
Inhibition of the Renin–Angiotensin System Improves Hemodynamic Function of the Diabetic Rat Heart by Restoring Intracellular Calcium Regulation


Abstract
1. Introduction
2. Materials and Methods
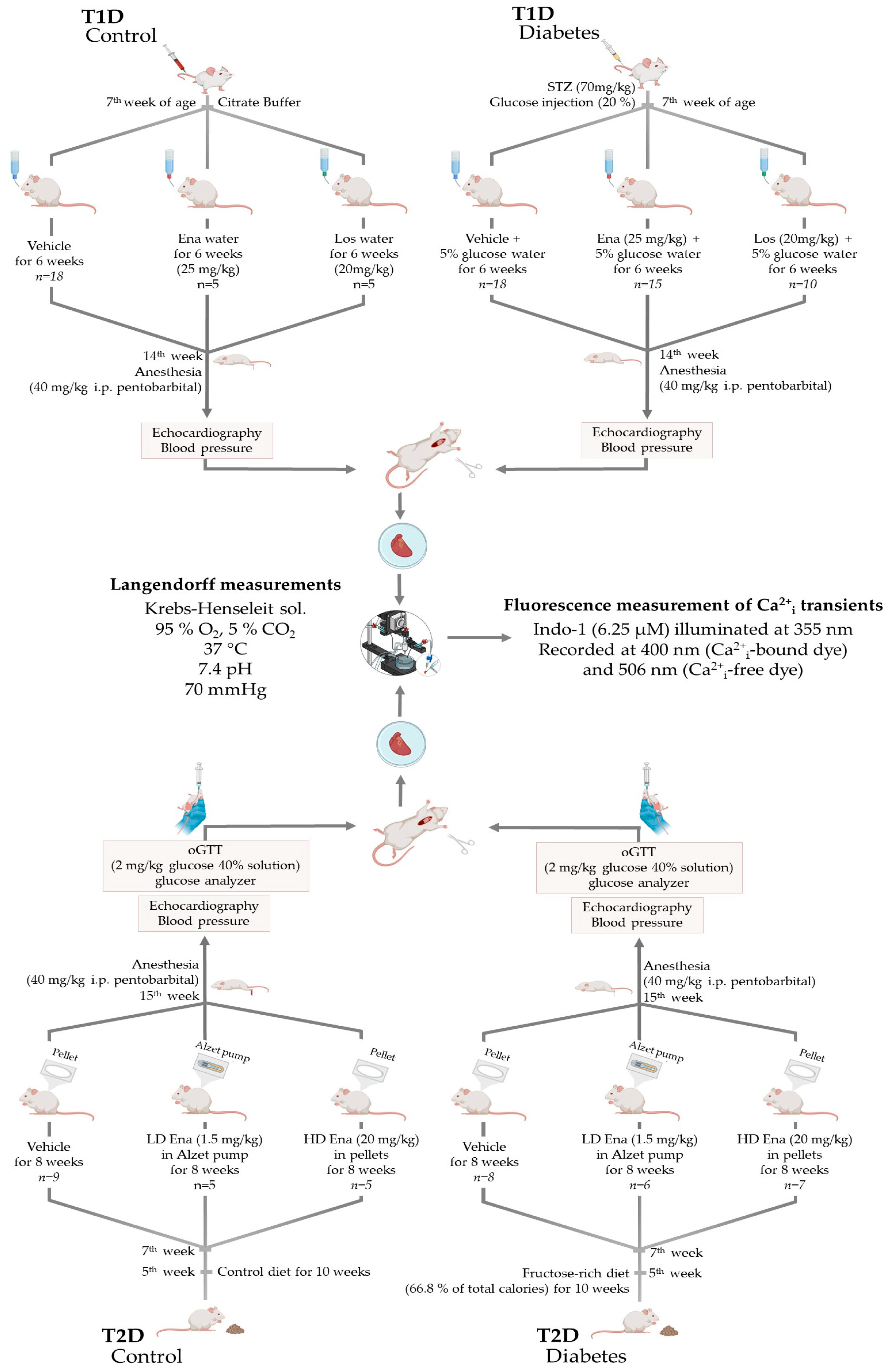
2.1. Study Design
2.1.1. Animal Models and Ethical Considerations
2.1.2. Type 1 Diabetic Model (T1D)—Experimental Design
- The drinking solution of 10 animals was supplemented with AT1R antagonist Los (Losartan Potassium, generous gift from Richter Gedeon Plc., Budapest, Hungary) at a dosage of ~20 mg/kg [36] (T1D + Los group, n = 10).
- Twenty rats consumed the glucose solution without drug supplementation (T1D Control, n = 20). However, two animals did not survive due to acute T1D complications, leaving 18 for analysis (T1D Control group, n = 18).
- Twenty animals served as the negative Control group; however, in 2 cases, the isolated heart preparation was not successful, reducing the final sample size to eighteen (Control, n = 18).
- To assess the potential effects of Ena and Los in healthy animals, 2 additional subgroups were created with 5 animals in each, where rats received the respective treatments (Control + Ena, n = 5 and Control + Los, n = 5).
2.1.3. Type 2 Diabetic Model (T2D)—Experimental Design
- One of the subgroups constituted animals (n = 8) treated with high-dose (HD) ACE inhibitor Ena (T2D + HD Ena group). This substance was liberated for 8 weeks continuously from subcutaneous biodegradation pellets. For this purpose, two 60-day release pellets of 200 mg Ena total dose (Innovative Research, Novi, MI, USA; Catalogue No.: SC-999) were implanted below the skin of the back to ensure an approximately 20 mg/kg mean daily dosage of the substance. Due to technical issues during the Langendorff experiment, one individual was excluded from the study, reducing the final sample size to 7.
- Another subgroup (n = 8) received Ena in low-dose (LD) (1.5 mg/kg [37,38], T2D + LD Ena group) via osmotic minipumps inserted below the skin of the back for 8 weeks. For this purpose, 4-week release 2 mL Alzet pumps (“2ML4” Alzet osmotic pump, DURECT Corporation, Cupertino, CA, USA) were filled with Ena (Enalapril maleate salt, Sigma-Aldrich Cas. No.: E-6888; Merck Group, Darmstadt, Germany) dissolved in distilled water to achieve the targeted mean daily release of 1.5 mg/kg. Pumps were replaced after 4 weeks to maintain a steady, even dose of the medicine. The LD Ena group was involved in the experimental protocol to inhibit local RAS activity by ACE inhibitor treatment independent of systemic blood pressure changes. The final sample size was 6, as 2 experiments were excluded due to cardiac arrest during isolated heart experiments.
- A subgroup of animals (n = 8) underwent the same operation procedure as the other subgroups, and subcutaneous pellets (n = 4) or osmotic minipumps (n = 4) loaded with vehicles were placed below their skins (T2D group). The experimental protocol was performed 10 weeks after the initiation of the fructose diet.
- T2D Control + HD Ena group (n = 5);
- T2D Control + LD Ena group (n = 5);
- T2D Control group (n = 9).
2.2. Experimental Protocol
2.2.1. Echocardiography
2.2.2. Blood Glucose Measurement, oGTT
2.2.3. Isolated Langendorff Perfused Heart—Fluorescent Measurement of Ca2+i Transients
2.2.4. Determination of Key Ca2+i Cycling Enzymes in T2D Groups by Western Blot Analysis
2.3. Statistical Analysis and Interpretation
3. Results
3.1. Biometric and Physiological Parameters in T1D and T2D Animals
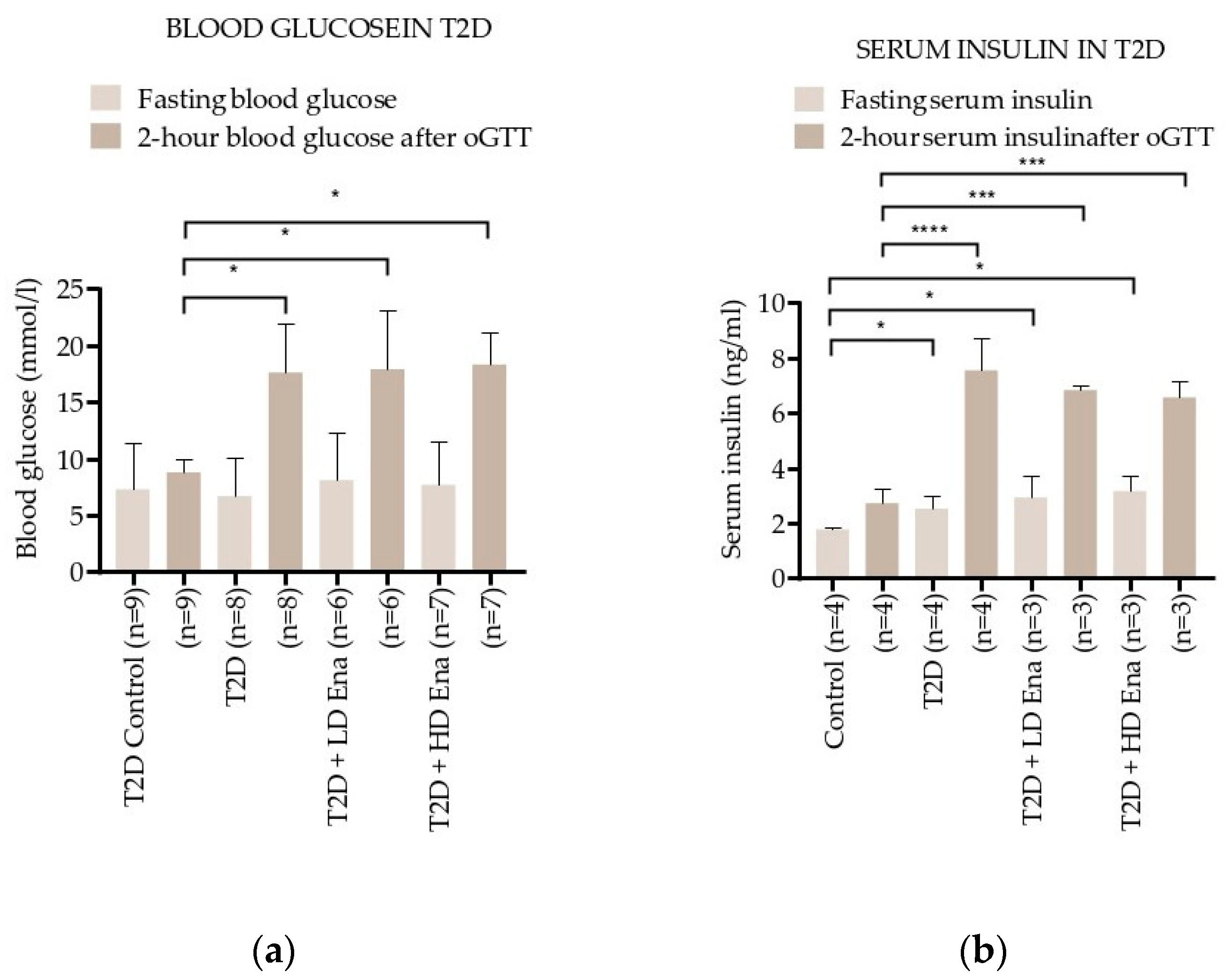
3.2. Blood Glucose and Insulin Levels in Response to oGTT in T2D Animals
3.3. Results of Echocardiography in T1D and T2D Animals
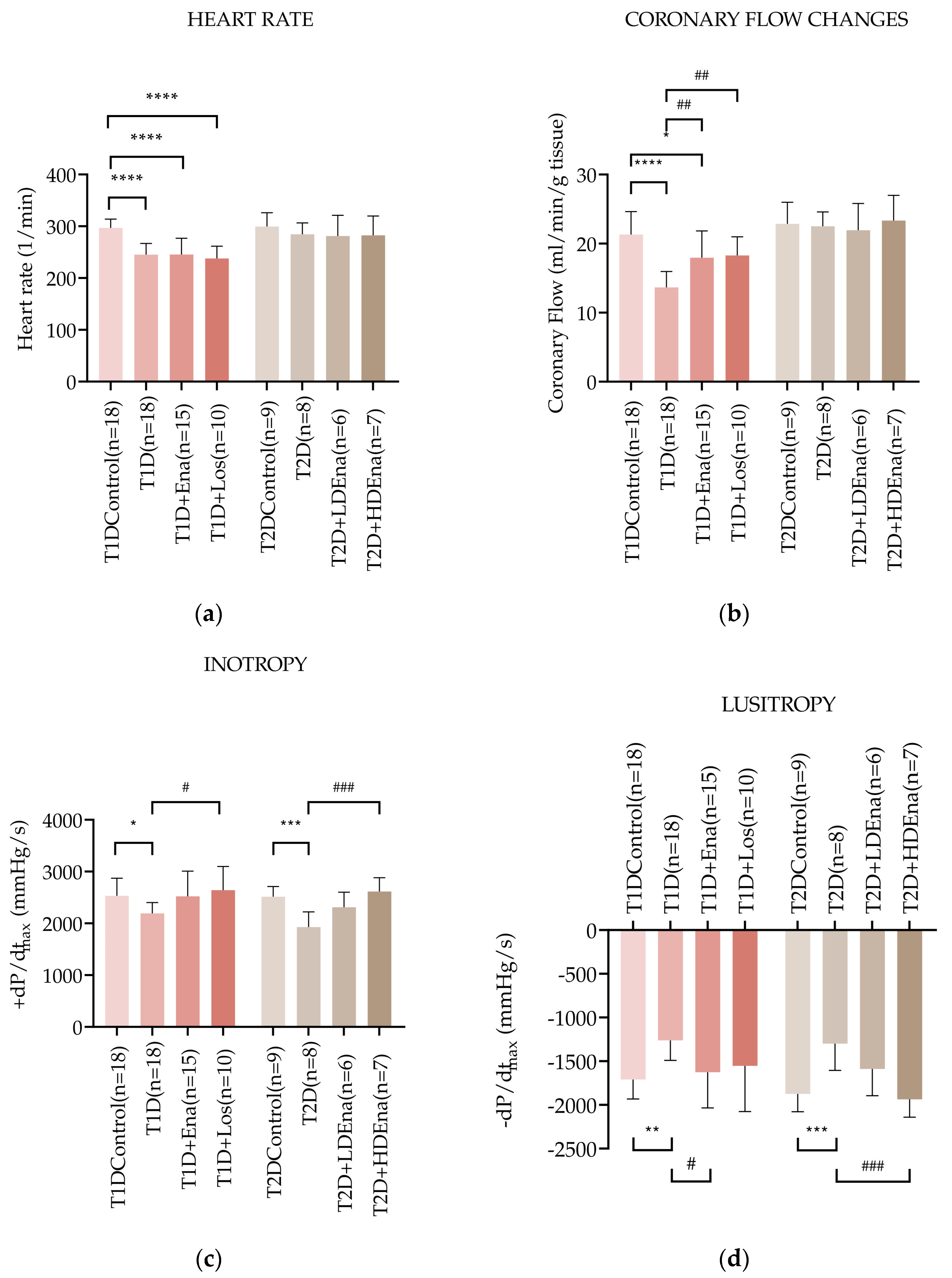
3.4. Hemodynamic Parameters in Langendorff Hearts of T1D and T2D Animals
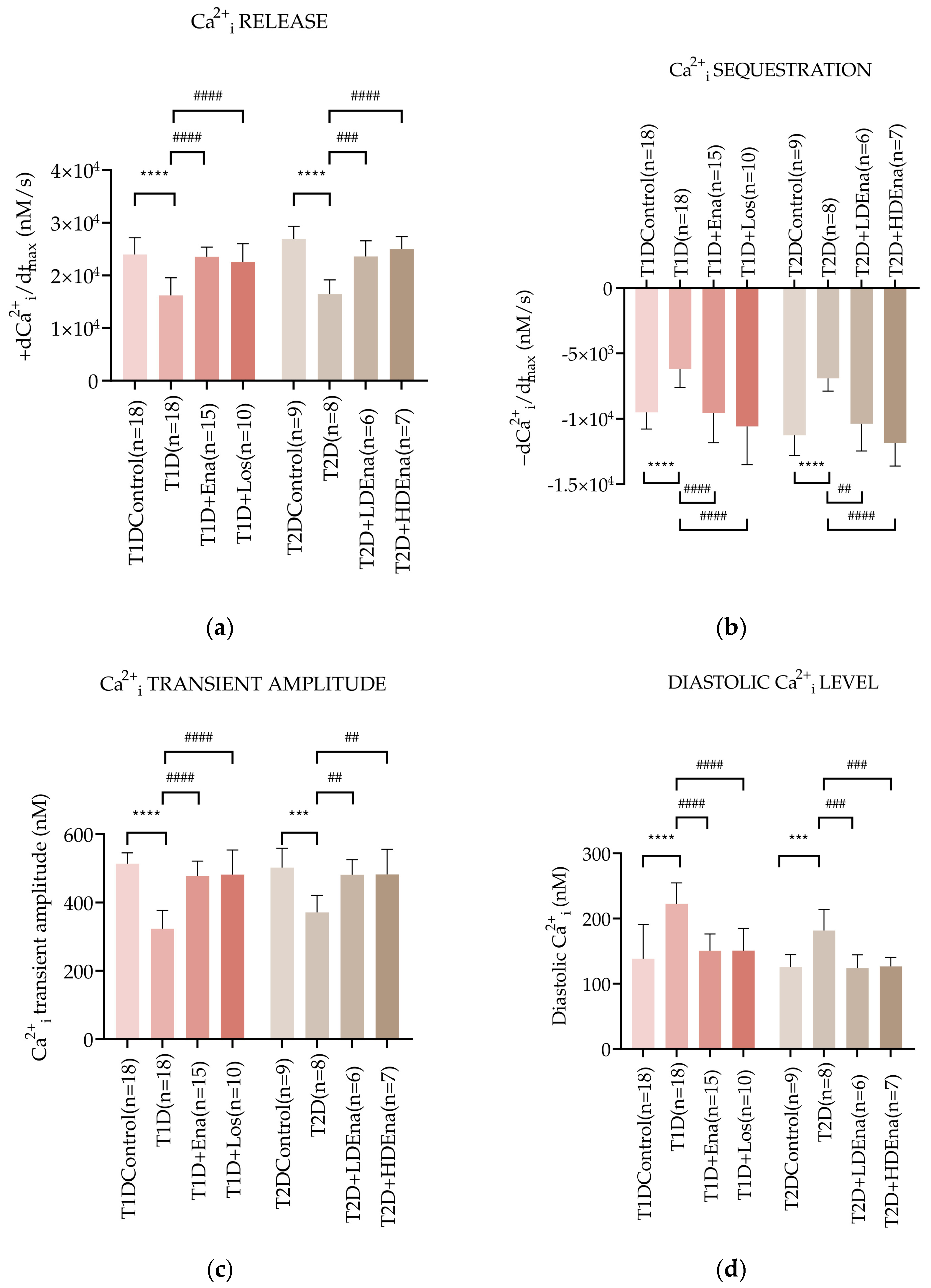
3.5. Myocardial Ca2+i Handling in Langendorff Hearts of T1D and T2D Animals
3.6. Expression of Key Ca2+i Cycling Enzymes in T2D Animals
4. Discussion
4.1. Isolated Pefused Heart Model
4.2. Validation of T1D and T2D Models
4.3. Local RAS Activation in Diabetes
4.4. Hemodynamic Parameters in Langendorff Hearts
4.5. Myocardial Ca2+i Transients in Langendorff Hearts
4.6. Therapeutic Implications
4.7. Limitations of the Study
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| +dCa2+i/dtmax | Ca2+i release |
| +dP/dtmax | Left ventricular inotropy |
| ACE | Angiotensin-converting enzyme |
| AT1R | Angiotensin type 1 receptor |
| ATII | Angiotensin II |
| Ca2+i | Intracellular calcium/intracellular calcium |
| −dCa2+i/dtmax | Ca2+i sequestration |
| −dP/dtmax | Left ventricular lusitropy |
| EDV | End-diastolic volume |
| EF | Ejection fraction |
| Ena | Enalapril |
| ESV | End-systolic volume |
| FS | Fractional shortening |
| HD Ena | High-dose enalapril |
| IVSd | Diastolic wall thickness |
| LD Ena | Low-dose enalapril |
| Los | Losartan |
| LVA | Left ventricular area |
| LVIDd | Left ventricular internal diameters in diastole |
| LVIDs | Left ventricular internal diameters in systole |
| LVL | Left ventricular length |
| MAPKs | Mitogen-activated protein kinase |
| NF-κB | Nuclear factor-kappa B |
| NOX | NADPH oxidase |
| oGTT | Oral glucose tolerance test |
| PLB | Phospholamban |
| PLC | Phospholipase C |
| P-PLB | 16Ser-phosphorylated phospholamban |
| RAS | Renin–angiotensin system |
| RyR2 | Ryanodine receptor type 2 |
| SERCA2a | Sarcoendoplasmic reticulum Ca2+-ATPase 2a |
| SR | Sarcoplasmic reticulum |
| STZ | Streptozotocin injection |
| T1D | Type 1 diabetes |
| T2D | Type 2 diabetes |
References
- Demographics: Total Adult Population (20-79 y), in 1,000s. Available online: https://diabetesatlas.org/data/ (accessed on 10 September 2024).
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Boonman-de Winter, L.J.M.; Rutten, F.H.; Cramer, M.J.M.; Landman, M.J.; Liem, A.H.; Rutten, G.E.H.M.; Hoes, A.W. High Prevalence of Previously Unknown Heart Failure and Left Ventricular Dysfunction in Patients with Type 2 Diabetes. Diabetologia 2012, 55, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Blecker, S.; Pazin-Filho, A.; Bertoni, A.; Chang, P.P.; Coresh, J.; Selvin, E. The Association of Hemoglobin A1c With Incident Heart Failure Among People Without Diabetes: The Atherosclerosis Risk in Communities Study. Diabetes 2010, 59, 2020–2026. [Google Scholar] [CrossRef]
- Nichols, G.A.; Gullion, C.M.; Koro, C.E.; Ephross, S.A.; Brown, J.B. The Incidence of Congestive Heart Failure in Type 2 Diabetes. Diabetes Care 2004, 27, 1879–1884. [Google Scholar] [CrossRef]
- Kannel, W.B. Diabetes and Cardiovascular Disease: The Framingham Study. JAMA 1979, 241, 2035. [Google Scholar] [CrossRef] [PubMed]
- Brunvand, L.; Fugelseth, D.; Stensaeth, K.H.; Dahl-Jørgensen, K.; Margeirsdottir, H.D. Early Reduced Myocardial Diastolic Function in Children and Adolescents with Type 1 Diabetes Mellitus a Population-Based Study. BMC Cardiovasc. Disord. 2016, 16, 103. [Google Scholar] [CrossRef]
- Lind, M.; Bounias, I.; Olsson, M.; Gudbjörnsdottir, S.; Svensson, A.-M.; Rosengren, A. Glycaemic Control and Incidence of Heart Failure in 20 985 Patients with Type 1 Diabetes: An Observational Study. Lancet 2011, 378, 140–146. [Google Scholar] [CrossRef]
- Suran, D.; Sinkovic, A.; Naji, F. Tissue Doppler Imaging Is a Sensitive Echocardiographic Technique to Detect Subclinical Systolic and Diastolic Dysfunction of Both Ventricles in Type 1 Diabetes Mellitus. BMC Cardiovasc. Disord. 2016, 16, 72. [Google Scholar] [CrossRef]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New Type of Cardiomyopathy Associated with Diabetic Glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Paolillo, S.; Marsico, F.; Prastaro, M.; Renga, F.; Esposito, L.; De Martino, F.; Di Napoli, P.; Esposito, I.; Ambrosio, A.; Ianniruberto, M.; et al. Diabetic Cardiomyopathy. Heart Fail. Clin. 2019, 15, 341–347. [Google Scholar] [CrossRef]
- Al Kury, L.T. Calcium Homeostasis in Ventricular Myocytes of Diabetic Cardiomyopathy. J. Diabetes Res. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Cai, L.; Li, W.; Wang, G.; Guo, L.; Jiang, Y.; Kang, Y.J. Hyperglycemia-Induced Apoptosis in Mouse Myocardium. Diabetes 2002, 51, 1938–1948. [Google Scholar] [CrossRef]
- Fiordaliso, F.; Li, B.; Latini, R.; Sonnenblick, E.H.; Anversa, P.; Leri, A.; Kajstura, J. Myocyte Death in Streptozotocin-Induced Diabetes in Rats Is Angiotensin II- Dependent. Lab. Investig. 2000, 80, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Fiordaliso, F.; Leri, A.; Cesselli, D.; Limana, F.; Safai, B.; Nadal-Ginard, B.; Anversa, P.; Kajstura, J. Hyperglycemia Activates P53 and P53-Regulated Genes Leading to Myocyte Cell Death. Diabetes 2001, 50, 2363–2375. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.Y.; Prins, J.B.; Marwick, T.H. Diabetic Cardiomyopathy: Evidence, Mechanisms, and Therapeutic Implications. Endocr. Rev. 2004, 25, 543–567. [Google Scholar] [CrossRef] [PubMed]
- Adeghate, E.; Singh, J. Structural Changes in the Myocardium during Diabetes-Induced Cardiomyopathy. Heart Fail. Rev. 2014, 19, 15–23. [Google Scholar] [CrossRef]
- Sayer, G.; Bhat, G. The Renin-Angiotensin-Aldosterone System and Heart Failure. Cardiol. Clin. 2014, 32, 21–32. [Google Scholar] [CrossRef]
- Kumar, R.; Yong, Q.C.; Thomas, C.M.; Baker, K.M. Review: Intracardiac Intracellular Angiotensin System in Diabetes. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2012, 302, R510–R517. [Google Scholar] [CrossRef]
- Bhullar, S.K.; Dhalla, N.S. Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy. Cells 2022, 11, 3336. [Google Scholar] [CrossRef]
- Lindholm, L.H.; Ibsen, H.; Dahlöf, B.; Devereux, R.B.; Beevers, G.; De Faire, U.; Fyhrquist, F.; Julius, S.; Kjeldsen, S.E.; Kristiansson, K.; et al. Cardiovascular Morbidity and Mortality in Patients with Diabetes in the Losartan Intervention For Endpoint Reduction in Hypertension Study (LIFE): A Randomised Trial against Atenolol. Lancet 2002, 359, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C. Reduction of Cardiovascular Events and Microvascular Complications in Diabetes with ACE Inhibitor Treatment: HOPE and MICRO-HOPE. Diabetes Metab. Res. Rev. 2002, 18, S82–S85. [Google Scholar] [CrossRef]
- Choi, K.M.; Zhong, Y.; Hoit, B.D.; Grupp, I.L.; Hahn, H.; Dilly, K.W.; Guatimosim, S.; Lederer, W.J.; Matlib, M.A. Defective Intracellular Ca 2+ Signaling Contributes to Cardiomyopathy in Type 1 Diabetic Rats. Am. J. Physiol.-Heart Circ. Physiol. 2002, 283, H1398–H1408. [Google Scholar] [CrossRef] [PubMed]
- Ligeti, L.; Szenczi, O.; Prestia, C.M.; Szabó, C.; Horváth, K.; Marcsek, Z.L.; van Stiphout, R.G.P.M.; van Riel, N.A.W.; Op den Buijs, J.; Van der Vusse, G.J.; et al. Altered Calcium Handling Is an Early Sign of Streptozotocin-Induced Diabetic Cardiomyopathy. Int. J. Mol. Med. 2006, 17, 1035–1043. [Google Scholar] [CrossRef]
- Pierce, G. Regulation of Intracellular Ca2+ in the Heart during Diabetes. Cardiovasc. Res. 1997, 34, 41–47. [Google Scholar] [CrossRef]
- Schaffer, S. Mechanisms Underlying Depressed Na+/Ca2+ Exchanger Activity in the Diabetic Heart. Cardiovasc. Res. 1997, 34, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Yu, J. Intracellular Calcium Levels Are Unchanged in the Diabetic Heart. Cardiovasc. Res. 1997, 34, 91–98. [Google Scholar] [CrossRef]
- Miklós, Z.; Kemecsei, P.; Bíró, T.; Marincsák, R.; Tóth, B.I.; Op den Buijs, J.; Benis, É.; Drozgyik, A.; Ivanics, T. Early Cardiac Dysfunction Is Rescued by Upregulation of SERCA 2a Pump Activity in a Rat Model of Metabolic Syndrome. Acta Physiol. 2012, 205, 381–393. [Google Scholar] [CrossRef]
- Yu, Z.; Tibbits, G.F.; McNeill, J.H. Cellular Functions of Diabetic Cardiomyocytes: Contractility, Rapid-Cooling Contracture, and Ryanodine Binding. Am. J. Physiol.-Heart Circ. Physiol. 1994, 266, H2082–H2089. [Google Scholar] [CrossRef]
- op den Buijs, J.; Miklós, Z.; van Riel, N.A.W.; Prestia, C.M.; Szenczi, O.; Tóth, A.; Van der Vusse, G.J.; Szabó, C.; Ligeti, L.; Ivanics, T. Beta-Adrenergic Activation Reveals Impaired Cardiac Calcium Handling at Early Stage of Diabetes. Life Sci. 2005, 76, 1083–1098. [Google Scholar] [CrossRef]
- Turan, B.; Désilets, M.; Açan, L.N.; Hotomaroglu, O.; Vannier, C.; Vassort, G. Oxidative Effects of Selenite on Rat Ventricular Contractility and Ca Movements. Cardiovasc. Res. 1996, 32, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Turan, B.; Fliss, H.; Désilets, M. Oxidants Increase Intracellular Free Zn2+ Concentration in Rabbit Ventricular Myocytes. Am. J. Physiol. 1997, 272, H2095–H2106. [Google Scholar] [CrossRef] [PubMed]
- Dukacz, S.A.; Adams, M.A.; Kline, R.L. The Persistent Effect of Long-Term Enalapril on Pressure Natriuresis in Spontaneously Hypertensive Rats. Am. J. Physiol.-Ren. Physiol. 1997, 273, F104–F112. [Google Scholar] [CrossRef]
- Kett, M.M.; Alcorn, D.; Bertram, J.F.; Anderson, W.P. Enalapril Does Not Prevent Renal Arterial Hypertrophy in Spontaneously Hypertensive Rats. Hypertension 1995, 25, 335–342. [Google Scholar] [CrossRef]
- Manni, M.E.; Bigagli, E.; Lodovici, M.; Zazzeri, M.; Raimondi, L. The Protective Effect of Losartan in the Nephropathy of the Diabetic Rat Includes the Control of Monoamine Oxidase Type A Activity. Pharmacol. Res. 2012, 65, 465–471. [Google Scholar] [CrossRef]
- Makino, N.; Sugano, M.; Hata, T.; Taguchi, S.; Yanaga, T. Chronic Low-Dose Treatment with Enalapril Induced Cardiac Regression of Left Ventricular Hypertrophy. Mol. Cell Biochem. 1996, 163–164, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Sakata, Y.; Yamamoto, K.; Mano, T.; Nishikawa, N.; Yoshida, J.; Hori, M.; Miwa, T.; Masuyama, T. Activation of Matrix Metalloproteinases Precedes Left Ventricular Remodeling in Hypertensive Heart Failure Rats: Its Inhibition as a Primary Effect of Angiotensin-Converting Enzyme Inhibitor. Circulation 2004, 109, 2143–2149. [Google Scholar] [CrossRef]
- Baumann, M.; Janssen, B.J.A.; Hermans, J.J.R.; Peutz-Kootstra, C.; Witzke, O.; Smits, J.F.M.; Struijker Boudier, H.A.J. Transient AT1 Receptor-Inhibition in Prehypertensive Spontaneously Hypertensive Rats Results in Maintained Cardiac Protection until Advanced Age. J. Hypertens. 2007, 25, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Ivanics, T.; Miklós, Z.; Dézsi, L.; Ikrényi, K.; Tóth, A.; Roemen, T.H.; Van der Vusse, G.J.; Ligeti, L. Concomitant Accumulation of Intracellular Free Calcium and Arachidonic Acid in the Ischemic-Reperfused Rat Heart. Mol. Cell Biochem. 2001, 226, 119–128. [Google Scholar] [CrossRef]
- Wafa, D.; Koch, N.; Kovács, J.; Kerék, M.; Proia, R.L.; Tigyi, G.J.; Benyó, Z.; Miklós, Z. Opposing Roles of S1P3 Receptors in Myocardial Function. Cells 2020, 9, 1770. [Google Scholar] [CrossRef]
- Miklós, Z.; Wafa, D.; Nádasy, G.L.; Tóth, Z.E.; Besztercei, B.; Dörnyei, G.; Laska, Z.; Benyó, Z.; Ivanics, T.; Hunyady, L.; et al. Angiotensin II-Induced Cardiac Effects Are Modulated by Endocannabinoid-Mediated CB1 Receptor Activation. Cells 2021, 10, 724. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A New Generation of Ca2+ Indicators with Greatly Improved Fluorescence Properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Diagnosis & Tests|ADA. Available online: https://diabetes.org/about-diabetes/diagnosis (accessed on 6 October 2024).
- Kumar, R.; Nandhini, L.P.; Kamalanathan, S.; Sahoo, J.; Vivekanadan, M. Evidence for Current Diagnostic Criteria of Diabetes Mellitus. World J. Diabetes 2016, 7, 396–405. [Google Scholar] [CrossRef]
- Gilbert, G.; Demydenko, K.; Dries, E.; Puertas, R.D.; Jin, X.; Sipido, K.; Roderick, H.L. Calcium Signaling in Cardiomyocyte Function. Cold Spring Harb. Perspect. Biol. 2020, 12, a035428. [Google Scholar] [CrossRef]
- Høydal, M.A.; Kirkeby-Garstad, I.; Karevold, A.; Wiseth, R.; Haaverstad, R.; Wahba, A.; Stølen, T.L.; Contu, R.; Condorelli, G.; Ellingsen, Ø.; et al. Human Cardiomyocyte Calcium Handling and Transverse Tubules in Mid-stage of Post-myocardial-infarction Heart Failure. ESC Heart Fail. 2018, 5, 332–342. [Google Scholar] [CrossRef]
- Luo, M.; Anderson, M.E. Ca2+ Cycling in Heart Failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef]
- Pierce, G.N.; Czubryt, M.P. The Contribution of Ionic Imbalance to Ischemia/Reperfusion-Induced Injury. J. Mol. Cell Cardiol. 1995, 27, 53–63. [Google Scholar] [CrossRef]
- Pedersen, T.M.; Boardman, N.T.; Hafstad, A.D.; Aasum, E. Isolated Perfused Working Hearts Provide Valuable Additional Information during Phenotypic Assessment of the Diabetic Mouse Heart. PLoS ONE 2018, 13, e0204843. [Google Scholar] [CrossRef]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. 2021, 1, e78. [Google Scholar] [CrossRef]
- Cosyns, B.; Droogmans, S.; Hernot, S.; Degaillier, C.; Garbar, C.; Weytjens, C.; Roosens, B.; Schoors, D.; Lahoutte, T.; Franken, P.R.; et al. Effect of Streptozotocin-Induced Diabetes on Myocardial Blood Flow Reserve Assessed by Myocardial Contrast Echocardiography in Rats. Cardiovasc. Diabetol. 2008, 7, 26. [Google Scholar] [CrossRef]
- Li, X.S.; Tanz, R.D.; Chang, K.S. Effect of Age and Methacholine on the Rate and Coronary Flow of Isolated Hearts of Diabetic Rats. Br. J. Pharmacol. 1989, 97, 1209–1217. [Google Scholar] [CrossRef]
- Ferrario, C.M. Cardiac Remodelling and RAS Inhibition. Ther. Adv. Cardiovasc. Dis. 2016, 10, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Matthes, J.; Schuster, I.; Valdivia, H.H.; Herzig, S.; Richard, S.; Gómez, A.M. Mechanisms of [Ca2+]i Transient Decrease in Cardiomyopathy of Db/Db Type 2 Diabetic Mice. Diabetes 2006, 55, 608–615. [Google Scholar] [CrossRef]
- Akula, A.; Kota, M.K.; Gopisetty, S.G.; Chitrapu, R.V.; Kalagara, M.; Kalagara, S.; Veeravalli, K.K.; Gomedhikam, J. Biochemical, Histological and Echocardiographic Changes during Experimental Cardiomyopathy in STZ-Induced Diabetic Rats. Pharmacol. Res. 2003, 48, 429–435. [Google Scholar] [CrossRef]
- Turan, B.; Ugur, M.; Ozdemir, S.; Yaras, N. Altered Mechanical and Electrical Activities of the Diabetic Heart: Possible Use of New Therapeutics? Exp. Clin. Cardiol. 2005, 10, 189–195. [Google Scholar]
- Singh, V.P.; Le, B.; Khode, R.; Baker, K.M.; Kumar, R. Intracellular Angiotensin II Production in Diabetic Rats Is Correlated With Cardiomyocyte Apoptosis, Oxidative Stress, and Cardiac Fibrosis. Diabetes 2008, 57, 3297–3306. [Google Scholar] [CrossRef]
- Giacchetti, G.; Sechi, L.A.; Rilli, S.; Carey, R.M. The Renin–Angiotensin–Aldosterone System, Glucose Metabolism and Diabetes. Trends Endocrinol. Metab. 2005, 16, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Price, D.A.; Porter, L.E.; Gordon, M.; Fisher, N.D.L.; De’Oliveira, J.M.F.; Laffel, L.M.B.; Passan, D.R.; Williams, G.H.; Hollenberg, N.K. The Paradox of the Low-Renin State in Diabetic Nephropathy. J. Am. Soc. Nephrol. 1999, 10, 2382–2391. [Google Scholar] [CrossRef]
- Anderson, S.; Jung, F.F.; Ingelfinger, J.R. Renal Renin-Angiotensin System in Diabetes: Functional, Immunohistochemical, and Molecular Biological Correlations. Am. J. Physiol.-Ren. Physiol. 1993, 265, F477–F486. [Google Scholar] [CrossRef]
- Raizada, V.; Skipper, B.; Luo, W.; Griffith, J. Intracardiac and Intrarenal Renin-Angiotensin Systems: Mechanisms of Cardiovascular and Renal Effects. J. Investig. Med. 2007, 55, 341–359. [Google Scholar] [CrossRef]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of Local Renin-Angiotensin Systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Baker, K.M.; Kumar, R. Activation of the Intracellular Renin-Angiotensin System in Cardiac Fibroblasts by High Glucose: Role in Extracellular Matrix Production. Am. J. Physiol. -Heart Circ. Physiol. 2008, 294, H1675–H1684. [Google Scholar] [CrossRef] [PubMed]
- Aboalgasm, H.; Petersen, M.; Gwanyanya, A. Improvement of Cardiac Ventricular Function by Magnesium Treatment in Chronic Streptozotocin-Induced Diabetic Rat Heart. Cardiovasc. J. Afr. 2021, 32, 141–148. [Google Scholar] [CrossRef]
- Sultan, A.; Jacobson, M.; Adeghate, E.; Oulhaj, A.; Shafiullah, M.; Qureshi, A.; Howarth, F.C. Effects of Obesity and Diabesity on Heart Rhythm in the Zucker Rat. Clin. Exp. Pharma Physio 2021, 48, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Le, B.; Bhat, V.B.; Baker, K.M.; Kumar, R. High-Glucose-Induced Regulation of Intracellular ANG II Synthesis and Nuclear Redistribution in Cardiac Myocytes. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H939–H948. [Google Scholar] [CrossRef]
- Garvin, A.M.; Khokhar, B.S.; Czubryt, M.P.; Hale, T.M. RAS Inhibition in Resident Fibroblast Biology. Cell. Signal. 2021, 80, 109903. [Google Scholar] [CrossRef]
- Tuleta, I.; Frangogiannis, N.G. Fibrosis of the Diabetic Heart: Clinical Significance, Molecular Mechanisms, and Therapeutic Opportunities. Adv. Drug Deliv. Rev. 2021, 176, 113904. [Google Scholar] [CrossRef]
- Pan, K.-L.; Hsu, Y.-C.; Chang, S.-T.; Chung, C.-M.; Lin, C.-L. The Role of Cardiac Fibrosis in Diabetic Cardiomyopathy: From Pathophysiology to Clinical Diagnostic Tools. IJMS 2023, 24, 8604. [Google Scholar] [CrossRef]
- Dong, B.; Yu, Q.T.; Dai, H.Y.; Gao, Y.Y.; Zhou, Z.L.; Zhang, L.; Jiang, H.; Gao, F.; Li, S.Y.; Zhang, Y.H.; et al. Angiotensin-Converting Enzyme-2 Overexpression Improves Left Ventricular Remodeling and Function in a Rat Model of Diabetic Cardiomyopathy. J. Am. Coll. Cardiol. 2012, 59, 739–747. [Google Scholar] [CrossRef]
- Russo, I.; Frangogiannis, N.G. Diabetes-Associated Cardiac Fibrosis: Cellular Effectors, Molecular Mechanisms and Therapeutic Opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef]
- Huynh, K.; McMullen, J.R.; Julius, T.L.; Tan, J.W.; Love, J.E.; Cemerlang, N.; Kiriazis, H.; Du, X.-J.; Ritchie, R.H. Cardiac-Specific IGF-1 Receptor Transgenic Expression Protects Against Cardiac Fibrosis and Diastolic Dysfunction in a Mouse Model of Diabetic Cardiomyopathy. Diabetes 2010, 59, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Ares-Carrasco, S.; Picatoste, B.; Benito-Martín, A.; Zubiri, I.; Sanz, A.B.; Sánchez-Niño, M.D.; Ortiz, A.; Egido, J.; Tuñón, J.; Lorenzo, O. Myocardial Fibrosis and Apoptosis, but Not Inflammation, Are Present in Long-Term Experimental Diabetes. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H2109–H2119. [Google Scholar] [CrossRef]
- Wang, M.; Li, Y.; Li, S.; Lv, J. Endothelial Dysfunction and Diabetic Cardiomyopathy. Front. Endocrinol. 2022, 13, 851941. [Google Scholar] [CrossRef] [PubMed]
- Pacurari, M.; Kafoury, R.; Tchounwou, P.B.; Ndebele, K. The Renin-Angiotensin-Aldosterone System in Vascular Inflammation and Remodeling. Int. J. Inflamm. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yaras, N.; Ugur, M.; Ozdemir, S.; Gurdal, H.; Purali, N.; Lacampagne, A.; Vassort, G.; Turan, B. Effects of Diabetes on Ryanodine Receptor Ca Release Channel (RyR2) and Ca2+ Homeostasis in Rat Heart. Diabetes 2005, 54, 3082–3088. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Nallani, K.; Besch, H.R.; Dincer, U.D. Streptozotocin-Induced Diabetes Increases Disulfide Bond Formation on Cardiac Ryanodine Receptor (RyR2). J. Pharmacol. Exp. Ther. 2003, 305, 989–998. [Google Scholar] [CrossRef]
- Ng, Y.H.; Lamberts, R.R.; Jones, P.P.; Sammut, I.A.; Diffee, G.M.; Wilkins, G.T.; Baldi, J.C. Sarco/Endoplasmic Reticulum Calcium ATPase Activity Is Unchanged despite Increased Myofilament Calcium Sensitivity in Zucker Type 2 Diabetic Fatty Rat Heart. Sci. Rep. 2022, 12, 16904. [Google Scholar] [CrossRef]
- Belke, D.D.; Swanson, E.A.; Dillmann, W.H. Decreased Sarcoplasmic Reticulum Activity and Contractility in Diabetic Db/Db Mouse Heart. Diabetes 2004, 53, 3201–3208. [Google Scholar] [CrossRef]
- Zhong, Y.; Ahmed, S.; Grupp, I.L.; Matlib, M.A. Altered SR Protein Expression Associated with Contractile Dysfunction in Diabetic Rat Hearts. Am. J. Physiol.-Heart Circ. Physiol. 2001, 281, H1137–H1147. [Google Scholar] [CrossRef]
- Sulaiman, M.; Matta, M.J.; Sunderesan, N.R.; Gupta, M.P.; Periasamy, M.; Gupta, M. Resveratrol, an Activator of SIRT1, Upregulates Sarcoplasmic Calcium ATPase and Improves Cardiac Function in Diabetic Cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 2010, 298, H833–H843. [Google Scholar] [CrossRef]
- Phillips, M.I.; De Oliveira, E.M. Brain Renin Angiotensin in Disease. J. Mol. Med. 2008, 86, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Wen, H. Oxidative Stress-Mediated Effects of Angiotensin II in the Cardiovascular System. World J. Hypertens. 2012, 2, 34. [Google Scholar] [CrossRef]
- Xu, K.Y.; Zweier, J.L.; Becker, L.C. Hydroxyl Radical Inhibits Sarcoplasmic Reticulum Ca2+ -ATPase Function by Direct Attack on the ATP Binding Site. Circ. Res. 1997, 80, 76–81. [Google Scholar] [CrossRef]
- Ying, J.; Sharov, V.; Xu, S.; Jiang, B.; Gerrity, R.; Schöneich, C.; Cohen, R.A. Cysteine-674 Oxidation and Degradation of Sarcoplasmic Reticulum Ca2+ ATPase in Diabetic Pig Aorta. Free Radic. Biol. Med. 2008, 45, 756–762. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Dinçer, Ü.D.; Besch, H.R. Ryanodine Receptor Dysfunction in Hearts of Streptozotocin-Induced Diabetic Rats. Mol. Pharmacol. 2001, 60, 1356–1364. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Nallani, K.; Yu, Y.; Cocklin, R.R.; Zhang, Y.; Wang, M.; Dincer, Ü.D.; Besch, H.R. Chronic Diabetes Increases Advanced Glycation End Products on Cardiac Ryanodine Receptors/Calcium-Release Channels. Diabetes 2003, 52, 1825–1836. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Zhang, Y.; Shao, C.H.; Wang, M.; Patel, K.P.; Dincer, Ü.D.; Besch, H.R. Diabetes Increases Formation of Advanced Glycation End Products on Sarco(Endo)Plasmic Reticulum Ca2+-ATPase. Diabetes 2004, 53, 463–473. [Google Scholar] [CrossRef]
- Tsai, C.-T.; Wu, C.-K.; Lee, J.-K.; Chang, S.-N.; Kuo, Y.-M.; Wang, Y.-C.; Lai, L.-P.; Chiang, F.-T.; Hwang, J.-J.; Lin, J.-L. TNF- down-Regulates Sarcoplasmic Reticulum Ca2+ ATPase Expression and Leads to Left Ventricular Diastolic Dysfunction through Binding of NF- B to Promoter Response Element. Cardiovasc. Res. 2015, 105, 318–329. [Google Scholar] [CrossRef]
- Heart Outcomes Prevention Evaluation Study Investigators. Effects of Ramipril on Cardiovascular and Microvascular Outcomes in People with Diabetes Mellitus: Results of the HOPE Study and MICRO-HOPE Substudy. Lancet 2000, 355, 253–259. [Google Scholar] [CrossRef]
- Dunlay, S.M.; Givertz, M.M.; Aguilar, D.; Allen, L.A.; Chan, M.; Desai, A.S.; Deswal, A.; Dickson, V.V.; Kosiborod, M.N.; Lekavich, C.L.; et al. Type 2 Diabetes Mellitus and Heart Failure: A Scientific Statement From the American Heart Association and the Heart Failure Society of America: This Statement Does Not Represent an Update of the 2017 ACC/AHA/HFSA Heart Failure Guideline Update. Circulation 2019, 140, e294–e324. [Google Scholar] [CrossRef]
- Shekelle, P.G.; Rich, M.W.; Morton, S.C.; Atkinson, C.S.W.; Tu, W.; Maglione, M.; Rhodes, S.; Barrett, M.; Fonarow, G.C.; Greenberg, B.; et al. Efficacy of Angiotensin-Converting Enzyme Inhibitors and Beta-Blockers in the Management of Left Ventricular Systolic Dysfunction According to Race, Gender, and Diabetic Status. J. Am. Coll. Cardiol. 2003, 41, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Braunwald, E.; Moyé, L.A.; Basta, L.; Brown, E.J.; Cuddy, T.E.; Davis, B.R.; Geltman, E.M.; Goldman, S.; Flaker, G.C.; et al. Effect of Captopril on Mortality and Morbidity in Patients with Left Ventricular Dysfunction after Myocardial Infarction: Results of the Survival and Ventricular Enlargement Trial. N. Engl. J. Med. 1992, 327, 669–677. [Google Scholar] [CrossRef] [PubMed]
- The SOLVD Investigators. Effect of Enalapril on Survival in Patients with Reduced Left Ventricular Ejection Fractions and Congestive Heart Failure. N. Engl. J. Med. 1991, 325, 293–302. [Google Scholar] [CrossRef]
- Køber, L.; Torp-Pedersen, C.; Carlsen, J.E.; Bagger, H.; Eliasen, P.; Lyngborg, K.; Videbæk, J.; Cole, D.S.; Auclert, L.; Pauly, N.C.; et al. A Clinical Trial of the Angiotensin-Converting–Enzyme Inhibitor Trandolapril in Patients with Left Ventricular Dysfunction after Myocardial Infarction. N. Engl. J. Med. 1995, 333, 1670–1676. [Google Scholar] [CrossRef]
- Konstam, M.A.; Neaton, J.D.; Dickstein, K.; Drexler, H.; Komajda, M.; Martinez, F.A.; Riegger, G.A.; Malbecq, W.; Smith, R.D.; Guptha, S.; et al. Effects of High-Dose versus Low-Dose Losartan on Clinical Outcomes in Patients with Heart Failure (HEAAL Study): A Randomised, Double-Blind Trial. Lancet 2009, 374, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; McMurray, J.J.V.; Velazquez, E.J.; Rouleau, J.-L.; Køber, L.; Maggioni, A.P.; Solomon, S.D.; Swedberg, K.; Van De Werf, F.; White, H.; et al. Valsartan, Captopril, or Both in Myocardial Infarction Complicated by Heart Failure, Left Ventricular Dysfunction, or Both. N. Engl. J. Med. 2003, 349, 1893–1906. [Google Scholar] [CrossRef]
- MacDonald, M.R.; Petrie, M.C.; Varyani, F.; Ostergren, J.; Michelson, E.L.; Young, J.B.; Solomon, S.D.; Granger, C.B.; Swedberg, K.; Yusuf, S.; et al. Impact of Diabetes on Outcomes in Patients with Low and Preserved Ejection Fraction Heart Failure: An Analysis of the Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity (CHARM) Programme. Eur. Heart J. 2008, 29, 1377–1385. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V. Sex and Gender Differences in Heart Failure. Int. J. Heart Fail. 2020, 2, 157. [Google Scholar] [CrossRef]
- Lala, A.; Tayal, U.; Hamo, C.E.; Youmans, Q.; Al-Khatib, S.M.; Bozkurt, B.; Davis, M.B.; Januzzi, J.; Mentz, R.; Sauer, A.; et al. Sex Differences in Heart Failure. J. Card. Fail. 2022, 28, 477–498. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T1D Control (n = 18) | T1D (n = 18) | T1D + Ena (n = 15) | T1D + Los (n = 10) | |
|---|---|---|---|---|
| Body weight (g) | 403 ± 18 | 194 ± 24 **** | 274 ± 53 ****,#### | 287 ± 66 ****,#### |
| Heart weight (g) | 1.6 ± 0.2 | 0.8 ± 0.1 **** | 1.2 ± 0.2 ****,#### | 1.2 ± 0.3 ****,### |
| Heart weight/Body weight (×10−3) | 4.1 ± 0.6 | 4.2 ± 0.9 | 4.4 ± 1.1 | 4.4 ± 1.0 |
| Mean arterial pressure (mmHg) | 87 ± 9 | 82 ± 7 | 80 ± 11 | 85 ± 9 |
| Blood glucose (mmol/L) | 8.6 ± 2.2 | 22.2 ± 3.4 **** | 21.4 ± 4.1 **** | 23.6 ± 6.8 **** |
| T2D Control (n = 9) | T2D (n = 8) | T2D + LD Ena (n = 6) | T2D + HD Ena (n = 7) | |
| Body weight (g) | 511 ± 27 | 507 ± 35 | 508 ± 33 | 493 ± 29 |
| Heart weight (g) | 1.8 ± 0.2 | 2.2 ± 0.2 ** | 2.1 ± 0.3 * | 1.7 ± 0.2 ##,$ |
| Heart weight/Body weight (×10−3) | 3.5 ± 0.4 | 4.4 ± 0.2 *** | 4.1 ± 0.4 * | 3.4 ± 0.3 ###,$$ |
| Mean arterial pressure (mmHg) | 88 ± 10 | 122 ± 7 **** | 117 ± 15 **** | 89 ± 9 ###,$$ |
| Fasting blood glucose (mmol/L) | 7.3 ± 4.1 | 6.7 ± 3.4 | 7.7 ± 3.8 | 8.1 ± 4.2 |
| T1D Control (n = 18) | T1D (n = 18) | T1D + Ena (n = 15) | T1D + Los (n = 10) | |
|---|---|---|---|---|
| Diastolic wall thickness (IVSd) (cm) | 0.18 ± 0.04 | 0.17 ± 0.04 | 0.19 ± 0.03 | 0.18 ± 0.03 |
| End-diastolic volume (EDV) (cm3) | 0.34 ± 0.07 | 0.30 ± 0.06 | 0.33 ± 0.05 | 0.32 ± 0.07 |
| Ejection fraction (EF: 100 × SV/EDV) (%) | 79 ± 7 | 54 ± 11 *** | 65 ± 10 *,# | 69 ± 10 *,# |
| Fractional shortening (FS: 100 × (LVIDd-LVIDs)/LVIDd) (%) | 50 ± 6 | 40 ± 5 ** | 45 ± 9 | 47 ± 6 # |
| T2D Control (n = 9) | T2D (n = 8) | T2D + LD Ena (n = 6) | T2D + HD Ena (n = 7) | |
| Diastolic wall thickness (IVSd) (cm) | 0.19 ± 0.04 | 0.26 ± 0.03 ** | 0.25 ± 0.04 ** | 0.21 ± 0.03 #,$ |
| End-diastolic volume (EDV) (cm3) | 0.40 ± 0.07 | 0.43 ± 0.10 | 0.42 ± 0.06 | 0.44 ± 0.08 |
| Ejection fraction (EF: 100 × SV/EDV) (%) | 73 ± 8 | 52 ± 6 ** | 62 ± 8 *,# | 76 ± 8 #,$ |
| Fractional shortening (FS: 100 × (LVIDd-LVIDs)/LVIDd)(%) | 48 ± 6 | 38 ± 7 *** | 41 ± 7 *,# | 47 ± 6 #,$ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulik, K.A.; Ivanics, T.; Dunay, G.A.; Fülöp, Á.; Kerék, M.; Takács, K.; Benyó, Z.; Miklós, Z. Inhibition of the Renin–Angiotensin System Improves Hemodynamic Function of the Diabetic Rat Heart by Restoring Intracellular Calcium Regulation. Biomedicines 2025, 13, 757. https://doi.org/10.3390/biomedicines13030757
Paulik KA, Ivanics T, Dunay GA, Fülöp Á, Kerék M, Takács K, Benyó Z, Miklós Z. Inhibition of the Renin–Angiotensin System Improves Hemodynamic Function of the Diabetic Rat Heart by Restoring Intracellular Calcium Regulation. Biomedicines. 2025; 13(3):757. https://doi.org/10.3390/biomedicines13030757
Chicago/Turabian StylePaulik, Krisztina Anna, Tamás Ivanics, Gábor A. Dunay, Ágnes Fülöp, Margit Kerék, Klára Takács, Zoltán Benyó, and Zsuzsanna Miklós. 2025. "Inhibition of the Renin–Angiotensin System Improves Hemodynamic Function of the Diabetic Rat Heart by Restoring Intracellular Calcium Regulation" Biomedicines 13, no. 3: 757. https://doi.org/10.3390/biomedicines13030757
APA StylePaulik, K. A., Ivanics, T., Dunay, G. A., Fülöp, Á., Kerék, M., Takács, K., Benyó, Z., & Miklós, Z. (2025). Inhibition of the Renin–Angiotensin System Improves Hemodynamic Function of the Diabetic Rat Heart by Restoring Intracellular Calcium Regulation. Biomedicines, 13(3), 757. https://doi.org/10.3390/biomedicines13030757