

Identifying MTHFD1 and LGALS4 as Potential Therapeutic Targets in Prostate Cancer Through Multi-Omics Mendelian Randomization Analysis

Abstract
1. Introduction
2. Materials and Methods
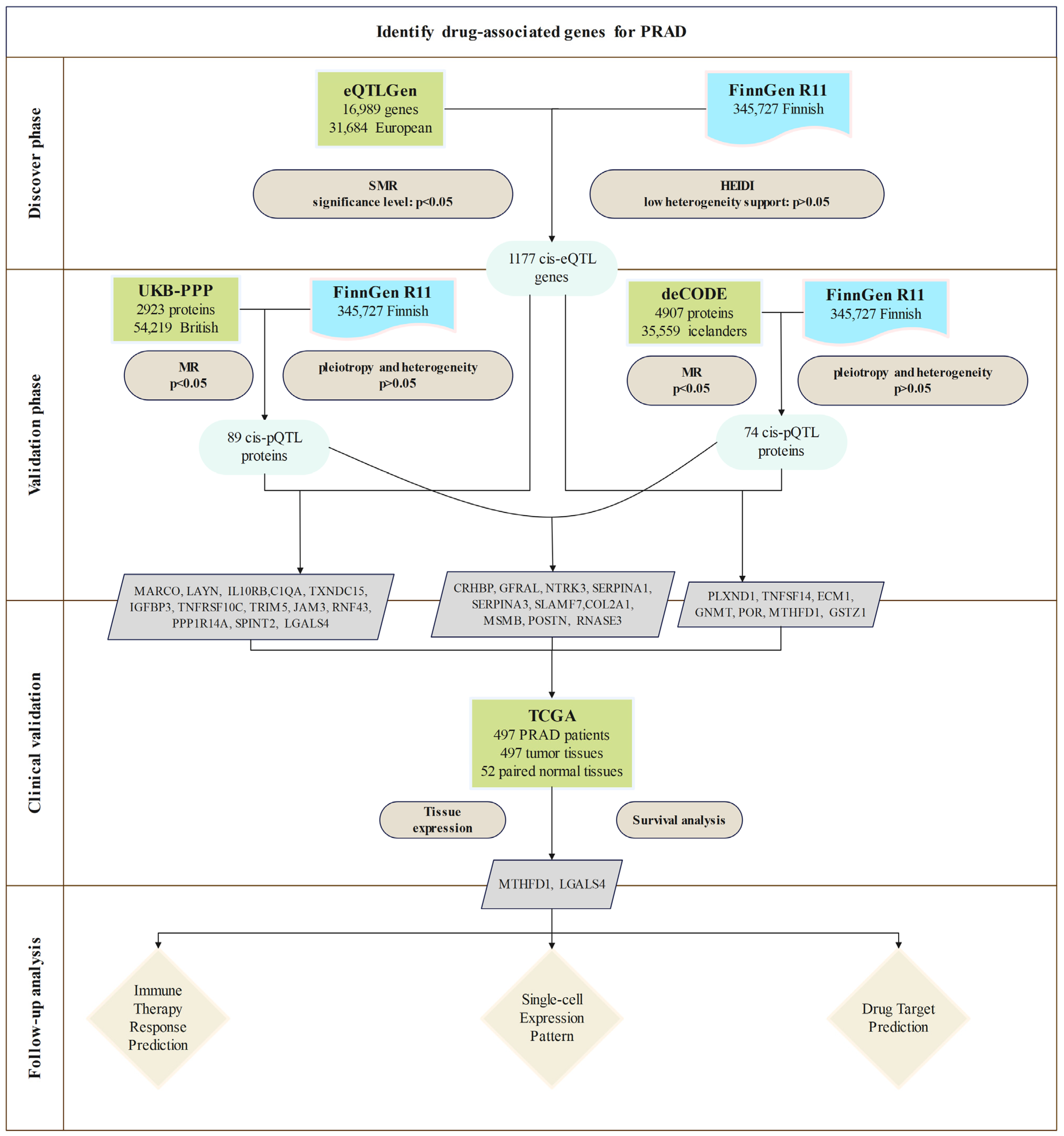
2.1. Study Design
2.2. Exposure Data
2.3. Outcome Data
2.4. SMR Analysis and HEIDI Test
2.5. Mendelian Randomization Analysis
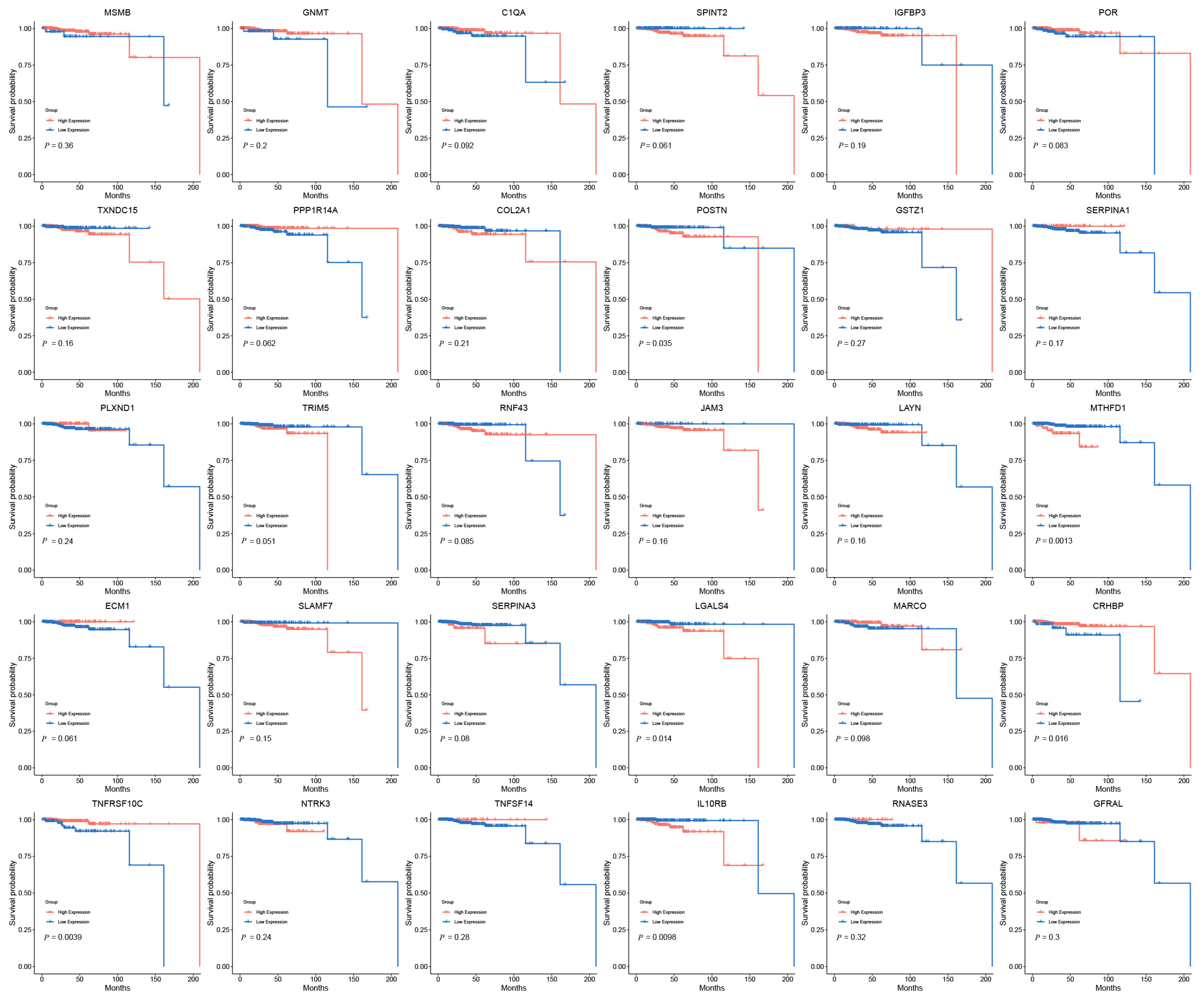
2.6. Clinical Expression and Survival Analysis
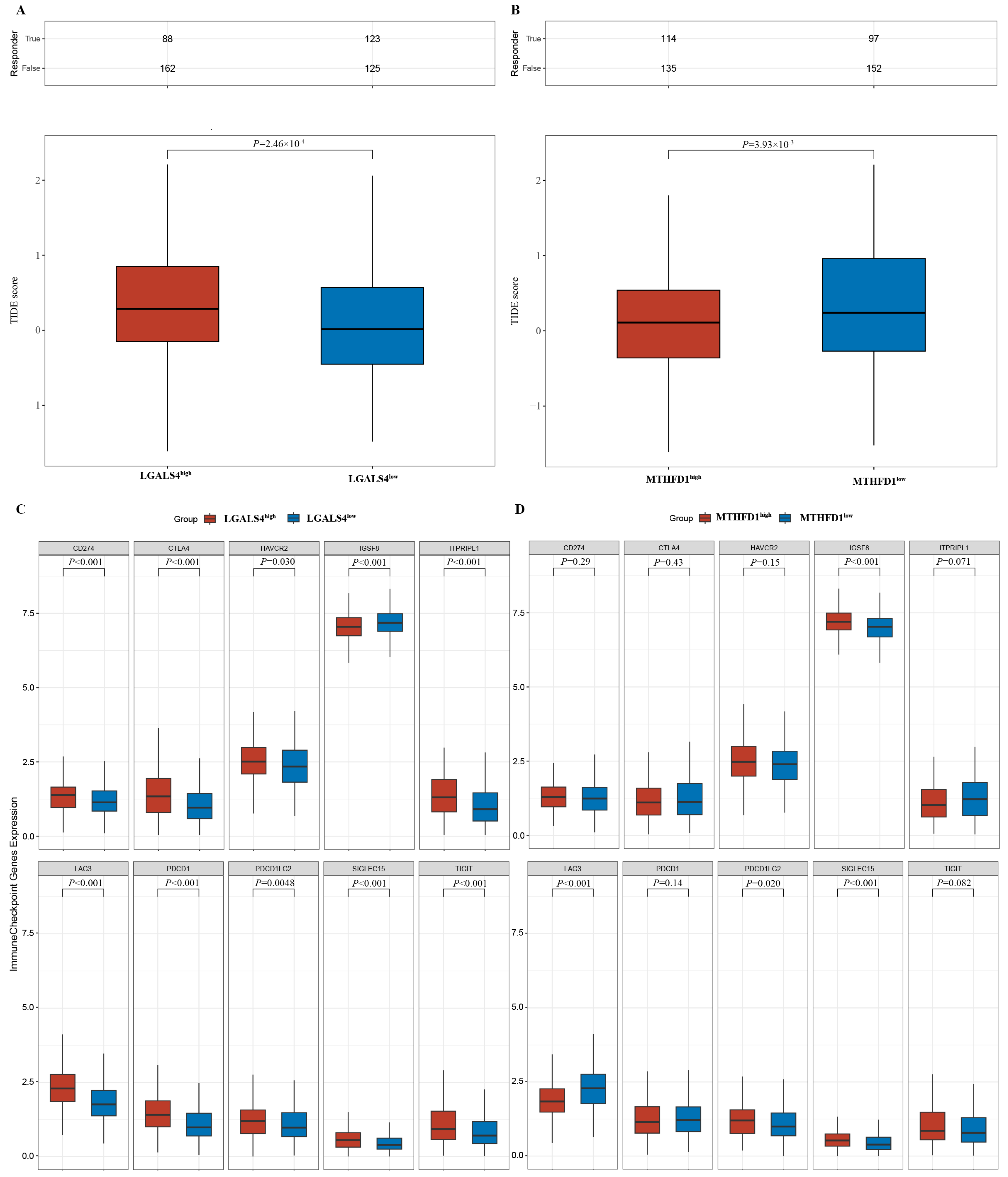
2.7. Immune Scoring and Immune Checkpoint Analysis
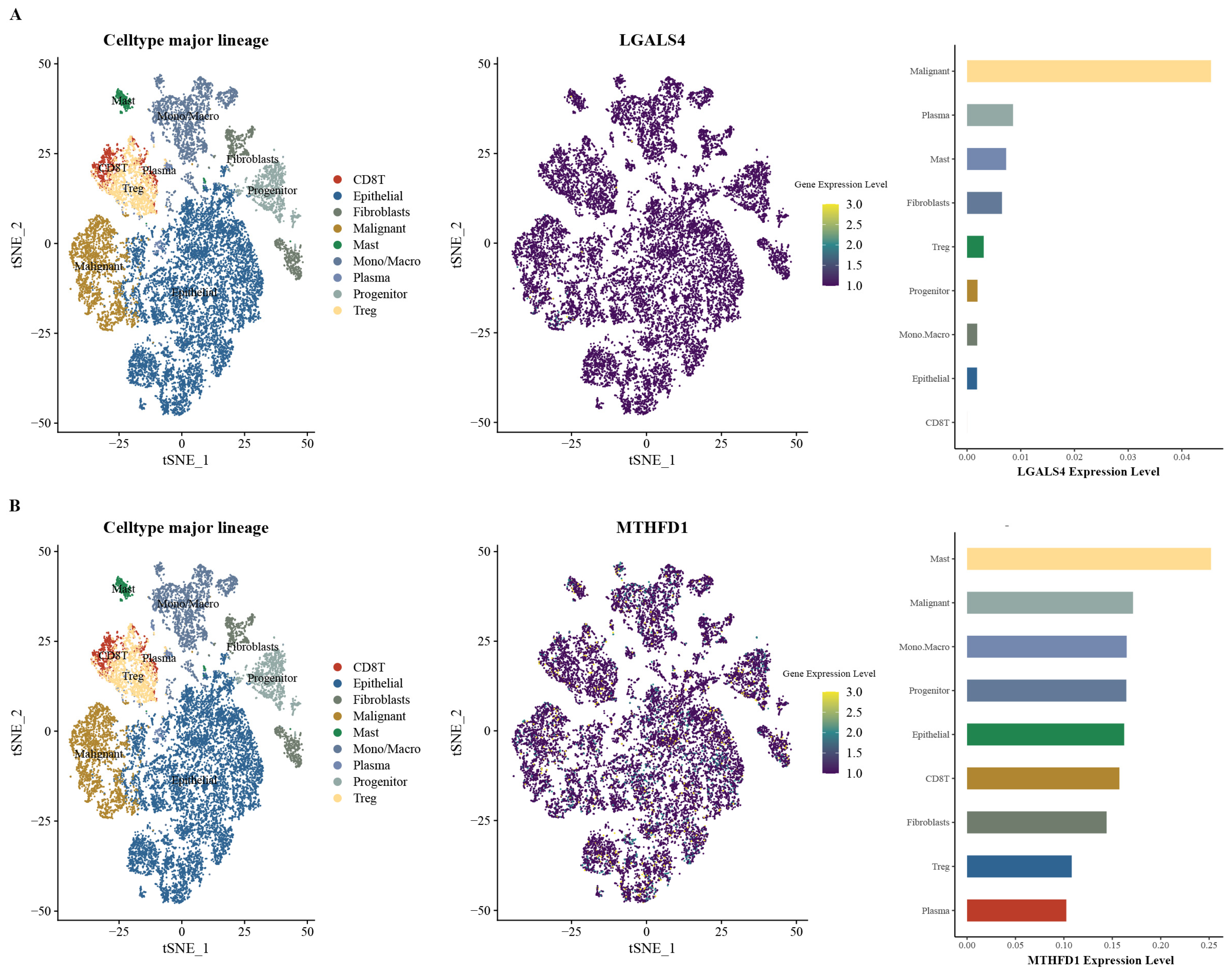
2.8. Analysis of MTHFD1 and LGALS4 Expression in PRAD
2.9. Drug Target Prediction
3. Results
3.1. Discovery of Potential cis-eQTL Genes and PRAD
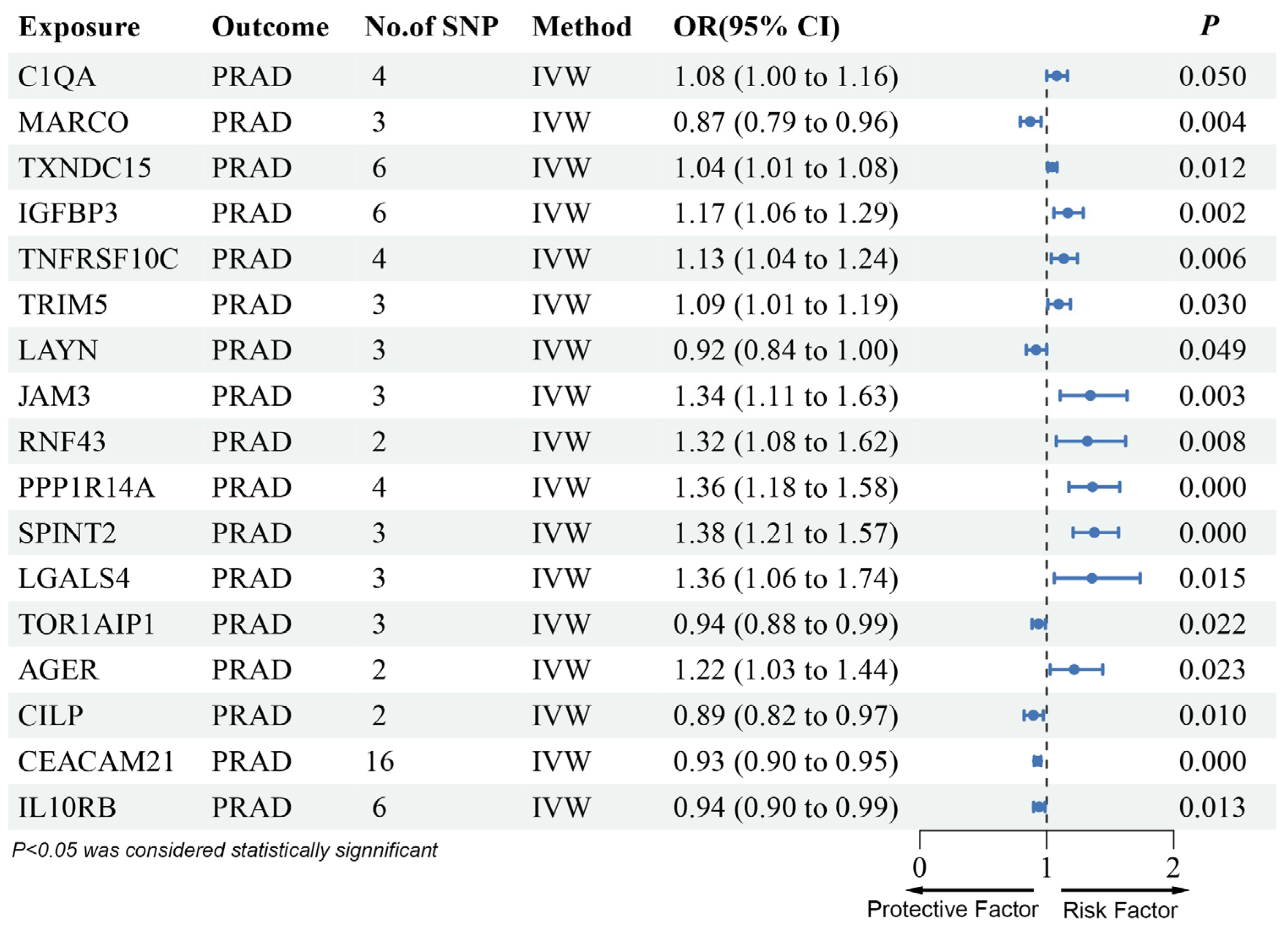
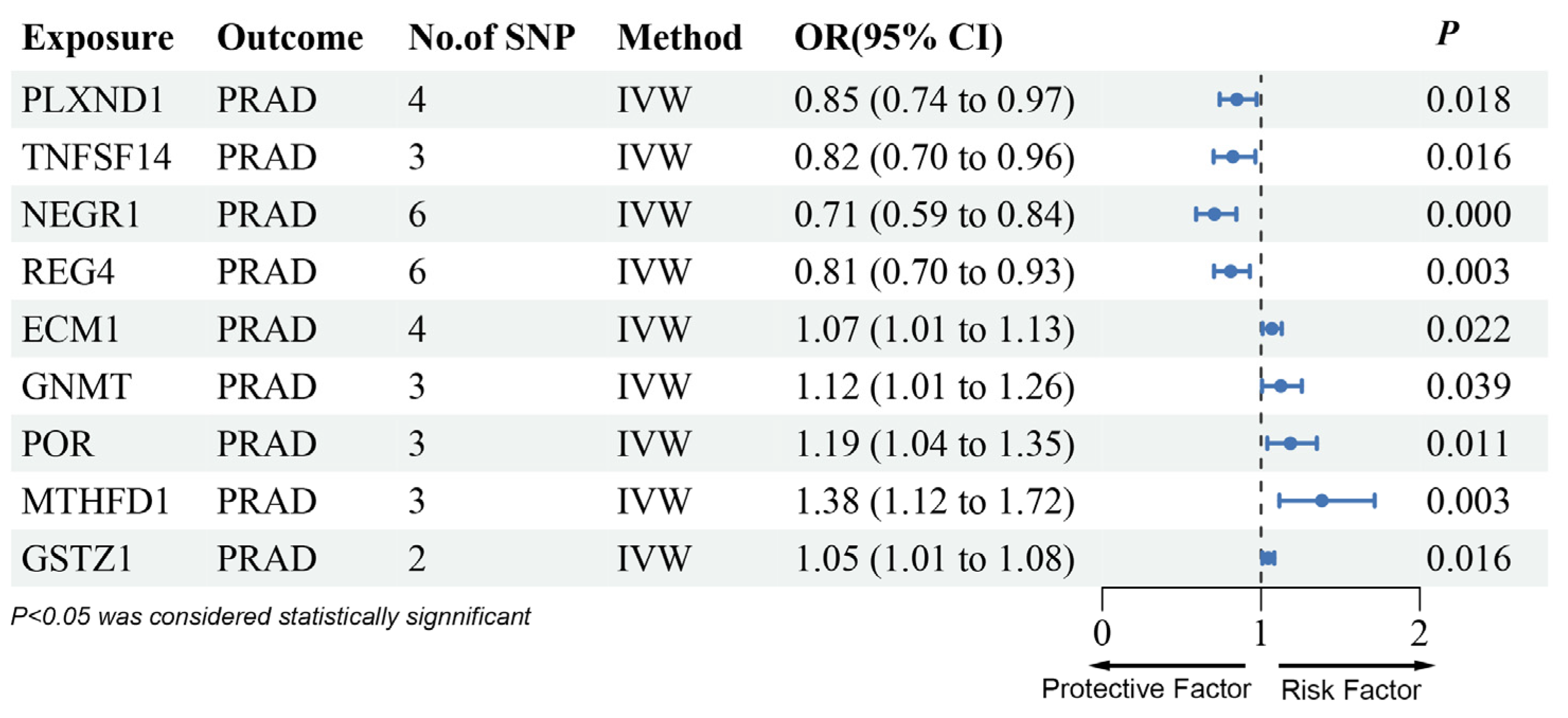
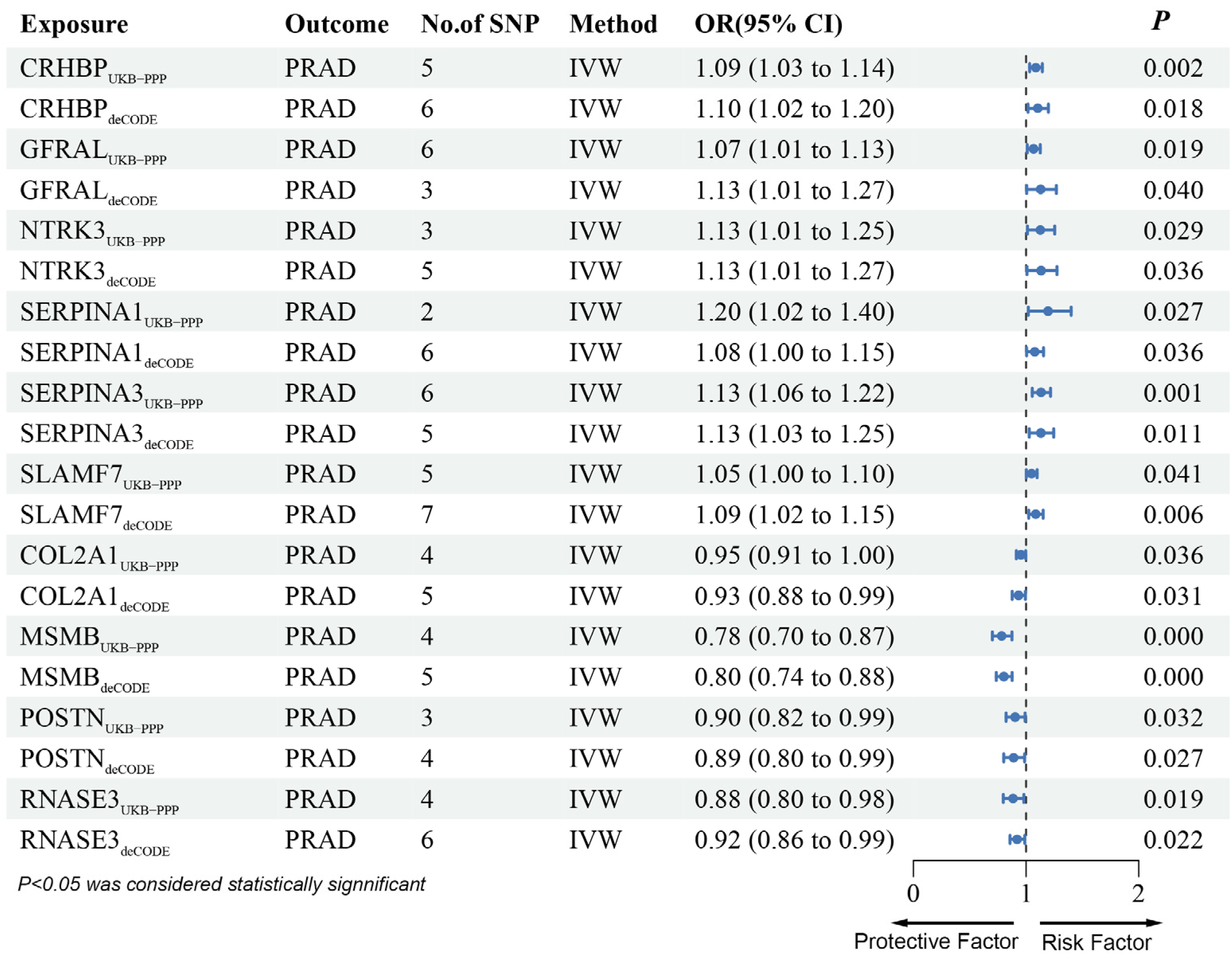
3.2. MR Analysis of pQTLs in Validation Phase
3.3. Clinical Expression and Survival Analysis
3.4. Potential Immune Therapy Response Prediction
3.5. Expression Patterns of MTHFD1 and LGALS4 in PRAD
3.6. Drug Target Prediction of MTHFD1 and LGALS4
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Kohaar, I.; Petrovics, G.; Srivastava, S. A Rich Array of Prostate Cancer Molecular Biomarkers: Opportunities and Challenges. Int. J. Mol. Sci. 2019, 20, 1813. [Google Scholar] [CrossRef] [PubMed]
- Pierre-Victor, D.; Parnes, H.L.; Andriole, G.L.; Pinsky, P.F. Prostate Cancer Incidence and Mortality Following a Negative Biopsy in a Population Undergoing PSA Screening. Urology 2021, 155, 62–69. [Google Scholar] [CrossRef]
- DE Nunzio, C.; Fiori, C.; Fusco, F.; Gregori, A.; Pagliarulo, V.; Alongi, F. Androgen Deprivation Therapy and Cardiovascular Risk in Prostate Cancer. Minerva Urol. Nephrol. 2022, 74, 508–517. [Google Scholar] [CrossRef]
- Cheng, H.H.; Lin, D.W.; Yu, E.Y. Advanced Clinical States in Prostate Cancer. Urol. Clin. N. Am. 2012, 39, 561–571. [Google Scholar] [CrossRef]
- Atiq, M.; Chandran, E.; Karzai, F.; Madan, R.A.; Aragon-Ching, J.B. Emerging Treatment Options for Prostate Cancer. Expert. Rev. Anticancer Ther. 2023, 23, 625–631. [Google Scholar] [CrossRef]
- Adamaki, M.; Zoumpourlis, V. Prostate Cancer Biomarkers: From Diagnosis to Prognosis and Precision-Guided Therapeutics. Pharmacol. Ther. 2021, 228, 107932. [Google Scholar] [CrossRef]
- Swami, U.; McFarland, T.R.; Nussenzveig, R.; Agarwal, N. Advanced Prostate Cancer: Treatment Advances and Future Directions. Trends Cancer 2020, 6, 702–715. [Google Scholar] [CrossRef]
- Sekhoacha, M.; Riet, K.; Motloung, P.; Gumenku, L.; Adegoke, A.; Mashele, S. Prostate Cancer Review: Genetics, Diagnosis, Treatment Options, and Alternative Approaches. Molecules 2022, 27, 5730. [Google Scholar] [CrossRef]
- Deluce, J.E.; Cardenas, L.; Lalani, A.-K.; Maleki Vareki, S.; Fernandes, R. Emerging Biomarker-Guided Therapies in Prostate Cancer. Curr. Oncol. 2022, 29, 5054–5076. [Google Scholar] [CrossRef]
- Haffner, M.C.; Zwart, W.; Roudier, M.P.; True, L.D.; Nelson, W.G.; Epstein, J.I.; De Marzo, A.M.; Nelson, P.S.; Yegnasubramanian, S. Genomic and Phenotypic Heterogeneity in Prostate Cancer. Nat. Rev. Urol. 2021, 18, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Farashi, S.; Kryza, T.; Clements, J.; Batra, J. Post-GWAS in Prostate Cancer: From Genetic Association to Biological Contribution. Nat. Rev. Cancer 2019, 19, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Towards the Genetic Architecture of Complex Gene Expression Traits: Challenges and Prospects for eQTL Mapping in Humans. Genes 2022, 13, 235. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; McCarthy, M.I.; Schwenk, J.M. Genetics Meets Proteomics: Perspectives for Large Population-Based Studies. Nat. Rev. Genet. 2021, 22, 19–37. [Google Scholar] [CrossRef]
- Emdin, C.A.; Khera, A.V.; Kathiresan, S. Mendelian Randomization. JAMA 2017, 318, 1925–1926. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Zhang, F.; Zhu, Z.; Qi, T.; Zheng, Z.; Lloyd-Jones, L.R.; Marioni, R.E.; Martin, N.G.; Montgomery, G.W.; et al. Integrative Analysis of Omics Summary Data Reveals Putative Mechanisms Underlying Complex Traits. Nat. Commun. 2018, 9, 918. [Google Scholar] [CrossRef]
- Hansen, S.B.; Unal, B.; Kuzu, O.F.; Saatcioglu, F. Immunological Facets of Prostate Cancer and the Potential of Immune Checkpoint Inhibition in Disease Management. Theranostics 2024, 14, 6913–6934. [Google Scholar] [CrossRef]
- Graham, M.K.; Wang, R.; Chikarmane, R.; Abel, B.; Vaghasia, A.; Gupta, A.; Zheng, Q.; Hicks, J.; Sysa-Shah, P.; Pan, X.; et al. Convergent Alterations in the Tumor Microenvironment of MYC-Driven Human and Murine Prostate Cancer. Nat. Commun. 2024, 15, 7414. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-Scale Cis- and Trans-eQTL Analyses Identify Thousands of Genetic Loci and Polygenic Scores That Regulate Blood Gene Expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- Sun, B.B.; Chiou, J.; Traylor, M.; Benner, C.; Hsu, Y.-H.; Richardson, T.G.; Surendran, P.; Mahajan, A.; Robins, C.; Vasquez-Grinnell, S.G.; et al. Plasma Proteomic Associations with Genetics and Health in the UK Biobank. Nature 2023, 622, 329–338. [Google Scholar] [CrossRef]
- Ferkingstad, E.; Sulem, P.; Atlason, B.A.; Sveinbjornsson, G.; Magnusson, M.I.; Styrmisdottir, E.L.; Gunnarsdottir, K.; Helgason, A.; Oddsson, A.; Halldorsson, B.V.; et al. Large-Scale Integration of the Plasma Proteome with Genetics and Disease. Nat. Genet. 2021, 53, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen Provides Genetic Insights from a Well-Phenotyped Isolated Population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of Summary Data from GWAS and eQTL Studies Predicts Complex Trait Gene Targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An Integrated Map of Genetic Variation from 1092 Human Genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef]
- Davey Smith, G.; Hemani, G. Mendelian Randomization: Genetic Anchors for Causal Inference in Epidemiological Studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian Randomization Analysis with Multiple Genetic Variants Using Summarized Data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef]
- Hartwig, F.P.; Davey Smith, G.; Bowden, J. Robust Inference in Summary Data Mendelian Randomization via the Zero Modal Pleiotropy Assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian Randomization with Invalid Instruments: Effect Estimation and Bias Detection through Egger Regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Verbanck, M.; Chen, C.-Y.; Neale, B.; Do, R. Detection of Widespread Horizontal Pleiotropy in Causal Relationships Inferred from Mendelian Randomization between Complex Traits and Diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug Signatures Database for Gene Set Analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Sparasci, D.; Napoli, I.; Rossi, L.; Pereira-Mestre, R.; Manconi, M.; Treglia, G.; Marandino, L.; Ottaviano, M.; Turco, F.; Mangan, D.; et al. Prostate Cancer and Sleep Disorders: A Systematic Review. Cancers 2022, 14, 1784. [Google Scholar] [CrossRef] [PubMed]
- Henríquez, I.; Roach, M.; Morgan, T.M.; Bossi, A.; Gómez, J.A.; Abuchaibe, O.; Couñago, F. Current and Emerging Therapies for Metastatic Castration-Resistant Prostate Cancer (mCRPC). Biomedicines 2021, 9, 1247. [Google Scholar] [CrossRef]
- Mizuno, K.; Beltran, H. Future Directions for Precision Oncology in Prostate Cancer. Prostate 2022, 82 (Suppl. S1), S86–S96. [Google Scholar] [CrossRef]
- Kapetanakis, N.-I.; Busson, P. Galectins as Pivotal Components in Oncogenesis and Immune Exclusion in Human Malignancies. Front. Immunol. 2023, 14, 1145268. [Google Scholar] [CrossRef]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a Glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar] [CrossRef]
- Liu, F.-T.; Stowell, S.R. The Role of Galectins in Immunity and Infection. Nat. Rev. Immunol. 2023, 23, 479–494. [Google Scholar] [CrossRef]
- Cao, Z.-Q.; Guo, X.-L. The Role of Galectin-4 in Physiology and Diseases. Protein Cell 2016, 7, 314–324. [Google Scholar] [CrossRef]
- Barrow, H.; Guo, X.; Wandall, H.H.; Pedersen, J.W.; Fu, B.; Zhao, Q.; Chen, C.; Rhodes, J.M.; Yu, L.-G. Serum Galectin-2, -4, and -8 Are Greatly Increased in Colon and Breast Cancer Patients and Promote Cancer Cell Adhesion to Blood Vascular Endothelium. Clin. Cancer Res. 2011, 17, 7035–7046. [Google Scholar] [CrossRef]
- Chen, C.; Duckworth, C.A.; Fu, B.; Pritchard, D.M.; Rhodes, J.M.; Yu, L.-G. Circulating Galectins -2, -4 and -8 in Cancer Patients Make Important Contributions to the Increased Circulation of Several Cytokines and Chemokines That Promote Angiogenesis and Metastasis. Br. J. Cancer 2014, 110, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-H.; Tzeng, S.-F.; Chao, T.-K.; Tsai, C.-Y.; Yang, Y.-C.; Lee, M.-T.; Hwang, J.-J.; Chou, Y.-C.; Tsai, M.-H.; Cha, T.-L.; et al. Metastatic Progression of Prostate Cancer Is Mediated by Autonomous Binding of Galectin-4-O-Glycan to Cancer Cells. Cancer Res. 2016, 76, 5756–5767. [Google Scholar] [CrossRef] [PubMed]
- Lidström, T.; Cumming, J.; Gaur, R.; Frängsmyr, L.; Pateras, I.S.; Mickert, M.J.; Franklin, O.; Forsell, M.N.E.; Arnberg, N.; Dongre, M.; et al. Extracellular Galectin 4 Drives Immune Evasion and Promotes T-Cell Apoptosis in Pancreatic Cancer. Cancer Immunol. Res. 2023, 11, 72–92. [Google Scholar] [CrossRef] [PubMed]
- Qing, L.; Li, Q.; Dong, Z. MUC1: An Emerging Target in Cancer Treatment and Diagnosis. Bull. Cancer 2022, 109, 1202–1216. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. MUC1: A Multifaceted Oncoprotein with a Key Role in Cancer Progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The Role of PD-1/PD-L1 and Application of Immune-Checkpoint Inhibitors in Human Cancers. Front. Immunol. 2022, 13, 964442. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-Small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362.e24. [Google Scholar] [CrossRef]
- Meng, Q.; Lu, Y.-X.; Wei, C.; Wang, Z.-X.; Lin, J.-F.; Liao, K.; Luo, X.-J.; Yu, K.; Han, Y.; Li, J.-J.; et al. Arginine Methylation of MTHFD1 by PRMT5 Enhances Anoikis Resistance and Cancer Metastasis. Oncogene 2022, 41, 3912–3924. [Google Scholar] [CrossRef]
- Green, A.C.; Marttila, P.; Kiweler, N.; Chalkiadaki, C.; Wiita, E.; Cookson, V.; Lesur, A.; Eiden, K.; Bernardin, F.; Vallin, K.S.A.; et al. Formate Overflow Drives Toxic Folate Trapping in MTHFD1 Inhibited Cancer Cells. Nat. Metab. 2023, 5, 642–659. [Google Scholar] [CrossRef]
- He, D.; Yu, Z.; Liu, S.; Dai, H.; Xu, Q.; Li, F. Methylenetetrahydrofolate Dehydrogenase 1 (MTHFD1) Is Underexpressed in Clear Cell Renal Cell Carcinoma Tissue and Transfection and Overexpression in Caki-1 Cells Inhibits Cell Proliferation and Increases Apoptosis. Med. Sci. Monit. 2018, 24, 8391–8400. [Google Scholar] [CrossRef]
- Zheng, B.S.; Wang, S.D.; Zhang, J.Y.; Ge, C.G. Expression, Prognostic Value, and Immune Infiltration of MTHFD Family in Bladder Cancer. Curr. Cancer Drug Targets 2024, 24, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wang, H.; Xu, H.-R.; Zhang, Y.-C.; Yu, X.-B.; Wu, M.-C.; Jin, G.-Z.; Cong, W.-M. Overexpression of MTHFD1 in Hepatocellular Carcinoma Predicts Poorer Survival and Recurrence. Future Oncol. 2019, 15, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Li, M.; Wang, Y.; Zhang, Y.; Que, Y.; Lu, S.; Wang, J.; Zhu, J.; Huang, J.; Zhen, Z.; et al. MTHFD1 Regulates the NADPH Redox Homeostasis in MYCN-Amplified Neuroblastoma. Cell Death Dis. 2024, 15, 124. [Google Scholar] [CrossRef]
- Li, Z.; Ke, H.; Cai, J.; Ye, S.; Huang, J.; Zhang, C.; Yuan, M.; Lan, P.; Wu, X. MTHFD1 Regulates Autophagy to Promote Growth and Metastasis in Colorectal Cancer via the PI3K-AKT-mTOR Signaling Pathway. Cancer Med. 2024, 13, e70267. [Google Scholar] [CrossRef]
- Ding, K.; Jiang, J.; Chen, L.; Xu, X. Methylenetetrahydrofolate Dehydrogenase 1 Silencing Expedites the Apoptosis of Non-Small Cell Lung Cancer Cells via Modulating DNA Methylation. Med. Sci. Monit. 2018, 24, 7499–7507. [Google Scholar] [CrossRef]
- Hao, Y.; Lu, R.; Guo, Y.; Bao, P. Specific Association of MTHFD1 Expressions with Small Cell Lung Cancer Development and Chemoradiotherapy Outcome. Saudi Med. J. 2024, 45, 783–790. [Google Scholar] [CrossRef]
- Loh, Z.N.; Wang, M.-E.; Wan, C.; Asara, J.M.; Ji, Z.; Chen, M. Nuclear PTEN Regulates Thymidylate Biosynthesis in Human Prostate Cancer Cell Lines. Metabolites 2023, 13, 939. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- MacFarlane, A.J.; Perry, C.A.; Girnary, H.H.; Gao, D.; Allen, R.H.; Stabler, S.P.; Shane, B.; Stover, P.J. Mthfd1 Is an Essential Gene in Mice and Alters Biomarkers of Impaired One-Carbon Metabolism. J. Biol. Chem. 2009, 284, 1533–1539. [Google Scholar] [CrossRef]
- Field, M.S.; Kamynina, E.; Agunloye, O.C.; Liebenthal, R.P.; Lamarre, S.G.; Brosnan, M.E.; Brosnan, J.T.; Stover, P.J. Nuclear Enrichment of Folate Cofactors and Methylenetetrahydrofolate Dehydrogenase 1 (MTHFD1) Protect de Novo Thymidylate Biosynthesis during Folate Deficiency. J. Biol. Chem. 2014, 289, 29642–29650. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | p Value | Odds Ratio | Combined Score | Genes |
|---|---|---|---|---|
| etoposide BOSS | 0.003 | 688.6 | 4000.6 | LGALS4 |
| 1-(5-deoxypentofuranosyl)-5-fluoropyrimidine- 2,4(1h,3h)-dione CTD 00001171 | 0.005 | 399.0 | 2106.4 | LGALS4 |
| choline CTD 00005662 | 0.005 | 399.0 | 2106.4 | MTHFD1 |
| capecitabine CTD 00003557 | 0.006 | 362.6 | 1880.6 | LGALS4 |
| lactose BOSS | 0.010 | 197.0 | 903.8 | LGALS4 |
| choline hydroxide BOSS | 0.011 | 185.9 | 842.3 | MTHFD1 |
| dl-methionine BOSS | 0.017 | 119.5 | 489.4 | MTHFD1 |
| folic acid BOSS | 0.018 | 110.1 | 442.2 | MTHFD1 |
| methotrexate BOSS | 0.018 | 109.5 | 439.1 | MTHFD1 |
| dihydroergocristine HL60 DOWN | 0.019 | 103.7 | 410.4 | MTHFD1 |
| zinc sulfate CTD 00007264 | 0.028 | 68.9 | 245.2 | MTHFD1 |
| paclitaxel CTD 00007144 | 0.049 | 39.6 | 119.8 | LGALS4 |
| alprostadil HL60 DOWN | 0.049 | 39.1 | 117.6 | MTHFD1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, H.; Su, H.; Lv, Z.; Zhu, C.; Huang, J. Identifying MTHFD1 and LGALS4 as Potential Therapeutic Targets in Prostate Cancer Through Multi-Omics Mendelian Randomization Analysis. Biomedicines 2025, 13, 185. https://doi.org/10.3390/biomedicines13010185
Han H, Su H, Lv Z, Zhu C, Huang J. Identifying MTHFD1 and LGALS4 as Potential Therapeutic Targets in Prostate Cancer Through Multi-Omics Mendelian Randomization Analysis. Biomedicines. 2025; 13(1):185. https://doi.org/10.3390/biomedicines13010185
Chicago/Turabian StyleHan, Huan, Hanwen Su, Zhihua Lv, Chengliang Zhu, and Jingtao Huang. 2025. "Identifying MTHFD1 and LGALS4 as Potential Therapeutic Targets in Prostate Cancer Through Multi-Omics Mendelian Randomization Analysis" Biomedicines 13, no. 1: 185. https://doi.org/10.3390/biomedicines13010185
APA StyleHan, H., Su, H., Lv, Z., Zhu, C., & Huang, J. (2025). Identifying MTHFD1 and LGALS4 as Potential Therapeutic Targets in Prostate Cancer Through Multi-Omics Mendelian Randomization Analysis. Biomedicines, 13(1), 185. https://doi.org/10.3390/biomedicines13010185