signaling in mammals. eLife 2022, 11, e77740. [Google Scholar] [CrossRef]
- Zheng, J.H.; Lin, S.R.; Tseng, F.J.; Tsai, M.J.; Lue, S.I.; Chia, Y.C.; Woon, M.; Fu, Y.S.; Weng, C.F. Clerodane Diterpene Ameliorates Inflammatory Bowel Disease and Potentiates Cell Apoptosis of Colorectal Cancer. Biomolecules 2019, 9, 762. [Google Scholar] [CrossRef]
- Rao, S.; Gurbani, D.; Du, G.; Everley, R.A.; Browne, C.M.; Chaikuad, A.; Tan, L.; Schröder, M.; Gondi, S.; Ficarro, S.B.; et al. Leveraging Compound Promiscuity to Identify Targetable Cysteines within the Kinome. Cell Chem. Biol. 2019, 26, 818–829.e819. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, J.; Wang, Y.; Lee, Y.; Zhao, X.; Lu, H.; Zhu, G.; Wang, H.; Jiang, Y.; Liu, F.; et al. The FoxO4/DKK3 axis represses IFN-γ expression by Th1 cells and limits antimicrobial immunity. J. Clin. Investig. 2022, 132, e147566. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zhou, L.; Liu, H.; Shan, Y.; Zhang, X. MicroRNA-224 Promotes Pancreatic Cancer Cell Proliferation and Migration by Targeting the TXNIP-Mediated HIF1α Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 48, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- Fei, Q.; Jin, K.; Shi, S.; Li, T.; Guo, D.; Lin, M.; Yu, X.; Wu, W.; Ye, L. Suppression of pancreatic cancer proliferation through TXNIP-mediated inhibition of the MAPK signaling pathway. Acta Biochim. Biophys. Sin. 2024, 56, 513–524. [Google Scholar] [CrossRef]
- Uota, A.; Okuno, Y.; Fukuhara, A.; Sasaki, S.; Kobayashi, S.; Shimomura, I. ARMC5 selectively degrades SCAP-free SREBF1 and is essential for fatty acid desaturation in adipocytes. J. Biol. Chem. 2024, 300, 107953. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, X.; Liu, Y.; Liu, P.; Qin, J.; Zhu, Y.; Zhai, S.; Jiang, Y.; Liu, Y.; Han, L.; et al. BHLHE40 Inhibits Ferroptosis in Pancreatic Cancer Cells via Upregulating SREBF1. Adv. Sci. 2024, 11, e2306298. [Google Scholar] [CrossRef]
- Seager, R.J.; Hajal, C.; Spill, F.; Kamm, R.D.; Zaman, M.H. Dynamic interplay between tumour, stroma and immune system can drive or prevent tumour progression. Converg. Sci. Phys. Oncol. 2017, 3, 034002. [Google Scholar] [CrossRef]
- Alonso-Nocelo, M.; Raimondo, T.M.; Vining, K.H.; López-López, R.; de la Fuente, M.; Mooney, D.J. Matrix stiffness and tumor-associated macrophages modulate epithelial to mesenchymal transition of human adenocarcinoma cells. Biofabrication 2018, 10, 035004. [Google Scholar] [CrossRef]
- Wang, Y.; Peng, B.; Ning, C.; He, S.; Yang, H.; Mao, Y.; Sun, L. Characterization of immune features and immunotherapy response in subtypes of hepatocellular carcinoma based on mitophagy. Front. Immunol. 2022, 13, 966167. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- van der Reest, J.; Lilla, S.; Zheng, L.; Zanivan, S.; Gottlieb, E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, S.; Bai, Y.; Li, G.; Wang, S.; Chen, J.; Liu, X.; Yin, H. Development and Validation of Ferroptosis-Related LncRNA Biomarker in Bladder Carcinoma. Front. Cell Dev. Biol. 2022, 10, 809747. [Google Scholar] [CrossRef]
- Wang, J.J.; Siu, M.K.; Jiang, Y.X.; Leung, T.H.; Chan, D.W.; Wang, H.G.; Ngan, H.Y.; Chan, K.K. A Combination of Glutaminase Inhibitor 968 and PD-L1 Blockade Boosts the Immune Response against Ovarian Cancer. Biomolecules 2021, 11, 1749. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Qi, G.; Han, F.; Lu, W.; Peng, J.; Li, R.; Yan, S.; Yuan, C.; Kong, B. HMGB3 promotes PARP inhibitor resistance through interacting with PARP1 in ovarian cancer. Cell Death Dis. 2022, 13, 263. [Google Scholar] [CrossRef]
- Ma, H.; Qi, G.; Han, F.; Gai, P.; Peng, J.; Kong, B. HMGB3 promotes the malignant phenotypes and stemness of epithelial ovarian cancer through the MAPK/ERK signaling pathway. Cell Commun. Signal. CCS 2023, 21, 144. [Google Scholar] [CrossRef]
- Mukherjee, A.; Huynh, V.; Gaines, K.; Reh, W.A.; Vasquez, K.M. Targeting the High-Mobility Group Box 3 Protein Sensitizes Chemoresistant Ovarian Cancer Cells to Cisplatin. Cancer Res. 2019, 79, 3185–3191. [Google Scholar] [CrossRef]
- Kitami, K.; Yoshihara, M.; Koya, Y.; Sugiyama, M.; Iyoshi, S.; Uno, K.; Mogi, K.; Tano, S.; Fujimoto, H.; Nawa, A.; et al. Microphthalmia-Associated Transcription Factor-Dependent Melanoma Cell Adhesion Molecule Activation Promotes Peritoneal Metastasis of Ovarian Cancer. Int. J. Mol. Sci. 2020, 21, 9776. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, L. CKAP2 Promotes Ovarian Cancer Proliferation and Tumorigenesis Through the FAK-ERK Pathway. DNA Cell Biol. 2017, 36, 983–990. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, W.; Wang, H.; Yang, W. Identification of CKAP2 as a Potential Target for Prevention of Gastric Cancer Progression: A Multi-Omics Study. Int. J. Mol. Sci. 2025, 26, 1557. [Google Scholar] [CrossRef]
- Paim, L.M.G.; Lopez-Jauregui, A.A.; McAlear, T.S.; Bechstedt, S. The spindle protein CKAP2 regulates microtubule dynamics and ensures faithful chromosome segregation. Proc. Natl. Acad. Sci. USA 2024, 121, e2318782121. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, F.; Liu, F.; Xu, G. Predicting STAT1 as a prognostic marker in patients with solid cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920917558. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, S.; Gaire, B.; Zou, Y.; Uddin, M.M.; Vancurova, I. IFNγ-induced PD-L1 expression in ovarian cancer cells is regulated by JAK1, STAT1 and IRF1 signaling. Cell. Signal. 2022, 97, 110400. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, S.; Xu, J.; Wei, J.; Wu, Y. WTAP contributes to platinum resistance in high-grade serous ovarian cancer by up-regulating malic acid: Insights from liquid chromatography and mass spectrometry analysis. Cancer Metab. 2025, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Sowamber, R.; Nelson, O.; Dodds, L.; DeCastro, V.; Paudel, I.; Milea, A.; Considine, M.; Cope, L.; Pinto, A.; Schlumbrecht, M.; et al. Integrative Transcriptome Analyses of the Human Fallopian Tube: Fimbria and Ampulla-Site of Origin of Serous Carcinoma of the Ovary. Cancers 2020, 12, 1090. [Google Scholar] [CrossRef]
- Ketterer, S.; Mitschke, J.; Ketscher, A.; Schlimpert, M.; Reichardt, W.; Baeuerle, N.; Hess, M.E.; Metzger, P.; Boerries, M.; Peters, C.; et al. Cathepsin D deficiency in mammary epithelium transiently stalls breast cancer by interference with mTORC1 signaling. Nat. Commun. 2020, 11, 5133. [Google Scholar] [CrossRef]
- Scheidemann, E.R.; Demas, D.M.; Hou, C.; Ma, J.; He, W.; Sharma, G.; Schultz, E.; Weilbaecher, K.N.; Shajahan-Haq, A.N. Resistance to abemaciclib is associated with increased metastatic potential and lysosomal protein deregulation in breast cancer cells. Mol. Carcinog. 2024, 63, 209–223. [Google Scholar] [CrossRef]
- Taraszka, K.; Groha, S.; King, D.; Tell, R.; White, K.; Ziv, E.; Zaitlen, N.; Gusev, A. A comprehensive analysis of clinical and polygenic germline influences on somatic mutational burden. Am. J. Hum. Genet. 2024, 111, 242–258. [Google Scholar] [CrossRef]
- Stenzinger, A.; Allen, J.D.; Maas, J.; Stewart, M.D.; Merino, D.M.; Wempe, M.M.; Dietel, M. Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer 2019, 58, 578–588. [Google Scholar] [CrossRef]
- Wang, X.; Li, M. Correlate tumor mutation burden with immune signatures in human cancers. BMC Immunol. 2019, 20, 4. [Google Scholar] [CrossRef]
- Qiu, G.H.; Yu, B.; Ma, M. G protein-coupled receptor-mediated signaling of immunomodulation in tumor progression. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2024, 38, e23829. [Google Scholar] [CrossRef]
- Liu, Y.; An, S.; Ward, R.; Yang, Y.; Guo, X.X.; Li, W.; Xu, T.R. G protein-coupled receptors as promising cancer targets. Cancer Lett. 2016, 376, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.K.; Mukherjee, S. Consequences of Extracellular Matrix Remodeling in Headway and Metastasis of Cancer along with Novel Immunotherapies: A Great Promise for Future Endeavor. Anti-Cancer Agents Med. Chem. 2022, 22, 1257–1271. [Google Scholar] [CrossRef]
- Du, W.; Xia, X.; Hu, F.; Yu, J. Extracellular matrix remodeling in the tumor immunity. Front. Immunol. 2023, 14, 1340634. [Google Scholar] [CrossRef]
- Scott, E.N.; Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Regulatory T Cells: Barriers of Immune Infiltration Into the Tumor Microenvironment. Front. Immunol. 2021, 12, 702726. [Google Scholar] [CrossRef]
- Lorusso, G.; Rüegg, C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem. Cell Biol. 2008, 130, 1091–1103. [Google Scholar] [CrossRef]
- Huang, Y.K.; Busuttil, R.A.; Boussioutas, A. The Role of Innate Immune Cells in Tumor Invasion and Metastasis. Cancers 2021, 13, 5885. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, A.; Xu, D. Integrative Bioinformatic Analysis of Cellular Senescence Genes in Ovarian Cancer: Molecular Subtyping, Prognostic Risk Stratification, and Chemoresistance Prediction. Biomedicines 2025, 13, 877. https://doi.org/10.3390/biomedicines13040877
Li A, Xu D. Integrative Bioinformatic Analysis of Cellular Senescence Genes in Ovarian Cancer: Molecular Subtyping, Prognostic Risk Stratification, and Chemoresistance Prediction. Biomedicines. 2025; 13(4):877. https://doi.org/10.3390/biomedicines13040877
Chicago/Turabian StyleLi, Ailian, and Dianbo Xu. 2025. "Integrative Bioinformatic Analysis of Cellular Senescence Genes in Ovarian Cancer: Molecular Subtyping, Prognostic Risk Stratification, and Chemoresistance Prediction" Biomedicines 13, no. 4: 877. https://doi.org/10.3390/biomedicines13040877
APA StyleLi, A., & Xu, D. (2025). Integrative Bioinformatic Analysis of Cellular Senescence Genes in Ovarian Cancer: Molecular Subtyping, Prognostic Risk Stratification, and Chemoresistance Prediction. Biomedicines, 13(4), 877. https://doi.org/10.3390/biomedicines13040877