

The Role of Kidney Biopsy in Fabry Disease

 , , ,
, , ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. The Kidney Biopsy in the Diagnosis of Fabry Disease
2.1. Algoritm of Diagnosis of Fabry Disease
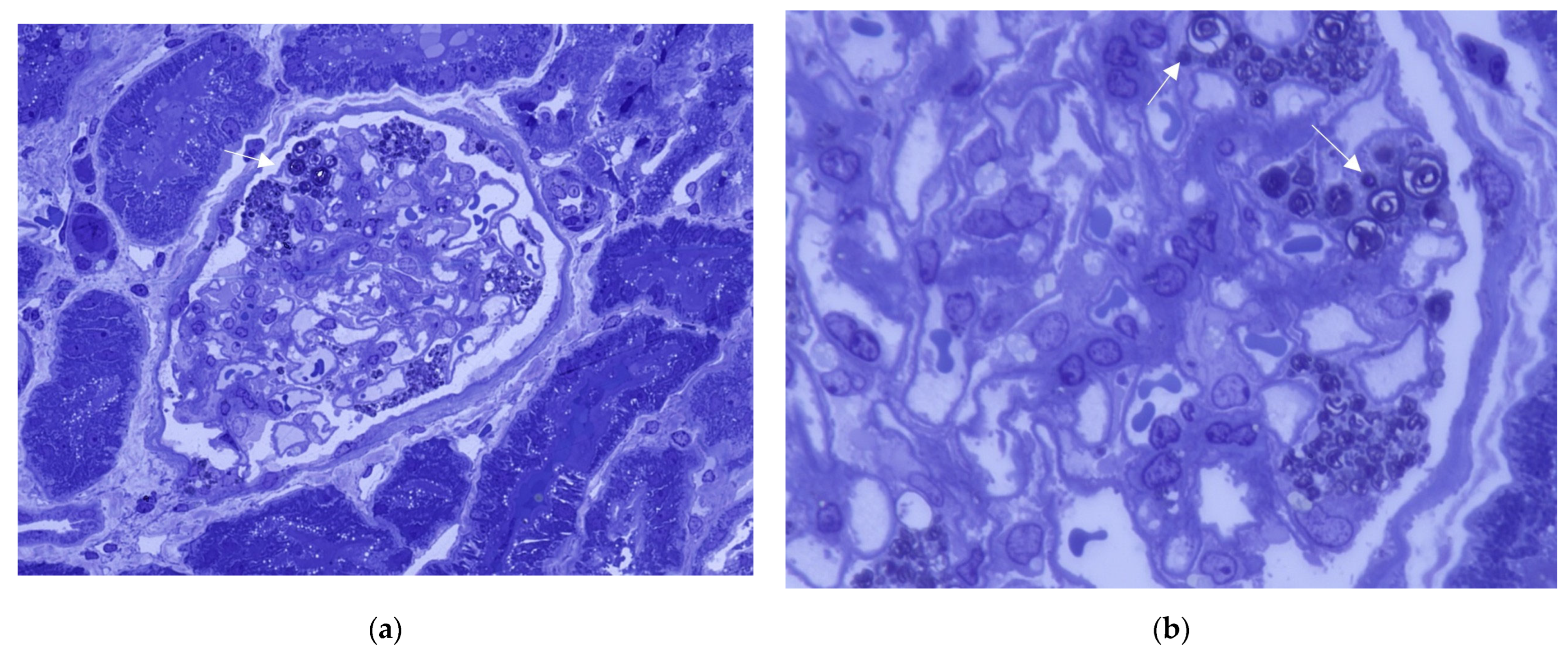
2.2. Diagnostic Finding on Kidney Biopsy in FD
2.3. The Histological Scoring
2.4. The Overlapping Diseases and Misdiagnosis
2.5. The Use of Kidney Biopsy in the Definition of VUS Pathogenicity and in the Female Patients
3. The Kidney Biopsy in Monitoring
4. Kidney Biopsy and Evaluation of Disease Progression
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
| FD | Fabry disease |
| Gb3 | Globotriaosylceramide |
| Lyso-Gb3 | Globotriaosylsphingosine |
| VUS | Variants of uncertain significance |
| XCI | X-chromosome inactivation |
| LM | Light microscopy |
| IF | Immunofluorescence |
| EM | Electron microscopy |
| H&E | Hematoxylin and eosin |
| PAS | Periodic acid-Schiff |
| GBM | Glomerular basement membrane |
| ECM | Extracellular matrix |
| ISGFN | International Study Group of Fabry Nephropathy |
| MCD | Minimal change disease |
| IgAN | IgA nephropathy |
| DIP | Drug-induced phospholipidosis |
| NPS | Nail–patella syndrome |
| ESRD | End-stage renal disease |
| NPLRD | Nail–patella-like renal disease |
| GL-3 | Globotriaosylceramide |
| FPW | Foot process width |
References
- Germain, D.P.; Altarescu, G.; Barriales-Villa, R.; Mignani, R.; Pawlaczyk, K.; Pieruzzi, F.; Terryn, W.; Vujkovac, B.; Ortiz, A. An expert consensus on practical clinical recommendations and guidance for patients with classic Fabry disease. Mol. Genet. Metab. 2022, 137, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliot, P.; Linthorst, G.; Wijburg, F.; Biegstraaten, M.; et al. Characterization of classical and nonclassical fabry disease: A multicenter study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [PubMed]
- Hughes, D.A.; Aguiar, P.; Deegan, P.B.; Ezgu, F.; Frustaci, A.; Lidove, O.; Linhart, A.; Lubanda, J.; Moon, J.; Nicholss, K.; et al. Early indicators of disease progression in Fabry disease that may indicate the need for disease-specific treatment initiation: Findings from the opinion-based PREDICT-FD modified Delphi consensus initiative. BMJ Open 2020, 10, e035182. [Google Scholar] [PubMed]
- Mignani, R.; Moschella, M.; Cenacchi, G.; Donati, I.; Flachi, M.; Grimaldi, D.; Cerretani, D.; Giovanni, P.; Montevecchi, M.; Rigotti, A.; et al. Different renal phenotypes in related adult males with Fabry disease with the same classic genotype. Mol. Genet. Genom. Med. 2017, 5, 438–442. [Google Scholar]
- Berti, G.M.; Aiello, V.; Vischini, G.; Lerario, S.; Ciurli, F.; Santostefano, M.; Donadio, V.; Biagini, E.; Fresina, M.; Fabbrizio, B.; et al. The importance of a multidisciplinary approach in two tricky cases: The perfect match for Fabry disease. BMC Nephrol. 2025, 26, 77. [Google Scholar]
- Mignani, R.; Gallieni, M.; Feriozzi, S.; Pisani, A.; Marziliano, N.; Morrone, A. La nefropatia in corso di malattia di Anderson-Fabry: Nuove raccomandazioni sulla diagnosi, il follow up e la terapia. G. Ital. Nefrol. 2015, 4, 1–13. [Google Scholar]
- van der Tol, L.; Cassiman, D.; Houge, G.; Janssen, M.C.; Lachmann, R.H.; Linthorst, G.E.; Ramaswami, U.; Sommer, C.; Tøndel, C.; West, M.; et al. Uncertain diagnosis of fabry disease in patients with neuropathic pain, angiokeratoma or cornea verticillata: Consensus on the approach to diagnosis and follow-up. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2014; pp. 83–90. [Google Scholar]
- Smid, B.E.; Van Der Tol, L.; Cecchi, F.; Elliott, P.M.; Hughes, D.A.; Linthorst, G.E.; Timmermans, J.; Weidemann, F.; West, M.L.; Biegstraaten, M.; et al. Uncertain diagnosis of Fabry disease: Consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int. J. Cardiol. 2014, 177, 400–408. [Google Scholar]
- Mignani, R. La nefropatia in corso di malattia di Fabry: Nuove evidenze sulla diagnosi, il monitoraggio e la terapia. G. Ital. Nefrol. 2019, 4, 41–52. [Google Scholar]
- Esposito, P.; Caputo, C.; Repetto, M.; Somaschini, A.; Pietro, B.; Colomba, P.; Zizzo, C.; Parodi, A.; Zanetti, V.; Canepa, M.; et al. Diagnosing Fabry nephropathy: The challenge of multiple kidney disease. BMC Nephrol. 2023, 24, 344. [Google Scholar]
- Tøndel, C.; Vikse, B.E.; Bostad, L.; Svarstad, E. Safety and complications of percutaneous kidney biopsies in 715 children and 8573 adults in Norway 1988–2010. Clin. J. Am. Soc. Nephrol. 2012, 7, 1591–1597. [Google Scholar] [PubMed]
- Tøndel, C.; Kanai, T.; Larsen, K.K.; Ito, S.; Politei, J.M.; Warnock, D.G.; Svarstad, E. Foot process effacement is an early marker of nephropathy in young classic fabry patients without albuminuria. Nephron 2015, 129, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Valbuena, C.; Leitão, D.; Carneiro, F.; Oliveira, J.P. Immunohistochemical diagnosis of Fabry nephropathy and localisation of globotriaosylceramide deposits in paraffin-embedded kidney tissue sections. Virchows Arch. 2012, 460, 211–221. [Google Scholar] [CrossRef]
- Alroy, J.; Sabnis, S.; Kopp, J.B. Renal pathology in Fabry disease. J. Am. Soc. Nephrol. 2002, 13 (Suppl. S2), S134–S138. [Google Scholar] [CrossRef]
- Abensur, H.; Dos Reis, M.A. Renal involvement in Fabry disease. J. Bras. Nefrol. ’Orgao Of. De Soc. Bras. Lat.-Am. Nefrol. 2016, 38, 245–254. [Google Scholar]
- Weidemann, F.; Sanchez-Niño, M.D.; Politei, J.; Oliveira, J.P.; Wanner, C.; Warnock, D.G.; Ortiz, A. Fibrosis: A key feature of Fabry disease with potential therapeutic implications. Orphanet J. Rare Dis. 2013, 8, 116. [Google Scholar] [PubMed]
- Valbuena, C.; Carvalho, E.; Bustorff, M.; Ganhão, M.; Relvas, S.; Nogueira, R.; Carneiro, F.; Olveira, J.P. Kidney biopsy findings in heterozygous Fabry disease females with early nephropathy. Virchows Arch. 2008, 453, 329–338. [Google Scholar]
- Sessa, A.; Toson, A.; Nebuloni, M.; Pallotti, F.; Giordano, F.; Battini, G.; Maglio, A.; Meroni, M.; Calconi, G.; Bertolone, G.; et al. Renal ultrastructural findings in Anderson-Fabry disease. J. Nephrol. 2002, 15, 109–112. [Google Scholar]
- Liguori, R.; Incensi, A.; De Pasqua, S.; Mignani, R.; Fileccia, E.; Santostefano, M.; Biagini, E.; Rapezzi, C.; Palmieri, S.; Romani, I.; et al. Skin globotriaosylceramide 3 deposits are specific to Fabry disease with classical mutations and associated with small fibre neuropathy. PLoS ONE 2017, 12, e0180581. [Google Scholar]
- Fogo, A.B.; Bostad, L.; Svarstad, E.; Cook, W.J.; Moll, S.; Barbey, F.; Geldenhuys, L.; West, M.; Ferluga, D.; Vujkovac, B.; et al. Scoring system for renal pathology in Fabry disease: Report of the International Study Group of Fabry Nephropathy (ISGFN). Nephrol. Dial. Transplant. 2010, 25, 2168–2177. [Google Scholar]
- Desnick, R.J. Fabry Disease: α-Galactosidase A Deficiency. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 5th ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 419–430. [Google Scholar]
- Manabe, S.; Mochizuki, T.; Sato, M.; Kataoka, H.; Taneda, S.; Honda, K.; Uchida, K.; Nitta, K. Lupus Nephritis and Hydroxychloroquine-Associated Zebra Bodies: Not Just in Fabry Disease. Kidney Med. 2021, 3, 442–446. [Google Scholar]
- Kanai, T.; Ito, T.; Aoyagi, J.; Yamagata, T. Membranous nephropathy without vacuolated podocytes in Fabry disease treated with agalsidase-β and carbamazepine: A case report. Medicine 2022, 101, E28830. [Google Scholar]
- Ronsin, C.; Jacquemont, L.; Testa, A.; Renaudin, K.; Ville, S. ANCA-negative pauci-immune crescentic glomerulonephritis associated with Fabry disease: Lesson for the clinical nephrologist. J. Nephrol. 2023, 36, 635–638. [Google Scholar]
- Wu, H.; Behera, T.R.; Gong, J.; Shen, Q. Coexistence of Fabry disease with IgM nephropathy: A case report. Medicine 2019, 98, e17566. [Google Scholar]
- Maixnerová, D.; Tesař, V.; Ryšavá, R.; Reiterová, J.; Poupětová, H.; Dvořáková, L.; Goláň, L.; Neprašová, M.; Kidorová, J.; Merta, M.; et al. The coincidence of IgA nephropathy and Fabry disease. BMC Nephrol. 2013, 14, 6. [Google Scholar]
- Zhou, L.N.; Dong, S.S.; Zhang, S.Z.; Huang, L.W.; Huang, W. Concurrent fabry disease and immunoglobulin a nephropathy: A case report. BMC Nephrol. 2023, 24, 324. [Google Scholar] [CrossRef]
- Choi, H.; Yim, H.; Park, I. Concurrent Fabry disease and IgA nephropathy in a patient with Crohn’s disease: A case report. Nephrology 2025, 30, e14419. [Google Scholar]
- Rusu, E.E.; Zilisteanu, D.S.; Ciobotaru, L.M.; Gherghiceanu, M.; Procop, A.; Jurcut, R.O.; Dulamea, A.O.; Sorohan, B.M. The Impact of Kidney Biopsy for Fabry Nephropathy Evaluation on Patients’ Management and Long-Term Outcomes: Experience of a Single Center. Biomedicines 2022, 10, 1520. [Google Scholar] [CrossRef]
- Pinto EVairo, F.; Pichurin, P.N.; Fervenza, F.C.; Nasr, S.H.; Mills, K.; Schmitz, C.T.; Klee, E.W.; Hermann, S.M. Nail-patella-like renal disease masquerading as Fabry disease on kidney biopsy: A case report. BMC Nephrol. 2020, 21, 341. [Google Scholar]
- Pintavorn, P.; Munie, S.; Munagapati, S. Lamellar Bodies in Podocytes Associated with Compound Heterozygous Mutations for Niemann Pick Type C1 Mimicking Fabry Disease, a Case Report. Can. J. Kidney Health Dis. 2022, 9, 20543581221124635. [Google Scholar]
- Choung, H.Y.G.; Jean-Gilles, J.; Goldman, B. Myeloid bodies is not an uncommon ultrastructural finding. Ultrastruct. Pathol. 2022, 46, 130–138. [Google Scholar] [CrossRef]
- Lei, L.; Oh, G.; Sutherland, S.; Abra, G.; Higgins, J.; Sibley, R.; Troxell, M.; Kambham, N. Myelin bodies in LMX1B-associated nephropathy: Potential for misdiagnosis. Pediatr. Nephrol. 2020, 35, 1647–1657. [Google Scholar] [CrossRef]
- Chen, J.; Bai, L.; He, Y. A possible case of carbamazepine-induced renal phospholipidosis mimicking Fabry disease. Clin. Exp. Nephrol. 2022, 26, 303–304. [Google Scholar] [CrossRef]
- Sen, R.; Borghoff, K.; Foster, K.W.; Radio, S.J.; Erickson, A.; Hearth-Holmes, M. Hydroxychloroquine and Fabry Disease: Three Case Reports Examining an Unexpected Pathologic Link and a Review of the Literature. Case Rep. Rheumatol. 2022, 2022, 2930103. [Google Scholar] [CrossRef]
- Germain, D.P.; Levade, T.; Hachulla, E.; Knebelmann, B.; Lacombe, D.; Seguin, V.L.; Nguyen, K.; Noël, E.; Rabès, J.P. Challenging the traditional approach for interpreting genetic variants: Lessons from Fabry disease. Clin. Genet. 2022, 101, 390–402. [Google Scholar] [CrossRef]
- Izhar, R.; Borriello, M.; La Russa, A.; Di Paola, R.; De, A.; Capasso, G.; Ingrosso, D.; Perna, A.F.; Simeoni, M. Fabry Disease in Women: Genetic Basis, Available Biomarkers, and Clinical Manifestations. Genes 2024, 15, 37. [Google Scholar] [CrossRef]
- Wilcox, W.R.; Oliveira, J.P.; Hopkin, R.J.; Ortiz, A.; Banikazemi, M.; Feldt-Rasmussen, U.; Sims, K.; Waldek, S.; Pastores, G.M.; Lee, P.; et al. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Mol. Genet. Metab. 2008, 93, 112–128. [Google Scholar] [CrossRef]
- Cerón-Rodríguez, M.; Ramón-García, G.; Barajas-Colón, E.; Franco-Álvarez, I.; Salgado-Loza, J.L. Renal globotriaosylceramide deposits for Fabry disease linked to uncertain pathogenicity gene variant c.352C>T/p.Arg118Cys: A family study. Mol. Genet. Genom. Med. 2019, 7, e981. [Google Scholar] [CrossRef]
- Lau, K.; Üçeyler, N.; Cairns, T.; Lorenz, L.; Sommer, C.; Schindehütte, M.; Armann, K.; Wanner, C.; Nordebeck, P. Gene variants of unknown significance in Fabry disease: Clinical characteristics of c.376A>G (p.Ser126Gly). Mol. Genet. Genom. Med. 2022, 10, e1912. [Google Scholar] [CrossRef]
- Santostefano, M.; Cappuccilli, M.; Gibertoni, D.; Fabbrizio, B.; Malvi, D.; Demetri, M.; Capelli, I.; Tringali, E.; Papa, V.; Biagini, E.; et al. Fabry Disease Nephropathy: Histological Changes with Nonclassical Mutations and Genetic Variants of Unknown Significance. Am. J. Kidney Dis. 2023, 82, 581–596.e0. [Google Scholar] [CrossRef]
- Mauer, M.; Sokolovskiy, A.; Barth, J.A.; Castelli, J.P.; Williams, H.N.; Benjamin, E.R.; Najafian, B. Reduction of podocyte globotriaosylceramide content in adult male patients with Fabry disease with amenable GLA mutations following 6 months of migalastat treatment. J. Med. Genet. 2017, 54, 781–786. [Google Scholar] [CrossRef]
- Najafian, B.; Svarstad, E.; Bostad, L.; Gubler, M.C.; Tøndel, C.; Whitley, C.; Mauer, M. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011, 79, 663–670. [Google Scholar]
- Najafian, B.; Silvestroni, A.; Sokolovskiy, A.; Tøndel, C.; Svarstad, E.; Obrisca, B.; Ismail, G.; Holida, M.D.; Mauer, M. A novel unbiased method reveals progressive podocyte globotriaosylceramide accumulation and loss with age in females with Fabry disease. Kidney Int. 2022, 102, 173–182. [Google Scholar] [CrossRef]
- Mauer, M.; Glynn, E.; Svarstad, E.; Tøndel, C.; Gubler, M.C.; West, M.; Sokolovskiy, A.; Whitley, C.; Najafian, B. Mosaicism of podocyte involvement is related to podocyte injury in females with Fabry disease. PLoS ONE 2014, 9, e112188. [Google Scholar]
- Smerkous, D.; Mauer, M.; Tøndel, C.; Svarstad, E.; Gubler, M.C.; Nelson, R.G.; Oliveira, J.P.; Sargolzaeiaval, F.; Najafian, B. Development of an automated estimation of foot process width using deep learning in kidney biopsies from patients with Fabry, minimal change, and diabetic kidney diseases. Kidney Int. 2024, 105, 165–176. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fabry Disease | Nail–Patella Syndrome | Niemann-Pick Disease Types A/B and C | |
|---|---|---|---|
| Gene | GLA | LMX1B | NPC1-NPC2 |
| Clinical features | Neuropathic pain Hypertrophic cardiomyopathy Cornea verticillata Gastrointestinal symptoms Hypohidrosis Angiokeratoma | Skeletal abnormalities (kneecap, iliac, patellae) Nail abnormalities Ocular (glaucoma) Neurological issues | Jaundice Hepatosplenomegaly Neurological issues (supranuclear vertical gaze palsy, cerebellar ataxia) Gelastic cataplexy |
| Renal involvement | Hematuria Proteinuria CKD | Hematuria (30–50 cases) Proteinuria ESRD (5% of patients) | Rare Hematuria Proteinuria CKD |
| Kidney biopsy findings | Myeloid bodies are widespread in all renal cellular compartments | Minor modifications Moth-eaten GBM (electron-lucent regions thickened GBM) Deposition of curvilinear type III collagen fibrils on GBM | Foamy podocyte’s cytoplasm on LM lamellar bodies in podocytes on EM |
| Study/Reference | Histopathological Findings | Clinical Prognostic Correlation |
|---|---|---|
| Fogo et al., 2010 [21] (ISGFN) | Semi-quantitative histological scoring of fibrosis, sclerosis, vascular changes | Used for risk stratification and prognosis assessment |
| Najafian et al., 2011 [44] | Progressive podocyte injury and Gb3 accumulation on EM | Associated with worsening renal function and proteinuria |
| Weidemann et al., 2013 [17] | Renal fibrosis on LM | Marker of disease progression and resistance to therapy |
| Tøndel et al., 2015 [13] | Podocyte foot process effacement on EM | Early marker of nephropathy, even before proteinuria onset |
| Mauer et al., 2017 [43] | Reduction of Gb3 inclusion in podocyte on EM with treatment | Correlates with improved renal outcomes in treated patients |
| Rusu et al., 2022 [30] | Glomerular sclerosis on LM | Predicts kidney disease progression and mortality |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capelli, I.; Martano, L.; Berti, G.M.; Vischini, G.; Lerario, S.; Donadio, V.; Incensi, A.; Aiello, V.; Ciurli, F.; Fabbrizio, B.; et al. The Role of Kidney Biopsy in Fabry Disease. Biomedicines 2025, 13, 767. https://doi.org/10.3390/biomedicines13040767
Capelli I, Martano L, Berti GM, Vischini G, Lerario S, Donadio V, Incensi A, Aiello V, Ciurli F, Fabbrizio B, et al. The Role of Kidney Biopsy in Fabry Disease. Biomedicines. 2025; 13(4):767. https://doi.org/10.3390/biomedicines13040767
Chicago/Turabian StyleCapelli, Irene, Laura Martano, Gian Marco Berti, Gisella Vischini, Sarah Lerario, Vincenzo Donadio, Alex Incensi, Valeria Aiello, Francesca Ciurli, Benedetta Fabbrizio, and et al. 2025. "The Role of Kidney Biopsy in Fabry Disease" Biomedicines 13, no. 4: 767. https://doi.org/10.3390/biomedicines13040767
APA StyleCapelli, I., Martano, L., Berti, G. M., Vischini, G., Lerario, S., Donadio, V., Incensi, A., Aiello, V., Ciurli, F., Fabbrizio, B., Chilotti, S., Mignani, R., Pasquinelli, G., & La Manna, G. (2025). The Role of Kidney Biopsy in Fabry Disease. Biomedicines, 13(4), 767. https://doi.org/10.3390/biomedicines13040767