
From Discovery to Innovative Translational Approaches in 80 Years of Fragile X Syndrome Research



Abstract
1. Introduction
2. From Discovery to Translation: 80 Years of Progress in FXS Research
3. Understanding the Clinical Phenotype of FXS: Cognitive and Physical Manifestations
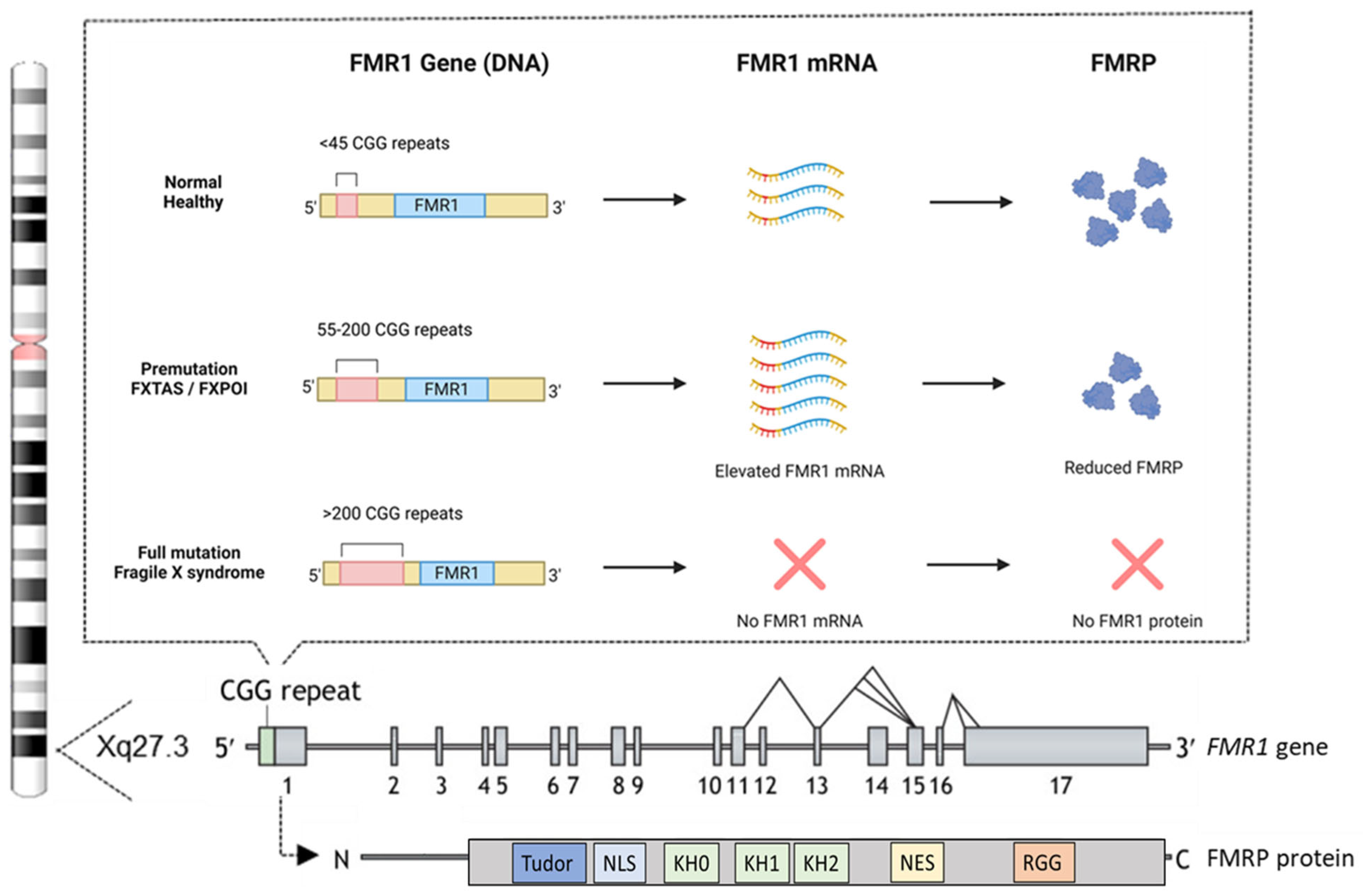
4. Genetic Basis of FXS: The Role of the FMR1 Gene and FMRP Protein
5. Modeling FXS in Mice: Insights into the Pathophysiology of FXS
6. Disrupted Pathways in FXS: A Complex Network of Affected Pathways and Targets
6.1. Modulating the Inhibitory GABAergic System in FXS: GABAA and GABAB Receptors
6.2. The Excitatory Glutamatergic System in FXS: From Potential Cure to Failure
6.3. Phosphodiesterase-4D (PDE4D) in FXS: From Discovery in 1990s to Phase 3 Clinical Trials
6.4. Exploring the Potential of CBD Transdermal Gel in FXS
6.5. Metformin: Can the Repurposed “Wonder Drug” Also Benefit FXS?
6.6. Unlocking New Possibilities: BK Channel Modulators in FXS
7. Novel Innovative Approaches: Combination and Gene Therapies
7.1. AI-Driven Combination Therapies Targeting Multiple Distinct Affected Pathways in FXS
7.2. Adeno-Associated Virus (AAV) Vectors to Restore FMR1 Function
7.3. R-Loop Driven Reactivation of FMR1 in FXS
7.4. Advances in CRISPR-Based Reactivation of FMR1 in FXS
7.5. ASO Therapy for FXS
7.6. Challenges of Brain-Targeted Gene Therapy in FXS
8. Bridging the Gap: Overcoming Challenges in Translating FXS Treatments
8.1. Preclinical Testing Protocols
8.2. Clinical Trial Designs
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Keith, K.; Albizua, I.; Malone, T.; Mowrey, J.; Sherman, S.L.; Warren, S.T. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am. J. Hum. Genet. 2009, 85, 503–514. [Google Scholar] [PubMed]
- Genovese, A.C.; Butler, M.G. Systematic Review: Fragile X Syndrome Across the Lifespan with a Focus on Genetics, Neurodevelopmental, Behavioral and Psychiatric Associations. Genes 2025, 16, 149. [Google Scholar] [CrossRef]
- Martin, J.P.; Bell, J. A pedigree of mental defect showing sex-linkage. J. Neurol. Psychiatry 1943, 6, 154–157. [Google Scholar] [PubMed]
- Salvador-Carulla, L.; Reed, G.M.; Vaez-Azizi, L.M.; Cooper, S.-A.; Martinez-Leal, R.; Bertelli, M.; Bertelli, M.; Adnams, C.; Cooray, S.; Deb, S.; et al. Intellectual developmental disorders: Towards a new name, definition and framework for “mental retardation/intellectual disability” in ICD-11. World Psychiatry 2011, 10, 175–180. [Google Scholar]
- Lubs, H.A. A marker X chromosome. Am. J. Hum. Genet. 1969, 21, 231–244. [Google Scholar]
- Richards, B.W.; Sylvester, P.E.; Brooker, C. Fragile X-linked mental retardation: The Martin-Bell syndrome. J. Ment. Defic. Res. 1981, 25 Pt 4, 253–256. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar]
- Oberlé, I.; Rousseau, F.; Heitz, D.; Kretz, C.; Devys, D.; Hanauer, A.; Boué, J.; Bertheas, M.F.; Mandel, J.L. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991, 252, 1097–1102. [Google Scholar]
- Kremer, E.J.; Pritchard, M.; Lynch, M.; Yu, S.; Holman, K.; Baker, E.; Warren, S.T.; Schlessinger, D.; Sutherland, G.R.; Richards, R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 1991, 252, 1711–1714. [Google Scholar]
- Maddalena, A.; Richards, C.S.; McGinniss, M.J.; Brothman, A.; Desnick, R.J.; E Grier, R.; Hirsch, B.; Jacky, P.; A McDowell, G.; Popovich, B.; et al. Technical standards and guidelines for fragile X: The first of a series of disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics. Quality Assurance Subcommittee of the Laboratory Practice Committee. Genet. Med. 2001, 3, 200–205. [Google Scholar] [PubMed]
- The Dutch-Belgian Fragile X Consortium; Bakker, C.E.; Verheij, C.; Willemsen, R.; van der Helm, R.; Oerlemans, F.; Vermey, M.; Bygrave, A.; Hoogeveen, A.; Oostra, B.A.; et al. Fmr1 knockout mice: A model to study fragile X mental retardation. Cell 1994, 78, 23–33. [Google Scholar]
- Mientjes, E.; Nieuwenhuizen, I.; Kirkpatrick, L.; Zu, T.; Hoogeveen-Westerveld, M.; Severijnen, L.; Rifé, M.; Willemsen, R.; Nelson, D.; Oostra, B. The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol. Dis. 2006, 21, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Kazdoba, T.M.; Leach, P.T.; Silverman, J.L.; Crawley, J.N. Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res. 2014, 3, 118–133. [Google Scholar] [CrossRef]
- Kooy, R.F. Of mice and the fragile X syndrome. Trends Genet. 2003, 19, 148–154. [Google Scholar] [CrossRef]
- Kooy, R.F.; Jin, P.; Bao, H.; Till, S.; Kind, P.; Willemsen, R. Animal models of fragile X syndrome. In Fragile X Syndrome; Elsevier: Amsterdam, The Netherlands, 2017; pp. 123–147. [Google Scholar]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef]
- D’Hulst, C.; Kooy, R.F. The GABAA receptor: A novel target for treatment of fragile X? Trends Neurosci. 2007, 30, 425–431. [Google Scholar]
- Braat, S.; Kooy, R.F. The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron 2015, 86, 1119–1130. [Google Scholar] [CrossRef]
- Braat, S.; Kooy, R.F. Fragile X syndrome neurobiology translates into rational therapy. Drug Discov. Today 2014, 19, 510–519. [Google Scholar] [CrossRef]
- Ligsay, A.; Hagerman, R.J. Review of targeted treatments in fragile X syndrome. Intractable Rare Dis. Res. 2016, 5, 158–167. [Google Scholar] [PubMed]
- Hagerman, R.J.; Berry-Kravis, E.; Kaufmann, W.E.; Ono, M.Y.; Tartaglia, N.; Lachiewicz, A.; Kronk, R.; Delahunty, C.; Hessl, D.; Visootsak, J.; et al. Advances in the treatment of fragile X syndrome. Pediatrics 2009, 123, 378–390. [Google Scholar] [PubMed]
- FRAXA Invited to The White House, Celebrating The Children’s Health Act • FRAXA Research Foundation—Finding a Cure for Fragile X Syndrome. Available online: https://www.fraxa.org/fraxa-invited-to-the-white-house-celebrating-the-childrens-health-act/ (accessed on 21 March 2025).
- Fragile X Awareness Day Origins and a Tribute • FRAXA Research Foundation—Finding a Cure for Fragile X Syndrome. Available online: https://www.fraxa.org/fragile-x-awareness-day-origins-tribute/ (accessed on 21 March 2025).
- National Fragile X Foundation | Our Mission, Vision and Priorities. Available online: https://fragilex.org/about-us/mission-vision/ (accessed on 21 March 2025).
- Finding Effective Treatments and Ultimately a Cure for Fragile X syndrome • FRAXA Research Foundation—Finding a Cure for Fragile X Syndrome. Available online: https://www.fraxa.org/about/#mission (accessed on 21 March 2025).
- Chadwick, W.; Angulo-Herrera, I.; Cogram, P.; Deacon, R.J.M.; Mason, D.J.; Brown, D.; Roberts, I.; O’donovan, D.J.; Tranfaglia, M.R.; Guilliams, T.; et al. A novel combination treatment for fragile X syndrome predicted using computational methods. Brain Commun. 2024, 6, fcad353. [Google Scholar]
- Shah, S.; Sharp, K.J.; Ponny, S.R.; Lee, J.; Watts, J.K.; Berry-Kravis, E.; Richter, J.D. Antisense oligonucleotide rescue of CGG expansion-dependent FMR1 mis-splicing in fragile X syndrome restores FMRP. Proc. Natl. Acad. Sci. USA 2023, 120, e2302534120. [Google Scholar]
- Lee, H.-G.; Imaichi, S.; Kraeutler, E.; Aguilar, R.; Lee, Y.-W.; Sheridan, S.D.; Lee, J.T. Site-specific R-loops induce CGG repeat contraction and fragile X gene reactivation. Cell 2023, 186, 2593–2609.e18. [Google Scholar]
- Garber, K.B.; Visootsak, J.; Warren, S.T. Fragile X syndrome. Eur. J. Hum. Genet. 2008, 16, 666–672. [Google Scholar]
- Kaufmann, W.E.; Kidd, S.A.; Andrews, H.F.; Budimirovic, D.B.; Esler, A.; Haas-Givler, B.; Stackhouse, T.; Riley, C.; Peacock, G.; Sherman, S.L.; et al. Autism spectrum disorder in fragile X syndrome: Cooccurring conditions and current treatment. Pediatrics 2017, 139 (Suppl. S3), S194–S206. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Hagerman, P.J.; Hagerman, R. Fragile X syndrome. Curr. Biol. 2021, 31, R273–R275. [Google Scholar]
- Heulens, I.; Suttie, M.; Postnov, A.; De Clerck, N.; Perrotta, C.S.; Mattina, T.; Faravelli, F.; Forzano, F.; Kooy, R.F. Craniofacial characteristics of fragile X syndrome in mouse and man. Eur. J. Hum. Genet. 2013, 21, 816–823. [Google Scholar]
- Kidd, S.A.; Lachiewicz, A.; Barbouth, D.; Blitz, R.K.; Delahunty, C.; McBrien, D.; Visootsak, J.; Berry-Kravis, E. Fragile X syndrome: A review of associated medical problems. Pediatrics 2014, 134, 995–1005. [Google Scholar]
- Berry-Kravis, E.; Raspa, M.; Loggin-Hester, L.; Bishop, E.; Holiday, D.; Bailey, D.B. Seizures in fragile X syndrome: Characteristics and comorbid diagnoses. Am. J. Intellect. Dev. Disabil. 2010, 115, 461–472. [Google Scholar] [PubMed]
- Heard, T.T.; Ramgopal, S.; Picker, J.; Lincoln, S.A.; Rotenberg, A.; Kothare, S.V. EEG abnormalities and seizures in genetically diagnosed Fragile X syndrome. Int. J. Dev. Neurosci. 2014, 38, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Kronk, R.; Bishop, E.E.; Raspa, M.; Bickel, J.O.; Mandel, D.A.; Bailey, D.B. Prevalence, nature, and correlates of sleep problems among children with fragile X syndrome based on a large scale parent survey. Sleep 2010, 33, 679–687. [Google Scholar] [PubMed]
- Herring, J.; Johnson, K.; Richstein, J. The use of “retardation” in FRAXA, FMRP, FMR1 and other designations. Cells 2022, 11, 1044. [Google Scholar] [CrossRef]
- Eichler, E.E.; Richards, S.; Gibbs, R.A.; Nelson, D.L. Fine structure of the human FMR1 gene. Hum. Mol. Genet. 1993, 2, 1147–1153. [Google Scholar] [CrossRef]
- Bakker, C.E.; de Diego Otero, Y.; Bontekoe, C.; Raghoe, P.; Luteijn, T.; Hoogeveen, A.T.; Oostra, B.A.; Willemsen, R. Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp. Cell Res. 2000, 258, 162–170. [Google Scholar]
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.P.; Mandel, J.L. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 1993, 4, 335–340. [Google Scholar] [CrossRef]
- Feng, Y.; Gutekunst, C.A.; Eberhart, D.E.; Yi, H.; Warren, S.T.; Hersch, S.M. Fragile X mental retardation protein: Nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neurosci. 1997, 17, 1539–1547. [Google Scholar]
- Nolin, S.L.; Brown, W.T.; Glicksman, A.; Houck, J.G.E.; Gargano, A.D.; Sullivan, A.; Biancalana, V.; Bröndum-Nielsen, K.; Hjalgrim, H.; Holinski-Feder, E.; et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 2003, 72, 454–464. [Google Scholar]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, 57, 127–130. [Google Scholar]
- Allingham-Hawkins, D.J.; Babul-Hirji, R.; Chitayat, D.; Holden, J.J.; Yang, K.T.; Lee, C.; Hudson, R.; Gorwill, H.; Nolin, S.L.; Glicksman, A.; et al. Fragile X premutation is a significant risk factor for premature ovarian failure: The International Collaborative POF in Fragile X study—Preliminary data. Am. J. Med. Genet. 1999, 83, 322–325. [Google Scholar] [PubMed]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.A.; Hall, D.A.; Levine, R.A.; Brunberg, J.A.; Zhang, L.; Jardini, T.; Gane, L.W.; Harris, S.W.; et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004, 291, 460–469. [Google Scholar] [PubMed]
- Salcedo-Arellano, M.J.; Dufour, B.; McLennan, Y.; Martinez-Cerdeno, V.; Hagerman, R. Fragile X syndrome and associated disorders: Clinical aspects and pathology. Neurobiol. Dis. 2020, 136, 104740. [Google Scholar]
- Hall, D.A.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Rice, C.D.; Leehey, M.A. Symptomatic treatment in the fragile X-associated tremor/ataxia syndrome. Mov. Disord. 2006, 21, 1741–1744. [Google Scholar]
- Sherman, S.L.; Curnow, E.C.; A Easley, C.; Jin, P.; Hukema, R.K.; Tejada, M.I.; Willemsen, R.; Usdin, K. Use of model systems to understand the etiology of fragile X-associated primary ovarian insufficiency (FXPOI). J. Neurodev. Disord. 2014, 6, 26. [Google Scholar]
- Fu, Y.-H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkert, A.J.; Holden, J.J.; Fenwick, R.G.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar]
- Pieretti, M.; Zhang, F.; Fu, Y.-H.; Warren, S.T.; Oostra, B.A.; Caskey, C.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar]
- Tabolacci, E.; Nobile, V.; Pucci, C.; Chiurazzi, P. Mechanisms of the FMR1 repeat instability: How does the CGG sequence expand? Int. J. Mol. Sci. 2022, 23, 5425. [Google Scholar] [CrossRef]
- Weiler, I.J.; Irwin, S.A.; Klintsova, A.Y.; Spencer, C.M.; Brazelton, A.D.; Miyashiro, K.; Comery, T.A.; Patel, B.; Eberwine, J.; Greenough, W.T. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. USA 1997, 94, 5395–5400. [Google Scholar]
- Willemsen, R.; Bontekoe, C.; Tamanini, F.; Galjaard, H.; Hoogeveen, A.; Oostra, B. Association of FMRP with ribosomal precursor particles in the nucleolus. Biochem. Biophys. Res. Commun. 1996, 225, 27–33. [Google Scholar]
- Bagni, C.; Zukin, R.S. A synaptic perspective of fragile X syndrome and autism spectrum disorders. Neuron 2019, 101, 1070–1088. [Google Scholar] [PubMed]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [PubMed]
- Richter, J.D.; Bassell, G.J.; Klann, E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci. 2015, 16, 595–605. [Google Scholar] [PubMed]
- Valverde, R.; Edwards, L.; Regan, L. Structure and function of KH domains. FEBS J. 2008, 275, 2712–2726. [Google Scholar]
- Myrick, L.K.; Hashimoto, H.; Cheng, X.; Warren, S.T. Human FMRP contains an integral tandem Agenet (Tudor) and KH motif in the amino terminal domain. Hum. Mol. Genet. 2015, 24, 1733–1740. [Google Scholar]
- Chowdhury, M.N.; Jin, H. The RGG motif proteins: Interactions, functions, and regulations. Wiley Interdiscip. Rev. RNA 2023, 14, e1748. [Google Scholar]
- Eberhart, D.E.; Malter, H.E.; Feng, Y.; Warren, S.T. The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum. Mol. Genet. 1996, 5, 1083–1091. [Google Scholar]
- He, Q.; Ge, W. The tandem Agenet domain of fragile X mental retardation protein interacts with FUS. Sci. Rep. 2017, 7, 962. [Google Scholar]
- Hering, H.; Sheng, M. Dendritic spines: Structure, dynamics and regulation. Nat. Rev. Neurosci. 2001, 2, 880–888. [Google Scholar]
- Comery, T.A.; Harris, J.B.; Willems, P.J.; Oostra, B.A.; Irwin, S.A.; Weiler, I.J.; Greenough, W.T. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 1997, 94, 5401–5404. [Google Scholar]
- He, C.X.; Portera-Cailliau, C. The trouble with spines in fragile X syndrome: Density, maturity and plasticity. Neuroscience 2013, 251, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Bassell, G.J.; Warren, S.T. Fragile X syndrome: Loss of local mRNA regulation alters synaptic development and function. Neuron 2008, 60, 201–214. [Google Scholar] [PubMed]
- Gantois, I.; Vandesompele, J.; Speleman, F.; Reyniers, E.; D’Hooge, R.; Severijnen, L.-A.; Willemsen, R.; Tassone, F.; Kooy, R.F. Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol. Dis. 2006, 21, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Braat, S.; D’Hulst, C.; Heulens, I.; De Rubeis, S.; Mientjes, E.; Nelson, D.L.; Willemsen, R.; Bagni, C.; Van Dam, D.; De Deyn, P.P.; et al. The GABAA receptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle 2015, 14, 2985–2995. [Google Scholar]
- D’Hulst, C.; De Geest, N.; Reeve, S.P.; Van Dam, D.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006, 1121, 238–245. [Google Scholar]
- Heulens, I.; D’Hulst, C.; Van Dam, D.; De Deyn, P.P.; Kooy, R.F. Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav. Brain Res. 2012, 229, 244–249. [Google Scholar]
- D’Hulst, C.; Heulens, I.; Brouwer, J.R.; Willemsen, R.; De Geest, N.; Reeve, S.P.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 2009, 1253, 176–183. [Google Scholar] [CrossRef]
- van der Lei, M.B.; Kooy, R.F. Therapeutic potential of GABAA receptor subunit expression abnormalities in fragile X syndrome. Expert. Rev. Precis. Med. Drug Dev. 2022, 7, 105–120. [Google Scholar] [CrossRef]
- Willemsen, R.; Kooy, R.F. Mouse models of fragile X-related disorders. Dis. Model. Mech. 2023, 16, dmm049485. [Google Scholar] [CrossRef]
- Drozd, M.; Bardoni, B.; Capovilla, M. Modeling fragile X syndrome in drosophila. Front. Mol. Neurosci. 2018, 11, 124. [Google Scholar]
- Trajković, J.; Makevic, V.; Pesic, M.; Pavković-Lučić, S.; Milojevic, S.; Cvjetkovic, S.; Hagerman, R.; Budimirovic, D.B.; Protic, D. Drosophila melanogaster as a Model to Study Fragile X-Associated Disorders. Genes 2022, 14, 87. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A.; Idupulapati, M.; Gilbert, M.E.; Harris, J.B.; Chakravarti, A.B.; Rogers, E.J.; Crisostomo, R.A.; Larsen, B.P.; Mehta, A.; Alcantara, C.; et al. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am. J. Med. Genet. 2002, 111, 140–146. [Google Scholar] [PubMed]
- Cruz-Martín, A.; Crespo, M.; Portera-Cailliau, C. Delayed stabilization of dendritic spines in fragile X mice. J. Neurosci. 2010, 30, 7793–7803. [Google Scholar]
- Booker, S.A.; Domanski, A.P.F.; Dando, O.R.; Jackson, A.D.; Isaac, J.T.R.; Hardingham, G.E.; Wyllie, D.J.A.; Kind, P.C. Altered dendritic spine function and integration in a mouse model of fragile X syndrome. Nat. Commun. 2019, 10, 4813. [Google Scholar]
- Nagaoka, A.; Takehara, H.; Hayashi-Takagi, A.; Noguchi, J.; Ishii, K.; Shirai, F.; Yagishita, S.; Akagi, T.; Ichiki, T.; Kasai, H. Abnormal intrinsic dynamics of dendritic spines in a fragile X syndrome mouse model in vivo. Sci. Rep. 2016, 6, 26651. [Google Scholar]
- Dölen, G.; Osterweil, E.; Rao, B.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of fragile X syndrome in mice. Neuron 2007, 56, 955–962. [Google Scholar]
- Muddashetty, R.S.; Kelić, S.; Gross, C.; Xu, M.; Bassell, G.J. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 2007, 27, 5338–5348. [Google Scholar]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar]
- Bernard, P.B.; Castano, A.M.; O’Leary, H.; Simpson, K.; Browning, M.D.; Benke, T.A. Phosphorylation of FMRP and alterations of FMRP complex underlie enhanced mLTD in adult rats triggered by early life seizures. Neurobiol. Dis. 2013, 59, 1–17. [Google Scholar]
- D’Hooge, R.; Nagels, G.; Franck, F.; Bakker, C.; Reyniers, E.; Storm, K.; Kooy, R.; Oostra, B.; Willems, P.; De Deyn, P. Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 1997, 76, 367–376. [Google Scholar] [CrossRef]
- Dobkin, C.; Rabe, A.; Dumas, R.; El Idrissi, A.; Haubenstock, H.; Brown, W.T. Fmr1 knockout mouse has a distinctive strain-specific learning impairment. Neuroscience 2000, 100, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Paradee, W.; Melikian, H.E.; Rasmussen, D.L.; Kenneson, A.; Conn, P.J.; Warren, S.T. Fragile X mouse: Strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 1999, 94, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Toth, M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 2001, 103, 1043–1050. [Google Scholar]
- de Esch, C.E.F.; Ghazvini, M.; Loos, F.; Schelling-Kazaryan, N.; Widagdo, W.; Munshi, S.T.; van der Wal, E.; Douben, H.; Gunhanlar, N.; Kushner, S.A.; et al. Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Rep. 2014, 3, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.M.; Alekseyenko, O.; Hamilton, S.M.; Thomas, A.M.; Serysheva, E.; Yuva-Paylor, L.A.; Paylor, R. Modifying behavioral phenotypes in Fmr1KO mice: Genetic background differences reveal autistic-like responses. Autism Res. 2011, 4, 40–56. [Google Scholar] [CrossRef]
- Musumeci, S.A.; Hagerman, R.J.; Ferri, R.; Bosco, P.; Bernardina, B.D.; Tassinari, C.A.; De Sarro, G.B.; Elia, M. Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia 1999, 40, 1092–1099. [Google Scholar]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar]
- Willemsen, R.; Bontekoe, C.J.M.; Severijnen, L.-A.; Oostra, B.A. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum. Genet. 2002, 110, 601–605. [Google Scholar]
- Dahlhaus, R. Of men and mice: Modeling the fragile X syndrome. Front. Mol. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Osterweil, E.K.; Krueger, D.D.; Reinhold, K.; Bear, M.F. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 2010, 30, 15616–15627. [Google Scholar]
- Choi, C.H.; Schoenfeld, B.P.; Bell, A.J.; Hinchey, J.; Rosenfelt, C.; Gertner, M.J.; Campbell, S.R.; Emerson, D.; Hinchey, P.; Kollaros, M.; et al. Multiple Drug Treatments That Increase cAMP Signaling Restore Long-Term Memory and Aberrant Signaling in Fragile X Syndrome Models. Front. Behav. Neurosci. 2016, 10, 136. [Google Scholar]
- Gurney, M.E.; Cogram, P.; Deacon, R.M.; Rex, C.; Tranfaglia, M. Multiple Behavior Phenotypes of the Fragile-X Syndrome Mouse Model Respond to Chronic Inhibition of Phosphodiesterase-4D (PDE4D). Sci. Rep. 2017, 7, 14653. [Google Scholar]
- Choi, C.H.; Schoenfeld, B.P.; Weisz, E.D.; Bell, A.J.; Chambers, D.B.; Hinchey, J.; Choi, R.J.; Hinchey, P.; Kollaros, M.; Gertner, M.J.; et al. PDE-4 inhibition rescues aberrant synaptic plasticity in Drosophila and mouse models of fragile X syndrome. J. Neurosci. 2015, 35, 396–408. [Google Scholar] [PubMed]
- Hu, H.; Qin, Y.; Bochorishvili, G.; Zhu, Y.; van Aelst, L.; Zhu, J.J. Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J. Neurosci. 2008, 28, 7847–7862. [Google Scholar]
- Gross, C.; Nakamoto, M.; Yao, X.; Chan, C.-B.; Yim, S.Y.; Ye, K.; Warren, S.T.; Bassell, G.J. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci. 2010, 30, 10624–10638. [Google Scholar]
- Lovelace, J.W.; Wen, T.H.; Reinhard, S.; Hsu, M.S.; Sidhu, H.; Ethell, I.M.; Binder, D.K.; Razak, K.A. Matrix metalloproteinase-9 deletion rescues auditory evoked potential habituation deficit in a mouse model of Fragile X Syndrome. Neurobiol. Dis. 2016, 89, 126–135. [Google Scholar]
- Gkogkas, C.G.; Khoutorsky, A.; Cao, R.; Jafarnejad, S.M.; Prager-Khoutorsky, M.; Giannakas, N.; Kaminari, A.; Fragkouli, A.; Nader, K.; Price, T.J.; et al. Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep. 2014, 9, 1742–1755. [Google Scholar]
- Lovelace, J.W.; Ethell, I.M.; Binder, D.K.; Razak, K.A. Minocycline treatment reverses sound evoked EEG abnormalities in a mouse model of fragile X syndrome. Front. Neurosci. 2020, 14, 771. [Google Scholar]
- Sharma, A.; Hoeffer, C.A.; Takayasu, Y.; Miyawaki, T.; McBride, S.M.; Klann, E.; Zukin, R.S. Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 2010, 30, 694–702. [Google Scholar] [CrossRef]
- Hoeffer, C.A.; Sanchez, E.; Hagerman, R.J.; Mu, Y.; Nguyen, D.V.; Wong, H.; Whelan, A.M.; Zukin, R.S.; Klann, E.; Tassone, F. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012, 11, 332–341. [Google Scholar]
- Bhattacharya, A.; Mamcarz, M.; Mullins, C.; Choudhury, A.; Boyle, R.G.; Smith, D.G.; Walker, D.W.; Klann, E. Targeting Translation Control with p70 S6 Kinase 1 Inhibitors to Reverse Phenotypes in Fragile X Syndrome Mice. Neuropsychopharmacology 2016, 41, 1991–2000. [Google Scholar] [PubMed]
- Tabet, R.; Moutin, E.; Becker, J.A.J.; Heintz, D.; Fouillen, L.; Flatter, E.; Krężel, W.; Alunni, V.; Koebel, P.; Dembélé, D.; et al. Fragile X Mental Retardation Protein (FMRP) controls diacylglycerol kinase activity in neurons. Proc. Natl. Acad. Sci. USA 2016, 113, E3619–E3628. [Google Scholar]
- Guo, W.; Murthy, A.C.; Zhang, L.; Johnson, E.B.; Schaller, E.G.; Allan, A.M.; Zhao, X. Inhibition of GSK3β improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum. Mol. Genet. 2012, 21, 681–691. [Google Scholar]
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q.; et al. Selective inhibition of glycogen synthase kinase 3α corrects pathophysiology in a mouse model of fragile X syndrome. Sci. Transl. Med. 2020, 12, eaam8572. [Google Scholar]
- Gkogkas, C.G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D.B.; Vasuta, C.; Yee, S.; Truitt, M.; Dallaire, P.; et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, 493, 371–377. [Google Scholar]
- Dolan, B.M.; Duron, S.G.; Campbell, D.A.; Vollrath, B.; Rao, B.S.S.; Ko, H.-Y.; Lin, G.G.; Govindarajan, A.; Choi, S.-Y.; Tonegawa, S. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. USA 2013, 110, 5671–5676. [Google Scholar]
- Zafarullah, M.; Tassone, F. Molecular biomarkers in fragile X syndrome. Brain Sci. 2019, 9, 96. [Google Scholar] [CrossRef]
- Erickson, C.A.; Davenport, M.H.; Schaefer, T.L.; Wink, L.K.; Pedapati, E.V.; Sweeney, J.A.; Fitzpatrick, S.E.; Brown, W.T.; Budimirovic, D.; Hagerman, R.J.; et al. Fragile X targeted pharmacotherapy: Lessons learned and future directions. J. Neurodev. Disord. 2017, 9, 7. [Google Scholar]
- Berry-Kravis, E.M.; Lindemann, L.; Jønch, A.E.; Apostol, G.; Bear, M.F.; Carpenter, R.L.; Crawley, J.N.; Curie, A.; Portes, V.D.; Hossain, F.; et al. Drug development for neurodevelopmental disorders: Lessons learned from fragile X syndrome. Nat. Rev. Drug Discov. 2018, 17, 280–299. [Google Scholar]
- Olsen, R.W.; Sieghart, W. GABA A receptors: Subtypes provide diversity of function and pharmacology. Neuropharmacology 2009, 56, 141–148. [Google Scholar]
- Olmos-Serrano, J.L.; Paluszkiewicz, S.M.; Martin, B.S.; Kaufmann, W.E.; Corbin, J.G.; Huntsman, M.M. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J. Neurosci. 2010, 30, 9929–9938. [Google Scholar] [PubMed]
- Paluszkiewicz, S.M.; Martin, B.S.; Huntsman, M.M. Fragile X syndrome: The GABAergic system and circuit dysfunction. Dev. Neurosci. 2011, 33, 349–364. [Google Scholar]
- D’hulst, C.; Heulens, I.; Van der Aa, N.; Goffin, K.; Koole, M.; Porke, K.; Van De Velde, M.; Rooms, L.; Van Paesschen, W.; Van Esch, H.; et al. Positron emission tomography (PET) quantification of GABAA receptors in the brain of fragile X patients. PLoS ONE 2015, 10, e0131486. [Google Scholar]
- Ligsay, A.; Van Dijck, A.; Nguyen, D.V.; Lozano, R.; Chen, Y.; Bickel, E.S.; Hessl, D.; Schneider, A.; Angkustsiri, K.; Tassone, F.; et al. A randomized double-blind, placebo-controlled trial of ganaxolone in children and adolescents with fragile X syndrome. J. Neurodev. Disord. 2017, 9, 26. [Google Scholar]
- Olmos-Serrano, J.L.; Corbin, J.G.; Burns, M.P. The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev. Neurosci. 2011, 33, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Cogram, P.; Deacon, R.M.J.; Warner-Schmidt, J.L.; von Schimmelmann, M.J.; Abrahams, B.S.; During, M.J. Gaboxadol normalizes behavioral abnormalities in a mouse model of fragile X syndrome. Front. Behav. Neurosci. 2019, 13, 141. [Google Scholar]
- Budimirovic, D.B.; Dominick, K.C.; Gabis, L.V.; Adams, M.; Adera, M.; Huang, L.; Ventola, P.; Tartaglia, N.R.; Berry-Kravis, E. Gaboxadol in Fragile X Syndrome: A 12-Week Randomized, Double-Blind, Parallel-Group, Phase 2a Study. Front. Pharmacol. 2021, 12, 757825. [Google Scholar]
- Bettler, B.; Kaupmann, K.; Mosbacher, J.; Gassmann, M. Molecular structure and physiological functions of GABA(B) receptors. Physiol. Rev. 2004, 84, 835–867. [Google Scholar]
- Henderson, C.; Wijetunge, L.; Kinoshita, M.N.; Shumway, M.; Hammond, R.S.; Postma, F.R.; Brynczka, C.; Rush, R.; Thomas, A.; Paylor, R.; et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 2012, 4, 152ra128. [Google Scholar] [CrossRef]
- Pacey, L.K.K.; Heximer, S.P.; Hampson, D.R. Increased GABA(B) receptor-mediated signaling reduces the susceptibility of fragile X knockout mice to audiogenic seizures. Mol. Pharmacol. 2009, 76, 18–24. [Google Scholar]
- Qin, M.; Huang, T.; Kader, M.; Krych, L.; Xia, Z.; Burlin, T.; Zeidler, Z.; Zhao, T.; Smith, C.B. R-Baclofen Reverses a Social Behavior Deficit and Elevated Protein Synthesis in a Mouse Model of Fragile X Syndrome. Int. J. Neuropsychopharmacol. 2015, 18, pyv034. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.; Featherstone, R.; Naschek, M.; Nam, J.; Du, A.; Wright, S.; Pance, K.; Melnychenko, O.; Weger, R.; Akuzawa, S.; et al. GABA-B Agonist Baclofen Normalizes Auditory-Evoked Neural Oscillations and Behavioral Deficits in the Fmr1 Knockout Mouse Model of Fragile X Syndrome. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Hagerman, R.; Visootsak, J.; Budimirovic, D.; Kaufmann, W.E.; Cherubini, M.; Zarevics, P.; Walton-Bowen, K.; Wang, P.; Bear, M.F.; et al. Arbaclofen in fragile X syndrome: Results of phase 3 trials. J. Neurodev. Disord. 2017, 9, 3. [Google Scholar] [CrossRef]
- McBride, S.M.; Choi, C.H.; Wang, Y.; Liebelt, D.; Braunstein, E.; Ferreiro, D.; Sehgal, A.; Siwicki, K.K.; Dockendorff, T.C.; Nguyen, H.T.; et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 2005, 45, 753–764. [Google Scholar] [CrossRef]
- Bhakar, A.L.; Dölen, G.; Bear, M.F. The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci. 2012, 35, 417–443. [Google Scholar] [CrossRef]
- Dölen, G.; Bear, M.F. Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J. Physiol. 2008, 586, 1503–1508. [Google Scholar] [CrossRef]
- Stoppel, L.J.; Osterweil, E.K.; Bear, M.F. The mGluR Theory of Fragile X: From Mice to Men. In Fragile X Syndrome; Elsevier: Amsterdam, The Netherlands, 2017; pp. 173–204. [Google Scholar]
- Michalon, A.; Sidorov, M.; Ballard, T.M.; Ozmen, L.; Spooren, W.; Wettstein, J.G.; Jaeschke, G.; Bear, M.F.; Lindemann, L. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 2012, 74, 49–56. [Google Scholar] [CrossRef]
- Zerbi, V.; Markicevic, M.; Gasparini, F.; Schroeter, A.; Rudin, M.; Wenderoth, N. Inhibiting mGluR5 activity by AFQ056/Mavoglurant rescues circuit-specific functional connectivity in Fmr1 knockout mice. Neuroimage 2019, 191, 392–402. [Google Scholar] [CrossRef]
- Gantois, I.; Pop, A.S.; de Esch, C.E.; Buijsen, R.A.; Pooters, T.; Gomez-Mancilla, B.; Gasparini, F.; Oostra, B.A.; D’hooge, R.; Willemsen, R. Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav. Brain Res. 2013, 239, 72–79. [Google Scholar] [CrossRef]
- Pop, A.S.; Levenga, J.; de Esch, C.E.F.; Buijsen, R.A.M.; Nieuwenhuizen, I.M.; Li, T.; Isaacs, A.; Gasparini, F.; Oostra, B.A. Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/Mavoglurant. Psychopharmacology 2014, 231, 1227–1235. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Des Portes, V.; Hagerman, R.; Jacquemont, S.; Charles, P.; Visootsak, J.; Brinkman, M.; Rerat, K.; Koumaras, B.; Zhu, L.; et al. Mavoglurant in fragile X syndrome: Results of two randomized, double-blind, placebo-controlled trials. Sci. Transl. Med. 2016, 8, 321ra5. [Google Scholar]
- Hagerman, R.; Jacquemont, S.; Berry-Kravis, E.; Portes, V.D.; Stanfield, A.; Koumaras, B.; Rosenkranz, G.; Murgia, A.; Wolf, C.; Apostol, G.; et al. Mavoglurant in Fragile X Syndrome: Results of two open-label, extension trials in adults and adolescents. Sci. Rep. 2018, 8, 16970. [Google Scholar] [CrossRef] [PubMed]
- Youssef, E.A.; FragXis Study Group; Berry-Kravis, E.; Czech, C.; Hagerman, R.J.; Hessl, D.; Wong, C.Y.; Rabbia, M.; Deptula, D.; John, A.; et al. Effect of the mGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacology 2018, 43, 503–512. [Google Scholar]
- Berry-Kravis, E.; Abbeduto, L.; Hagerman, R.; Coffey, C.S.; Cudkowicz, M.; Erickson, C.A.; McDuffie, A.; Hessl, D.; Ethridge, L.; Tassone, F.; et al. Effects of AFQ056 on language learning in fragile X syndrome. J. Clin. Investig. 2023, 134, e171723. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, function, and pharmacology of glutamate receptor ion channels. Pharmacol. Rev. 2021, 73, 298–487. [Google Scholar] [PubMed]
- Pilpel, Y.; Kolleker, A.; Berberich, S.; Ginger, M.; Frick, A.; Mientjes, E.; Oostra, B.A.; Seeburg, P.H. Synaptic ionotropic glutamate receptors and plasticity are developmentally altered in the CA1 field of Fmr1 knockout mice. J. Physiol. 2009, 587 Pt 4, 787–804. [Google Scholar]
- Erickson, C.A.; Mullett, J.E.; McDougle, C.J. Open-label memantine in fragile X syndrome. J. Autism Dev. Disord. 2009, 39, 1629–1635. [Google Scholar]
- Berry-Kravis, E.; Hicar, M.; Ciurlionis, R. Reduced cyclic AMP production in fragile X syndrome: Cytogenetic and molecular correlations. Pediatr. Res. 1995, 38, 638–643. [Google Scholar]
- Berry-Kravis, E.; Huttenlocher, P.R. Cyclic AMP metabolism in fragile X syndrome. Ann. Neurol. 1992, 31, 22–26. [Google Scholar]
- Berry-Kravis, E.; Sklena, P. Demonstration of abnormal cyclic AMP production in platelets from patients with fragile X syndrome. Am. J. Med. Genet. 1993, 45, 81–87. [Google Scholar] [CrossRef]
- Kelley, D.J.; Davidson, R.J.; Elliott, J.L.; Lahvis, G.P.; Yin, J.C.P.; Bhattacharyya, A. The cyclic AMP cascade is altered in the fragile X nervous system. PLoS ONE 2007, 2, e931. [Google Scholar] [CrossRef] [PubMed]
- Linglart, A.; Fryssira, H.; Hiort, O.; Holterhus, P.-M.; de Nanclares, G.P.; Argente, J.; Heinrichs, C.; Kuechler, A.; Mantovani, G.; Leheup, B.; et al. PRKAR1A and PDE4D mutations cause acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. J. Clin. Endocrinol. Metab. 2012, 97, E2328–E2338. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.M.; Harnett, M.D.; Reines, S.A.; Reese, M.A.; Ethridge, L.E.; Outterson, A.H.; Michalak, C.; Furman, J. Inhibition of phosphodiesterase-4D in adults with fragile X syndrome: A randomized, placebo-controlled, phase 2 clinical trial. Nat. Med. 2021, 27, 862–870. [Google Scholar] [CrossRef]
- Palumbo, J.M.; Thomas, B.F.; Budimirovic, D.; Siegel, S.; Tassone, F.; Hagerman, R.; Faulk, C.; O’quinn, S.; Sebree, T. Role of the endocannabinoid system in fragile X syndrome: Potential mechanisms for benefit from cannabidiol treatment. J. Neurodev. Disord. 2023, 15, 1. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Jung, K.-M.; Sepers, M.; Henstridge, C.M.; Lassalle, O.; Neuhofer, D.; Martin, H.; Ginger, M.; Frick, A.; DiPatrizio, N.V.; Mackie, K.; et al. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat. Commun. 2012, 3, 1080. [Google Scholar] [CrossRef]
- Busquets-Garcia, A.; Gomis-González, M.; Guegan, T.; Agustín-Pavón, C.; Pastor, A.; Mato, S.; Pérez-Samartín, A.; Matute, C.; de la Torre, R.; Dierssen, M.; et al. Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat. Med. 2013, 19, 603–607. [Google Scholar] [CrossRef]
- Heussler, H.; Cohen, J.; Silove, N.; Tich, N.; Bonn-Miller, M.O.; Du, W.; O’Neill, C.; Sebree, T. A phase 1/2, open-label assessment of the safety, tolerability, and efficacy of transdermal cannabidiol (ZYN002) for the treatment of pediatric fragile X syndrome. J. Neurodev. Disord. 2019, 11, 16. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Hagerman, R.; Budimirovic, D.; Erickson, C.; Heussler, H.; Tartaglia, N.; Cohen, J.; Tassone, F.; Dobbins, T.; Merikle, E.; et al. A randomized, controlled trial of ZYN002 cannabidiol transdermal gel in children and adolescents with fragile X syndrome (CONNECT-FX). J. Neurodev. Disord. 2022, 14, 56. [Google Scholar] [CrossRef]
- Klein, D.J.; Cottingham, E.M.; Sorter, M.; Barton, B.A.; Morrison, J.A. A randomized, double-blind, placebo-controlled trial of metformin treatment of weight gain associated with initiation of atypical antipsychotic therapy in children and adolescents. Am. J. Psychiatry 2006, 163, 2072–2079. [Google Scholar] [CrossRef]
- Park, M.H.; Kinra, S.; Ward, K.J.; White, B.; Viner, R.M. Metformin for obesity in children and adolescents: A systematic review. Diabetes Care 2009, 32, 1743–1745. [Google Scholar] [CrossRef] [PubMed]
- Handen, B.L.; Anagnostou, E.; Aman, M.G.; Sanders, K.B.; Chan, J.; Hollway, J.A.; Brian, J.; Arnold, L.E.; Capano, L.; Williams, C.; et al. A Randomized, Placebo-Controlled Trial of Metformin for the Treatment of Overweight Induced by Antipsychotic Medication in Young People With Autism Spectrum Disorder: Open-Label Extension. J. Am. Acad. Child. Adolesc. Psychiatry 2017, 56, 849–856.e6. [Google Scholar] [CrossRef] [PubMed]
- Top, W.M.C.; Kooy, A.; Stehouwer, C.D.A. Metformin: A narrative review of its potential benefits for cardiovascular disease, cancer and dementia. Pharmaceuticals 2022, 15, 312. [Google Scholar] [CrossRef] [PubMed]
- Monyak, R.E.; Emerson, D.; Schoenfeld, B.P.; Zheng, X.; Chambers, D.B.; Rosenfelt, C.; Langer, S.; Hinchey, P.; Choi, C.H.; McDonald, T.V.; et al. Insulin signaling misregulation underlies circadian and cognitive deficits in a Drosophila fragile X model. Mol. Psychiatry 2017, 22, 1140–1148. [Google Scholar] [CrossRef]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A.; et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med. 2017, 23, 674–677. [Google Scholar] [CrossRef]
- Gantois, I.; Popic, J.; Khoutorsky, A.; Sonenberg, N. Metformin for treatment of fragile X syndrome and other neurological disorders. Annu. Rev. Med. 2019, 70, 167–181. [Google Scholar] [CrossRef]
- Dy, A.B.C.; Tassone, F.; Eldeeb, M.; Salcedo-Arellano, M.J.; Tartaglia, N.; Hagerman, R. Metformin as targeted treatment in fragile X syndrome. Clin. Genet. 2018, 93, 216–222. [Google Scholar] [CrossRef]
- Biag, H.M.B.; Potter, L.A.; Wilkins, V.; Afzal, S.; Rosvall, A.; Salcedo-Arellano, M.J.; Rajaratnam, A.; Manzano-Nunez, R.; Schneider, A.; Tassone, F.; et al. Metformin treatment in young children with fragile X syndrome. Mol. Genet. Genom. Med. 2019, 7, e956. [Google Scholar] [CrossRef]
- Protic, D.; Aydin, E.Y.; Tassone, F.; Tan, M.M.; Hagerman, R.J.; Schneider, A. Cognitive and behavioral improvement in adults with fragile X syndrome treated with metformin-two cases. Mol. Genet. Genom. Med. 2019, 7, e00745. [Google Scholar] [CrossRef]
- Kshatri, A.; Cerrada, A.; Gimeno, R.; Bartolomé-Martín, D.; Rojas, P.; Giraldez, T. Differential regulation of BK channels by fragile X mental retardation protein. J. Gen. Physiol. 2020, 152, e201912502. [Google Scholar]
- Deng, P.-Y.; Rotman, Z.; Blundon, J.A.; Cho, Y.; Cui, J.; Cavalli, V.; Zakharenko, S.S.; Klyachko, V.A. FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 2013, 77, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.-Y.; Klyachko, V.A. Genetic upregulation of BK channel activity normalizes multiple synaptic and circuit defects in a mouse model of fragile X syndrome. J. Physiol. 2016, 594, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Myrick, L.K.; Deng, P.-Y.; Hashimoto, H.; Oh, Y.M.; Cho, Y.; Poidevin, M.J.; Suhl, J.A.; Visootsak, J.; Cavalli, V.; Jin, P.; et al. Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc. Natl. Acad. Sci. USA 2015, 112, 949–956. [Google Scholar]
- Spinogenix Completes Phase 2 Study of SPG601 for Treatment of Fragile X Syndrome, a Common Inherited Form of Autism, Showing Strong Efficacy Signal in Measures of Abnormal Brain Activity—Spinogenix. Available online: https://www.spinogenix.com/spinogenix-completes-phase-2-study-of-spg601-fortreatment-offragile-x-syndrome-a-common-inherited-form-of-autismshowing-strong-efficacy-signal-in-measures-of-abnormal-brain-activity/ (accessed on 21 March 2025).
- Kaerus Bioscience | Kaerus Bioscience Successfully Completes. Available online: https://www.kaerusbio.com/phase1trial (accessed on 21 March 2025).
- Nardi, A.; Olesen, S.-P. BK channel modulators: A comprehensive overview. Curr. Med. Chem. 2008, 15, 1126–1146. [Google Scholar] [CrossRef]
- Healx | AI Drug Discovery | Rare Disease Treatment. Available online: https://healx.ai/ (accessed on 21 March 2025).
- Kantify | Improving Human Health Through Artificial Intelligence. Available online: https://www.kantify.com/platform (accessed on 21 March 2025).
- Morin-Parent, F.; Champigny, C.; Côté, S.; Mohamad, T.; Hasani, S.A.; Çaku, A.; Corbin, F.; Lepage, J. Neurophysiological effects of a combined treatment of lovastatin and minocycline in patients with fragile X syndrome: Ancillary results of the LOVAMIX randomized clinical trial. Autism Res. 2024, 17, 1944–1956. [Google Scholar]
- Zeidler, S.; Severijnen, L.A.; de Boer, H.; van der Toorn, E.C.; Ruivenkamp, C.A.; Bijlsma, E.K.; Willemsen, R. A missense variant in the nuclear export signal of the FMR1 gene causes intellectual disability. Gene 2021, 768, 145298. [Google Scholar]
- Hampson, D.R.; Hooper, A.W.M.; Niibori, Y. The Application of Adeno-Associated Viral Vector Gene Therapy to the Treatment of Fragile X Syndrome. Brain Sci. 2019, 9, 32. [Google Scholar] [CrossRef]
- Shitik, E.M.; Velmiskina, A.A.; Dolskiy, A.A.; Yudkin, D.V. Reactivation of FMR1 gene expression is a promising strategy for fragile X syndrome therapy. Gene Ther. 2020, 27, 247–253. [Google Scholar] [CrossRef]
- Hooper, A.W.; Wong, H.; Niibori, Y.; Abdoli, R.; Karumuthil-Melethil, S.; Qiao, C.; Danos, O.; Bruder, J.T.; Hampson, D.R. Gene therapy using an ortholog of human fragile X mental retardation protein partially rescues behavioral abnormalities and EEG activity. Mol. Ther. Methods Clin. Dev. 2021, 22, 196–209. [Google Scholar]
- Gholizadeh, S.; Arsenault, J.; Xuan, I.C.Y.; Pacey, L.K.; Hampson, D.R. Reduced phenotypic severity following adeno-associated virus-mediated Fmr1 gene delivery in fragile X mice. Neuropsychopharmacology 2014, 39, 3100–3111. [Google Scholar]
- Zeier, Z.; Kumar, A.; Bodhinathan, K.; Feller, J.A.; Foster, T.C.; Bloom, D.C. Fragile X mental retardation protein replacement restores hippocampal synaptic function in a mouse model of fragile X syndrome. Gene Ther. 2009, 16, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, J.; Gholizadeh, S.; Niibori, Y.; Pacey, L.K.; Halder, S.K.; Koxhioni, E.; Konno, A.; Hirai, H.; Hampson, D.R. FMRP expression levels in mouse central nervous system neurons determine behavioral phenotype. Hum. Gene Ther. 2016, 27, 982–996. [Google Scholar] [PubMed]
- Yang, C.; Tian, Y.; Su, F.; Wang, Y.; Liu, M.; Wang, H.; Cui, Y.; Yuan, P.; Li, X.; Li, A.; et al. Restoration of FMRP expression in adult V1 neurons rescues visual deficits in a mouse model of fragile X syndrome. Protein Cell 2022, 13, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Han, L.; Meng, J.; Wang, Z.; Zhou, Y.; Yuan, H.; Xu, H.; Zhang, X.; Zhao, Y.; Lu, J.; et al. Gene therapy using human FMRP isoforms driven by the human FMR1 promoter rescues fragile X syndrome mouse deficits. Mol. Ther. Methods Clin. Dev. 2022, 27, 246–258. [Google Scholar] [CrossRef]
- Tabet, R.; Vitale, N.; Moine, H. Fragile X syndrome: Are signaling lipids the missing culprits? Biochimie 2016, 130, 188–194. [Google Scholar]
- Habbas, K.; Cakil, O.; Zámbó, B.; Tabet, R.; Riet, F.; Dembele, D.; Mandel, J.; Hocquemiller, M.; Laufer, R.; Piguet, F.; et al. AAV-delivered diacylglycerol kinase DGKk achieves long-term rescue of fragile X syndrome mouse model. EMBO Mol. Med. 2022, 14, e14649. [Google Scholar]
- Park, C.-Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.-W. Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Rep. 2015, 13, 234–241. [Google Scholar] [CrossRef]
- Xie, N.; Gong, H.; Suhl, J.A.; Chopra, P.; Wang, T.; Warren, S.T. Reactivation of FMR1 by CRISPR/Cas9-Mediated Deletion of the Expanded CGG-Repeat of the Fragile X Chromosome. PLoS ONE 2016, 11, e0165499. [Google Scholar] [CrossRef]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 2018, 172, 979–992.e6. [Google Scholar]
- de Chaumont, F.; Ey, E.; Torquet, N.; Lagache, T.; Dallongeville, S.; Imbert, A.; Legou, T.; Le Sourd, A.M.; Faure, P.; Bourgeron, T.; et al. Real-time analysis of the behaviour of groups of mice via a depth-sensing camera and machine learning. Nat. Biomed. Eng. 2019, 3, 930–942. [Google Scholar] [CrossRef]
- Stoppel, D.C.; McCamphill, P.K.; Senter, R.K.; Heynen, A.J.; Bear, M.F. mGluR5 Negative Modulators for Fragile X: Treatment Resistance and Persistence. Front. Psychiatry 2021, 12, 718953. [Google Scholar]
- Hessl, D.; Nguyen, D.V.; Green, C.; Chavez, A.; Tassone, F.; Hagerman, R.J.; Senturk, D.; Schneider, A.; Lightbody, A.; Reiss, A.L.; et al. A solution to limitations of cognitive testing in children with intellectual disabilities: The case of fragile X syndrome. J. Neurodev. Disord. 2009, 1, 33–45. [Google Scholar]
- Berry-Kravis, E.; Hessl, D.; Abbeduto, L.; Reiss, A.L.; Beckel-Mitchener, A.; Urv, T.K. Outcome measures for clinical trials in fragile X syndrome. J. Dev. Behav. Pediatr. 2013, 34, 508–522. [Google Scholar] [PubMed]
- Budimirovic, D.B.; Berry-Kravis, E.; Erickson, C.A.; Hall, S.S.; Hessl, D.; Reiss, A.L.; King, M.K.; Abbeduto, L.; Kaufmann, W.E. Updated report on tools to measure outcomes of clinical trials in fragile X syndrome. J. Neurodev. Disord. 2017, 9, 14. [Google Scholar] [PubMed]
- Kenny, A.; Wright, D.; Stanfield, A.C. EEG as a translational biomarker and outcome measure in fragile X syndrome. Transl. Psychiatry 2022, 12, 34. [Google Scholar]
- Liu, R.; Pedapati, E.V.; Schmitt, L.M.; Shaffer, R.C.; Smith, E.G.; Dominick, K.C.; DeStefano, L.A.; Westerkamp, G.; Horn, P.; Sweeney, J.A.; et al. Reliability of resting-state electrophysiology in fragile X syndrome. Biomark. Neuropsychiatry 2023, 9, 100070. [Google Scholar]
- Boggs, A.E.; Schmitt, L.M.; McLane, R.D.; Adayev, T.; LaFauci, G.; Horn, P.S.; Dominick, K.C.; Gross, C.; Erickson, C.A. Optimization, validation and initial clinical implications of a Luminex-based immunoassay for the quantification of Fragile X Protein from dried blood spots. Sci. Rep. 2022, 12, 5617. [Google Scholar]


{kind=link}
{kind=link}
| Phenotype | Symptoms |
|---|---|
| Cognitive Phenotype | Cognitive deficits |
| Intellectual disabilities | |
| Developmental delays (motor and/or language) | |
| Behavioral Phenotype | Hyperactivity |
| (Social) anxiety | |
| Aggression | |
| Impulsivity | |
| Attention deficits | |
| Hyperarousal to sensory stimuli | |
| Repetitive stereotypic behaviors (hand flapping) | |
| Echolalia (unusual speaking patterns) | |
| Impaired social skills (ASD) | |
| Gaze avoidance | |
| Physical Phenotype | Elongated face |
| Large ears | |
| Prominent jaw and forehead | |
| High-arched palate | |
| Hyperflexible joints | |
| Flat feet | |
| Macroorchidism (large testicles in teens/adults) | |
| Comorbid Health Issues | Recurrent otitis media (ear infection) |
| Strabismus (crossed eyes) | |
| Weight gain | |
| Seizures | |
| Gastrointestinal problems | |
| Sleep problems |
| Nct Number | Interventions | Sponsor |
|---|---|---|
| NCT06334419 | Gaboxadol (GABAA receptor agonist) | Children’s Hospital, Cincinnati |
| NCT05418049 | Baclofen (GABAB receptor agonist) | Children’s Hospital Cincinnati |
| NCT05418049 | Memantine (NMDA antagonist) | Children’s Hospital Cincinnati |
| NCT05418049 | Roflumilast (PDE4 inhibitor) | Children’s Hospital Cincinnati |
| NCT05367960 | Zatolmilast/BPN14770 (PDE4D inhibitor) | Tetra Discovery Partners |
| NCT05358886 | Zatolmilast/BPN14770 (PDE4D inhibitor) | Tetra Discovery Partners |
| NCT05163808 | Zatolmilast/BPN14770 (PDE4D inhibitor) | Tetra Discovery Partners |
| NCT05120505 | Metformin (activation of AMPK) | Children’s Hospital of Fudan University |
| NCT03862950 | Metformin (activation of AMPK) | University of Alberta |
| NCT04977986 | ZYN002 (modulate EC system) | Zynerba Pharmaceuticals, Inc. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Lei, M.B.; Kooy, R.F. From Discovery to Innovative Translational Approaches in 80 Years of Fragile X Syndrome Research. Biomedicines 2025, 13, 805. https://doi.org/10.3390/biomedicines13040805
van der Lei MB, Kooy RF. From Discovery to Innovative Translational Approaches in 80 Years of Fragile X Syndrome Research. Biomedicines. 2025; 13(4):805. https://doi.org/10.3390/biomedicines13040805
Chicago/Turabian Stylevan der Lei, Mathijs B., and R. Frank Kooy. 2025. "From Discovery to Innovative Translational Approaches in 80 Years of Fragile X Syndrome Research" Biomedicines 13, no. 4: 805. https://doi.org/10.3390/biomedicines13040805
APA Stylevan der Lei, M. B., & Kooy, R. F. (2025). From Discovery to Innovative Translational Approaches in 80 Years of Fragile X Syndrome Research. Biomedicines, 13(4), 805. https://doi.org/10.3390/biomedicines13040805