

Integrative Analysis of Gene Expression and Promoter Methylation to Differentiate High-Grade Serous Ovarian Cancer from Benign Tumors
 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection and Sample Collection
2.2. Nucleic Acid Extraction
2.3. Synthesis of cDNA and Quantitative PCR
2.4. Methylation Specific PCR
2.5. Statistical Analysis
3. Results
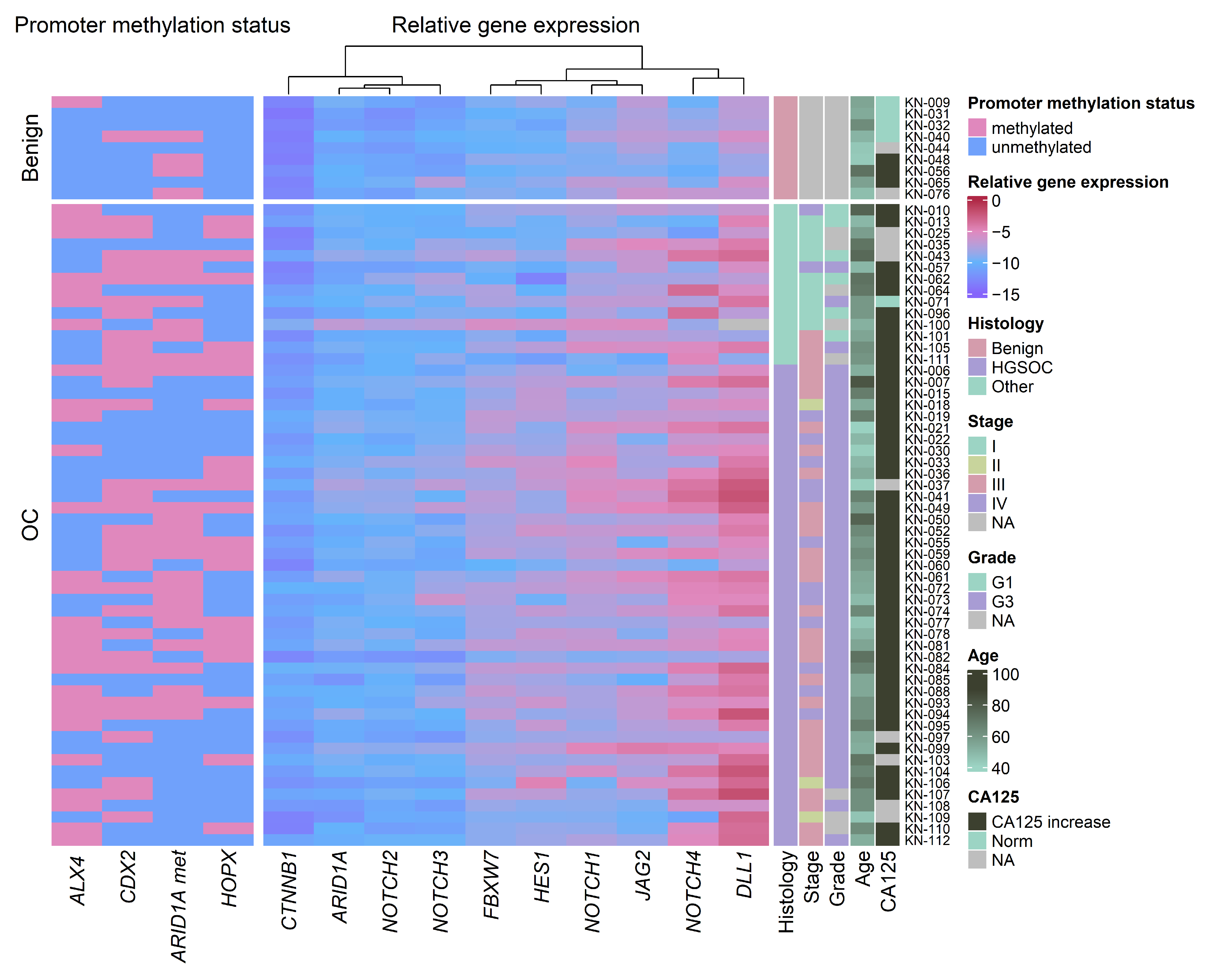
3.1. Promoter Methylation Changes in Ovarian Cancer Compared to Benign Gynecologic Tissues
3.2. Gene Expression Changes in Ovarian Cancer Compared to Benign Gynecologic Tissues
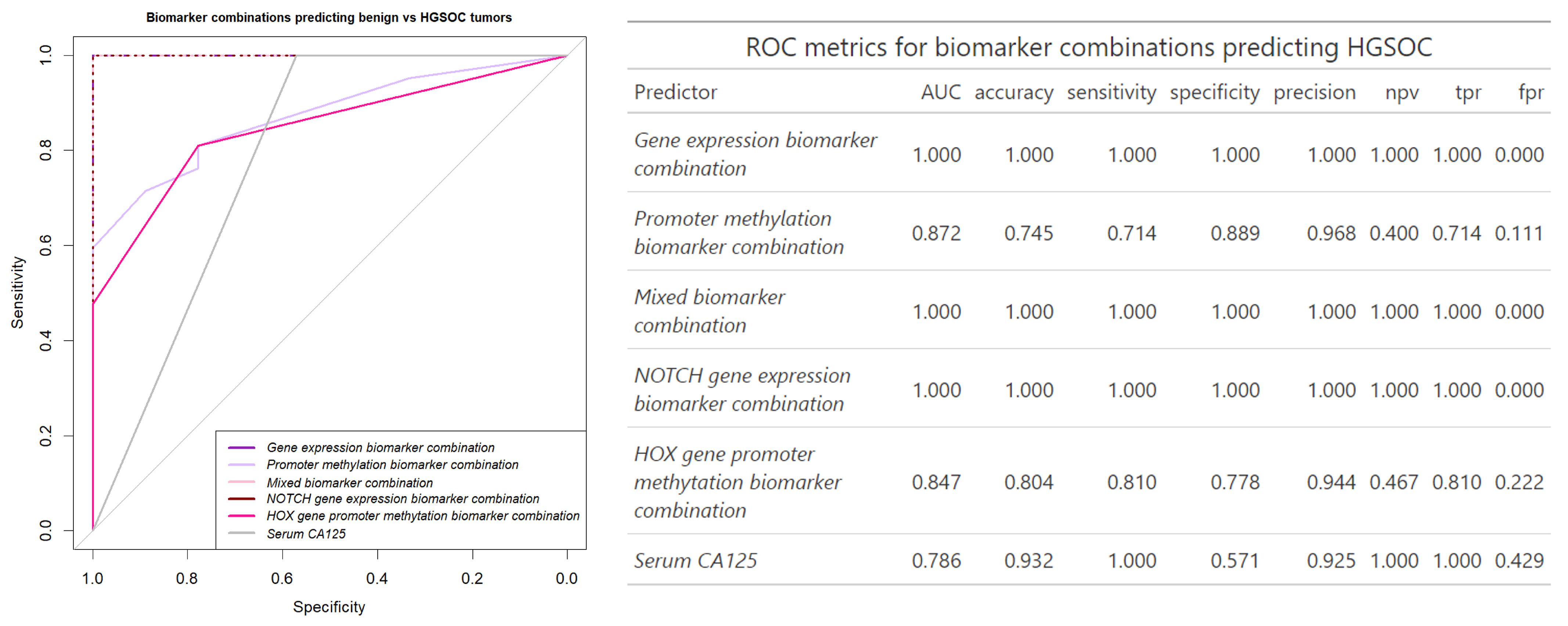
3.3. Building an Ovarian Cancer Diagnostic Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FC | Fold change |
| HGSOC | High-grade serous ovarian cancer |
| HOX | Homeobox domain |
| MSP | methylation specific PCR |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Matsuda, T.; Carlo, V.D.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [PubMed]
- Brincat, M.R.; Mira, A.R.; Lawrence, A. Current and Emerging Strategies for Tubo-Ovarian Cancer Diagnostics. Diagnostics 2023, 13, 3331. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Matias-Guiu, X.; Amant, F.; Concin, N.; Davidson, B.; Fotopoulou, C.; González-Martin, A.; Gourley, C.; Leary, A.; Lorusso, D.; et al. ESGO–ESMO–ESP consensus conference recommendations on ovarian cancer: Pathology and molecular biology and early, advanced and recurrent disease. Ann. Oncol. 2024, 35, 248–266. [Google Scholar] [CrossRef]
- Zhang, M.; Cheng, S.; Jin, Y.; Zhao, Y.; Wang, Y. Roles of CA125 in diagnosis, prediction, and oncogenesis of ovarian cancer. Biochim. Biophys. Acta-(BBA)-Rev. Cancer 2021, 1875, 188503. [Google Scholar] [CrossRef]
- Reyes, H.D.; Devor, E.J.; Warrier, A.; Newtson, A.M.; Mattson, J.; Wagner, V.; Duncan, G.N.; Leslie, K.K.; Gonzalez-Bosquet, J. Differential DNA methylation in high-grade serous ovarian cancer (HGSOC) is associated with tumor behavior. Sci. Rep. 2019, 9, 17996. [Google Scholar] [CrossRef]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; Disaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Teer, J.K.; Yoder, S.; Gjyshi, A.; Nicosia, S.V.; Zhang, C.; Monteiro, A.N. Mutational heterogeneity in non-serous ovarian cancers. Sci. Rep. 2017, 7, 9728. [Google Scholar] [CrossRef]
- Kessler, M.; Hoffmann, K.; Brinkmann, V.; Thieck, O.; Jackisch, S.; Toelle, B.; Berger, H.; Mollenkopf, H.J.; Mangler, M.; Sehouli, J.; et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 2015, 6, 8989. [Google Scholar] [CrossRef]
- Anusewicz, D.; Orzechowska, M.; Bednarek, A.K. Notch Signaling Pathway in Cancer-Review with Bioinformatic Analysis. Cancers 2021, 13, 768. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.F.S.D.; Esteves, C.M.; Xavier, F.C.A.; Nunes, F.D. Methylation status of homeobox genes in common human cancers. Genomics 2016, 108, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Paço, A.; de Bessa Garcia, S.A.; Freitas, R. Methylation in HOX Clusters and Its Applications in Cancer Therapy. Cells 2020, 9, 1613. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Chen, P.; Huang, H.W.; Liu, L.P.; Zhao, F. Reactive oxygen species downregulate ARID1A expression via its promoter methylation during the pathogenesis of endometriosis. Eur. Rev. Med. Pharm. Sci. 2017, 21, 4509–4515. [Google Scholar]
- Berns, K.; Caumanns, J.J.; Hijmans, E.M.; Gennissen, A.M.; Severson, T.M.; Evers, B.; Wisman, G.B.A.; Meersma, G.J.; Lieftink, C.; Beijersbergen, R.L.; et al. ARID1A mutation sensitizes most ovarian clear cell carcinomas to BET inhibitors. Oncogene 2018, 37, 4611–4625. [Google Scholar] [CrossRef]
- Zhao, L.; Guo, H.; Chen, X.; Zhang, W.; He, Q.; Ding, L.; Yang, B. Tackling drug resistance in ovarian cancer with epigenetic targeted drugs. Eur. J. Pharmacol. 2022, 927, 175071. [Google Scholar] [CrossRef]
- Mandal, J.; Mandal, P.; Wang, T.L.; Shih, I.M. Treating ARID1A mutated cancers by harnessing synthetic lethality and DNA damage response. J. Biomed. Sci. 2022, 29, 71. [Google Scholar] [CrossRef]
- Pearson, H.; Stirling, D. DNA Extraction from Tissue. Methods Mol. Biol. 2003, 226, 33–34. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb-prot5439. [Google Scholar] [CrossRef]
- Peterson, S.M.; Freeman, J.L. RNA isolation from embryonic zebrafish and cDNA synthesis for gene expression analysis. J. Vis. Exp. Jove 2009, 30, 1470. [Google Scholar] [CrossRef]
- Torrejón, D.; Cárdenas, J.; Juárez, D.; Espinoza, J.; Proleón, A.; Agurto-Arteaga, A.; Lazo, F.; Leguía, M.; Urra, F.A.; Sánchez, E.F.; et al. Comparison of Four Methods of RNA Extraction and cDNA Synthesis from The Venom of Peruvian Snakes of the Genus Bothrops of Clinical Importance. Int. J. Mol. Sci. 2023, 24, 11161. [Google Scholar] [CrossRef] [PubMed]
- Welch, B.L. The Significance of the Difference Between Two Means When the Population Variances Are Unequal. Biometrika 1938, 29, 350–362. [Google Scholar] [CrossRef]
- Delacre, M.; Lakens, D.; Leys, C. Why Psychologists Should by Default Use Welch’s t-test Instead of Student’s t-test. Int. Rev. Soc. Psychol. 2017, 30, 92–101. [Google Scholar] [CrossRef]
- Benjaminit, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bocchicchio, S.; Tesone, M.; Irusta, G. Convergence of Wnt and Notch signaling controls ovarian cancer cell survival. J. Cell. Physiol. 2019, 234, 22130–22143. [Google Scholar] [CrossRef]
- Kumar, V.; Vashishta, M.; Kong, L.; Wu, X.; Lu, J.J.; Guha, C.; Dwarakanath, B.S. The Role of Notch, Hedgehog, and Wnt Signaling Pathways in the Resistance of Tumors to Anticancer Therapies. Front. Cell Dev. Biol. 2021, 9, 650772. [Google Scholar] [CrossRef]
- Chen, B.; Jiang, K.; Wang, H.; Miao, L.; Lin, X.; Chen, Q.; Jing, L.; Lu, X. NOTCH Pathway Genes in Ovarian Cancer: Clinical Significance and Associations with Immune Cell Infiltration. Front. Biosci.-Landmark 2023, 28, 220. [Google Scholar] [CrossRef]
- AmeliMojarad, M.; AmeliMojarad, M.; Cui, X.; Shariati, P. Pan-cancer analysis of CTNNB1 with potential as a therapeutic target for human tumorigenesis. Inform. Med. Unlocked 2023, 42, 101331. [Google Scholar] [CrossRef]
- Fiore, R.D.; Suleiman, S.; Drago-Ferrante, R.; Subbannayya, Y.; Suleiman, S.; Vasileva-Slaveva, M.; Yordanov, A.; Pentimalli, F.; Giordano, A.; Calleja-Agius, J. The Role of FBXW7 in Gynecologic Malignancies. Cells 2023, 12, 1415. [Google Scholar] [CrossRef]
- Holland, P.W.; Booth, H.A.F.; Bruford, E.A. Classification and nomenclature of all human homeobox genes. BMC Biol. 2007, 5, 47. [Google Scholar] [CrossRef]
- Celik, S.; Logsdon, B.A.; Battle, S.; Drescher, C.W.; Rendi, M.; Hawkins, R.D.; Lee, S.I. Extracting a low-dimensional description of multiple gene expression datasets reveals a potential driver for tumor-associated stroma in ovarian cancer. Genome Med. 2016, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Ooki, A.; Yamashita, K.; Kikuchi, S.; Sakuramoto, S.; Katada, N.; Kokubo, K.; Kobayashi, H.; Kim, M.S.; Sidransky, D.; Watanabe, M. Potential utility of HOP homeobox gene promoter methylation as a marker of tumor aggressiveness in gastric cancer. Oncogene 2010, 29, 3263–3275. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Yamashita, K.; Waraya, M.; Margali, O.; Ooki, A.; Tamaki, H.; Sakagami, H.; Kokubo, K.; Sidransky, D.; Watanabe, M. Epigenetic silencing of HOPX promotes cancer progression in colorectal cancer. Neoplasia 2012, 14, 559–571. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Asanoma, K.; Takao, T.; Kato, K.; Wake, N. Homeobox gene HOPX is epigenetically silenced in human uterine endometrial cancer and suppresses estrogen-stimulated proliferation of cancer cells by inhibiting serum response factor. Int. J. Cancer 2009, 124, 2577–2588. [Google Scholar] [CrossRef]
- Yuan, H.; Kajiyama, H.; Ito, S.; Chen, D.; Shibata, K.; Hamaguchi, M.; Kikkawa, F.; Senga, T. HOXB13 and ALX4 induce SLUG expression for the promotion of EMT and cell invasion in ovarian cancer cells. Oncotarget 2015, 6, 13359. [Google Scholar] [CrossRef]
- Wang, Y.; Lei, L.; Chi, Y.G.; Liu, L.B.; Yang, B.P. A comprehensive understanding of ovarian carcinoma survival prognosis by novel biomarkers. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8257–8264. [Google Scholar] [CrossRef]
- Duan, C.; Yan, Z.; Wu, C.; Zhou, X.; Bao, W. DNA methylation characteristics associated with chemotherapy resistance in epithelial ovarian cancer. Heliyon 2024, 10, 27212. [Google Scholar] [CrossRef]
- Yang, J.; Han, F.; Liu, W.; Chen, H.; Hao, X.; Jiang, X.; Yin, L.; Huang, Y.; Cao, J.; Zhang, H.; et al. ALX4, an epigenetically down regulated tumor suppressor, inhibits breast cancer progression by interfering Wnt/β-catenin pathway. J. Exp. Clin. Cancer Res. 2017, 36, 170. [Google Scholar] [CrossRef]
- Salehi, R.; Atapour, N.; Vatandoust, N.; Farahani, N.; Ahangari, F.; Salehi, A. Methylation pattern of ALX4 gene promoter as a potential biomarker for blood-based early detection of colorectal cancer. Adv. Biomed. Res. 2015, 4, 252. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z.; Li, W.; Liu, S.; Han, B. Methylation of CDX2 gene promoter in the prediction of treatment efficacy in colorectal cancer. Oncol. Lett. 2018, 16, 195–198. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, X.; Zhan, Q.; Brock, M.V.; Herman, J.G.; Guo, M. CDX2 serves as a Wnt signaling inhibitor and is frequently methylated in lung cancer. Cancer Biol. Ther. 2012, 13, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Koh, I.; Hinoi, T.; Sentani, K.; Hirata, E.; Nosaka, S.; Niitsu, H.; Miguchi, M.; Adachi, T.; Yasui, W.; Ohdan, H.; et al. Regulation of multidrug resistance 1 expression by CDX2 in ovarian mucinous adenocarcinoma. Cancer Med. 2016, 5, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, N. A 10-gene expression signature of Notch pathway predicts recurrence in ovarian carcinoma. Oncol. Lett. 2015, 10, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Pi, Y.; Sun, F.; Zhang, Z.; Liu, X.; Lou, G. A Novel Notch-Related Gene Signature for Prognosis and Immune Response Prediction in Ovarian Cancer. Medicina 2023, 59, 1277. [Google Scholar] [CrossRef]
- Montavon, C.; Gloss, B.S.; Warton, K.; Barton, C.A.; Statham, A.L.; Scurry, J.P.; Tabor, B.; Nguyen, T.V.; Qu, W.; Samimi, G.; et al. Prognostic and diagnostic significance of DNA methylation patterns in high grade serous ovarian cancer. Gynecol. Oncol. 2012, 124, 582–588. [Google Scholar] [CrossRef]
- Tavares, V.; Marques, I.S.; de Melo, I.G.; Assis, J.; Pereira, D.; Medeiros, R. Paradigm Shift: A Comprehensive Review of Ovarian Cancer Management in an Era of Advancements. Int. J. Mol. Sci. 2024, 25, 1845. [Google Scholar] [CrossRef]
- Graule, J.; Uth, K.; Fischer, E.; Centeno, I.; Galván, J.A.; Eichmann, M.; Rau, T.T.; Langer, R.; Dawson, H.; Nitsche, U.; et al. CDX2 in colorectal cancer is an independent prognostic factor and regulated by promoter methylation and histone deacetylation in tumors of the serrated pathway. Clin. Epigenetics 2018, 10, 120. [Google Scholar] [CrossRef]
- Khalique, S.; Naidoo, K.; Attygalle, A.D.; Kriplani, D.; Daley, F.; Lowe, A.; Campbell, J.; Jones, T.; Hubank, M.; Fenwick, K.; et al. Optimised ARID1A immunohistochemistry is an accurate predictor of ARID1A mutational status in gynaecological cancers. J. Pathol. Clin. Res. 2018, 4, 154–166. [Google Scholar] [CrossRef]
- Englisz, A.; Smycz-Kubańska, M.; Mielczarek-Palacz, A. Sensitivity and Specificity of Selected Biomarkers and Their Combinations in the Diagnosis of Ovarian Cancer. Diagnostics 2024, 14, 949. [Google Scholar] [CrossRef]
- Clarke-Pearson, D.L. Clinical practice. Screening for ovarian cancer. N. Engl. J. Med. 2009, 361, 170–177. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Feature | Ovarian Cancer | Benign Gynecologic Tumors | All |
|---|---|---|---|
| n | 56 | 9 | 65 |
| Histology group | |||
| HGSOC | 42 (75.00%) | 42 (64.62%) | |
| Other gynecologic malignant tumors | 14 (25.00%) | 14 (21.54%) | |
| Benign gynecologic tumors | 9 (100.00%) | 9 (13.85%) | |
| Average age at diagnosis (±SD) | 59.16 (±9.69) | 53.67 (±9.33) | 58.40 (±9.76) |
| Median CA125 U/mL (min–max) | 478.50 (34.00–5384.00) | 18.22 (10.00–118.00) | 419.30 (10.00–5384.00) |
| FIGO stage | |||
| I | 9 (16.07%) | 9 (13.85%) | |
| II | 3 (5.36%) | 3 (4.62%) | |
| III | 30 (53.57 %) | 29 (44.62%) | |
| IV | 14 (25.00%) | 14 (21.54%) | |
| NA | 9 (100.00%) | 9 (13.85%) | |
| Grade | |||
| G1 | 6 (10.71%) | 7 (10.61%) | |
| G3 | 42 (75.00%) | 42 (63.64%) | |
| NA | 8 (14.29%) | 9 (100.00%) | 17 (25.76%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaicekauskaitė, I.; Kazlauskaitė, P.; Gineikaitė, R.; Čiurlienė, R.; Lazutka, J.R.; Sabaliauskaitė, R. Integrative Analysis of Gene Expression and Promoter Methylation to Differentiate High-Grade Serous Ovarian Cancer from Benign Tumors. Biomedicines 2025, 13, 441. https://doi.org/10.3390/biomedicines13020441
Vaicekauskaitė I, Kazlauskaitė P, Gineikaitė R, Čiurlienė R, Lazutka JR, Sabaliauskaitė R. Integrative Analysis of Gene Expression and Promoter Methylation to Differentiate High-Grade Serous Ovarian Cancer from Benign Tumors. Biomedicines. 2025; 13(2):441. https://doi.org/10.3390/biomedicines13020441
Chicago/Turabian StyleVaicekauskaitė, Ieva, Paulina Kazlauskaitė, Rugilė Gineikaitė, Rūta Čiurlienė, Juozas Rimantas Lazutka, and Rasa Sabaliauskaitė. 2025. "Integrative Analysis of Gene Expression and Promoter Methylation to Differentiate High-Grade Serous Ovarian Cancer from Benign Tumors" Biomedicines 13, no. 2: 441. https://doi.org/10.3390/biomedicines13020441
APA StyleVaicekauskaitė, I., Kazlauskaitė, P., Gineikaitė, R., Čiurlienė, R., Lazutka, J. R., & Sabaliauskaitė, R. (2025). Integrative Analysis of Gene Expression and Promoter Methylation to Differentiate High-Grade Serous Ovarian Cancer from Benign Tumors. Biomedicines, 13(2), 441. https://doi.org/10.3390/biomedicines13020441