
Comparison of Protective Effects of Antidepressants Mediated by Serotonin Receptor in Aβ-Oligomer-Induced Neurotoxicity

 ,
,
Abstract
1. Introduction
2. Materials and Methods
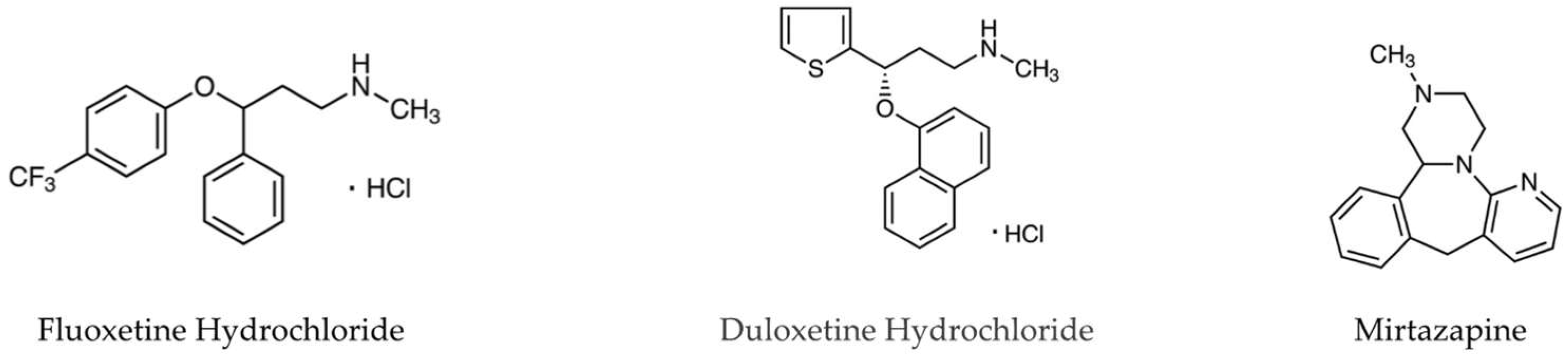
2.1. Drugs and Reagents
2.2. Preparation of Aβo
2.3. Cell Culture
2.4. Cell Viability Assay
MTT Assay
2.5. Oxidative Stress Assay
2.5.1. Detection of Reactive Oxygen Species (ROS)
2.5.2. Detection of Mitochondrial ROS
2.5.3. Phospholipid Peroxidation in Cell Membranes
2.5.4. Human Heme Oxygenase 1 (HO-1) Levels
2.5.5. Manganese Superoxide Dismutase (Mn-SOD) Levels
2.6. Statistical Analyses
3. Results
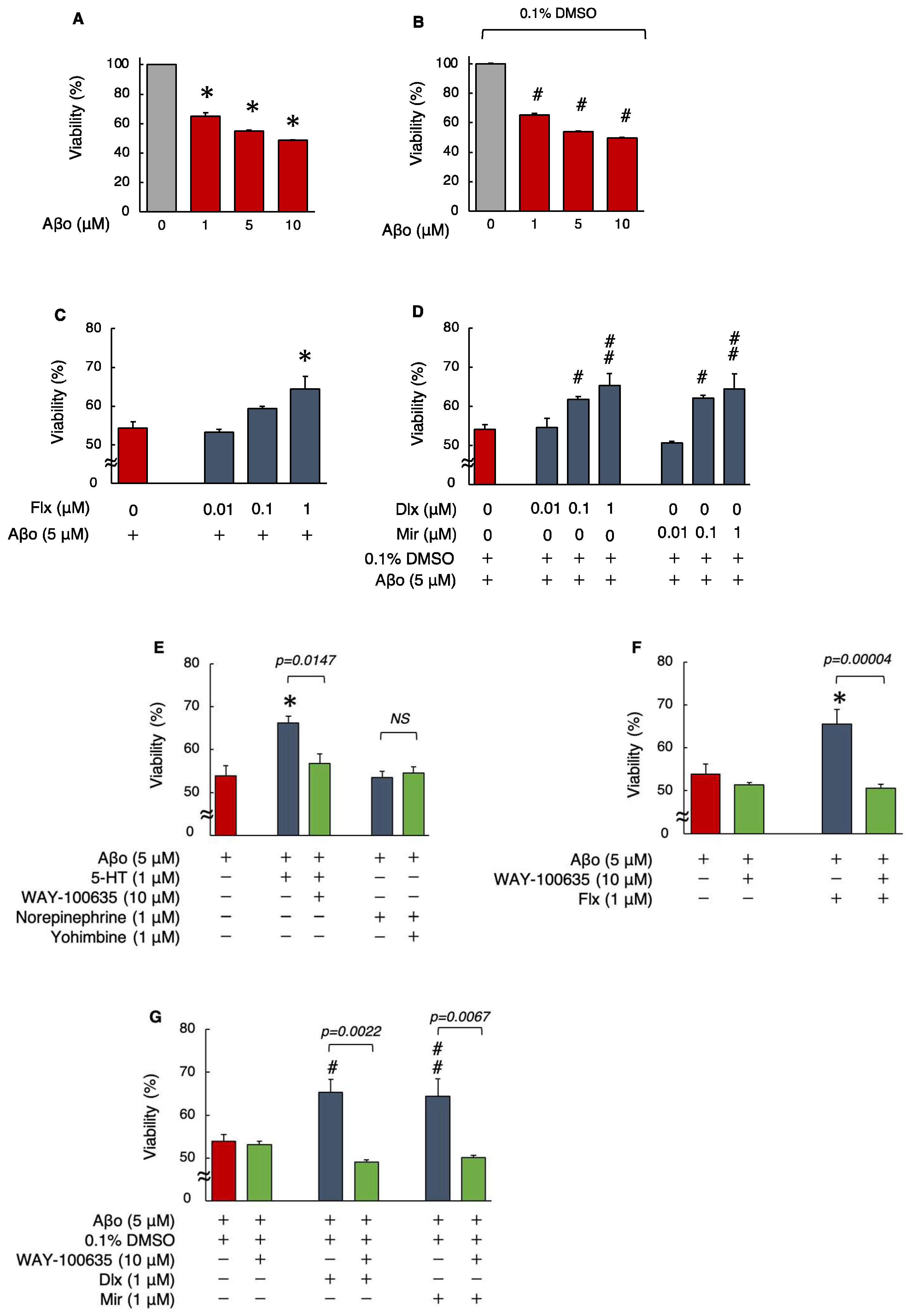
3.1. Effects of Antidepressants on Aβo-Induced Cytotoxicity
Detection of Cell Viability by MTT Assay
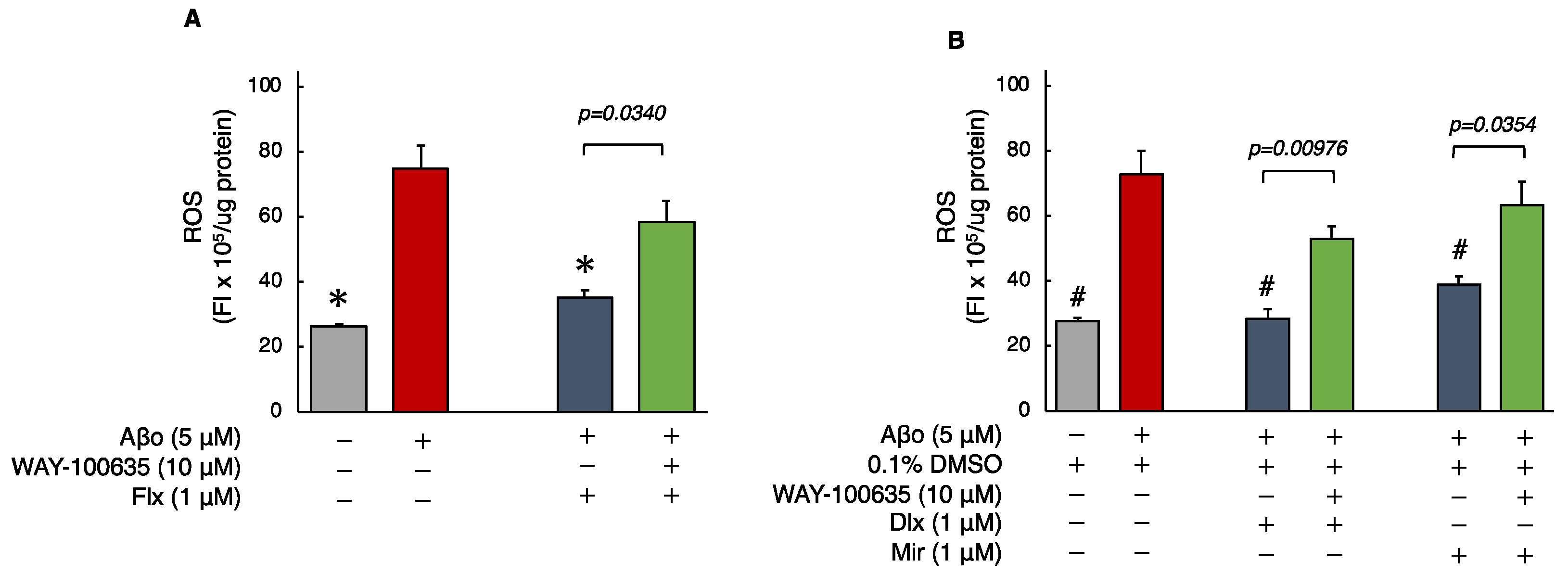
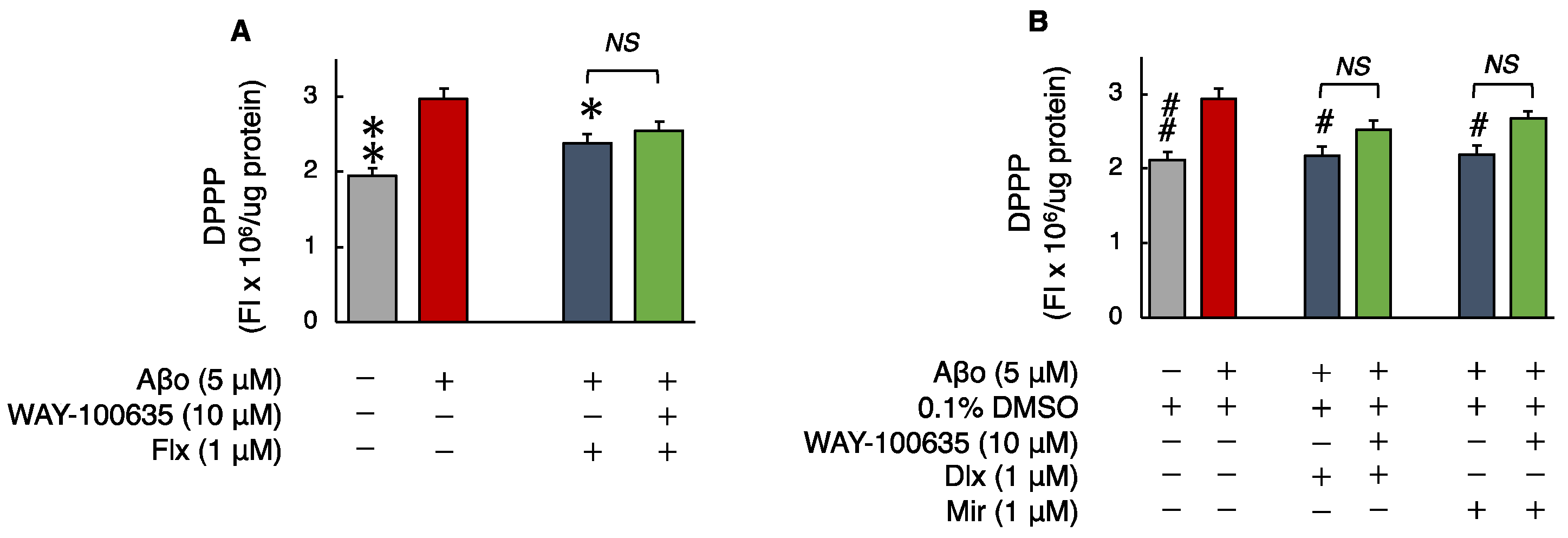
3.2. Effects of Antidepressants on Aβo-Induced Oxidative Stress
3.2.1. Detection of ROS
3.2.2. Detection of Mitochondrial ROS (mito-ROS)
3.2.3. Phospholipid Peroxidation in Cell Membranes
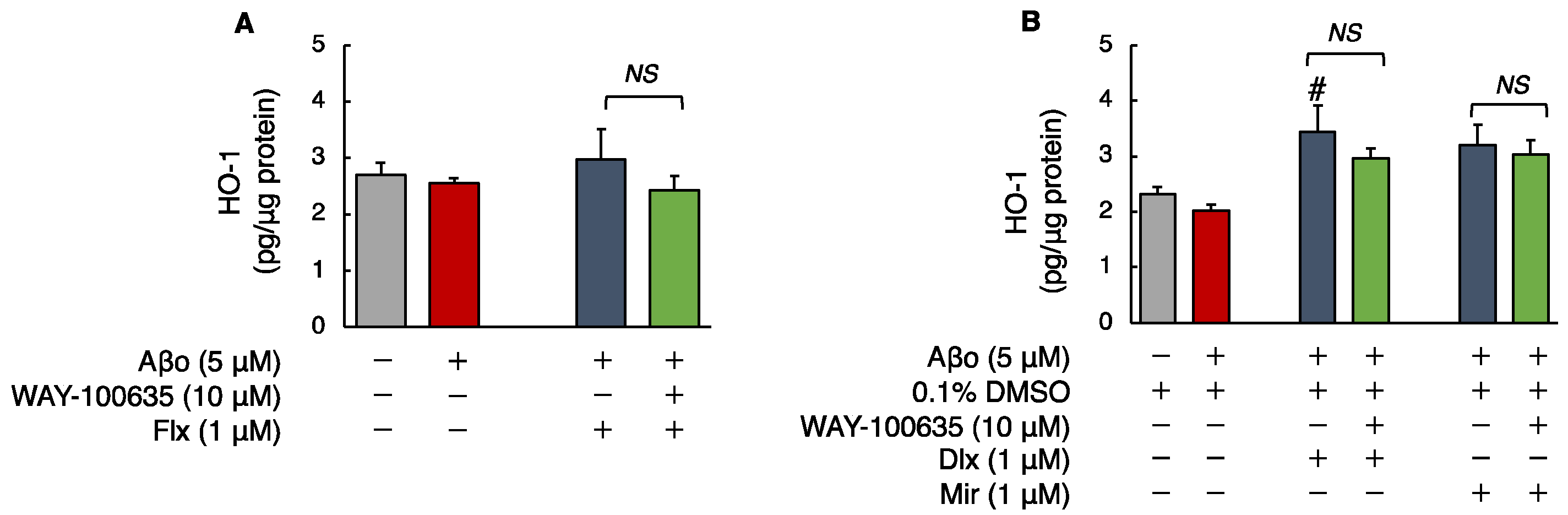
3.2.4. Human HO-1 Levels
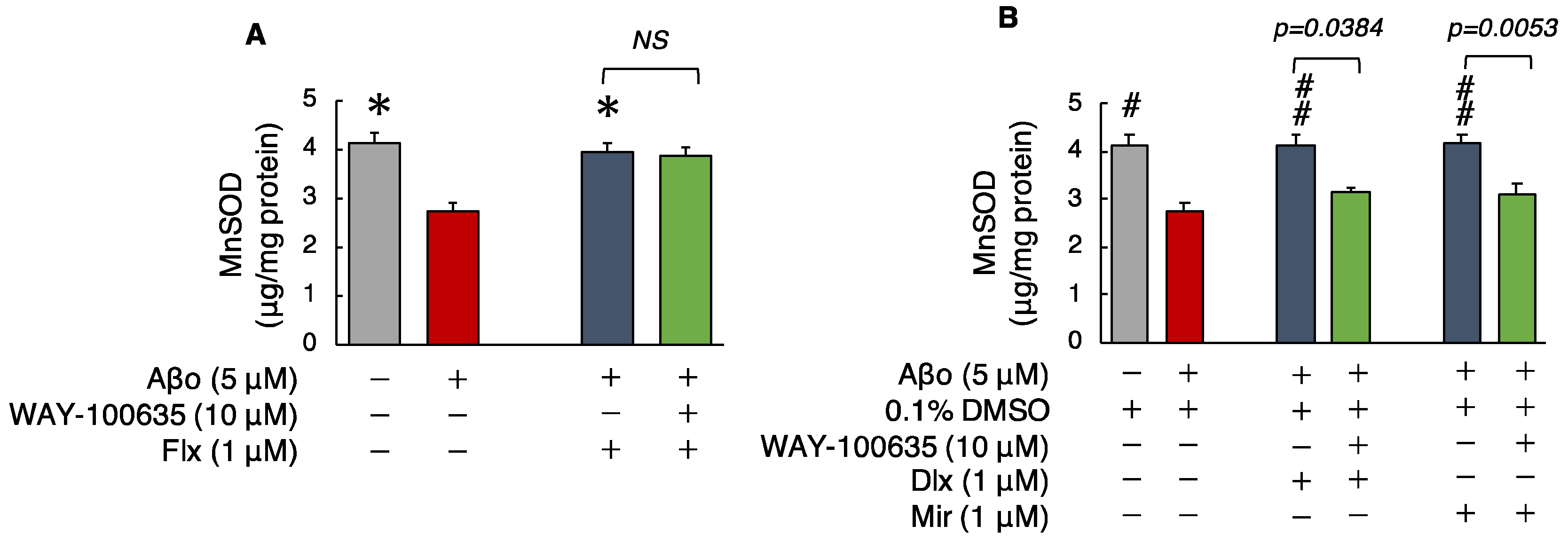
3.2.5. Manganese Superoxide Dismutase (MnSOD) Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Dementia. Available online: http://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 1 February 2024).
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Yasumoto, T.; Takamura, Y.; Tsuji, M.; Watanabe-Nakayama, T.; Imamura, K.; Inoue, H.; Nakamura, S.; Inoue, T.; Kimura, A.; Yano, S.; et al. High molecular weight amyloid β1-42 oligomers induce neurotoxicity via plasma membrane damage. FASEB J. 2019, 33, 9220–9234. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Mora, P.; Luna, R.; Colín-Barenque, L. Amyloid beta: Multiple mechanisms of toxicity and only some protective effects? Oxid Med. Cell. Longev. 2014, 2014, 795375. [Google Scholar] [CrossRef] [PubMed]
- Schwertner, E.; Pereira, J.B.; Xu, H.; Secnik, J.; Winblad, B.; Eriksdotter, M.; Nägga, K.; Religa, D. Behavioral and psychological Symptoms of dementia in different dementia disorders: A large-scale study of 10,000 individuals. J. Alzheimers Dis. 2022, 87, 1307–1318. [Google Scholar] [CrossRef]
- Richard, E.; Reitz, C.; Honig, L.H.; Schupf, N.; Tang, M.X.; Manly, J.J.; Mayeux, R.; Devanand, D.; Luchsinger, J.A. Late-life depression, mild cognitive impairment, and dementia. JAMA Neurol. 2013, 70, 374–382. [Google Scholar] [CrossRef]
- Leong, C. Antidepressants for depression in patients with dementia: A review of the literature. Consult. Pharm. 2014, 29, 254–263. [Google Scholar] [CrossRef]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Farina, N.; Morrell, L.; Banerjee, S. What is the therapeutic value of antidepressants in dementia? A narrative review. Int. J. Geriatr. Psychiatry 2017, 32, 32–49. [Google Scholar] [CrossRef]
- Pomara, N.; Sidtis, J. Possible therapeutic implication of Abeta disturbances in depression. Int. J. Geriatr. Psychiatry 2007, 22, 931–932. [Google Scholar] [CrossRef] [PubMed]
- NIH Medline Plus Magazine. Available online: https://magazine.medlineplus.gov/article/commonly-prescribed-antidepressants-and-how-they-work#:~:text=Selective%20serotonin%20reuptake%20inhibitors%20 (accessed on 12 April 2024).
- Anderson, I.M. Selective serotonin reuptake inhibitors versus tricyclic antidepressants: A meta-analysis of efficacy and tolerability. J. Affect. Disord. 2000, 58, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Aboukhatwa, M.; Luo, Y. Antidepressants modulate intracellular amyloid peptide species in N2a neuroblastoma cells. J. Alzheimers Dis. 2011, 24, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Fabbretti, E.; Antognolli, G.; Tongiorgi, E. Amyloid-β impairs dendritic trafficking of Golgi-like organelles in the early phase preceding neurite atrophy: Rescue by mirtazapine. Front. Mol. Neurosci. 2021, 14, 661728. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Xu, H.; Zhu, S.; Wang, H.; He, J.; Zhang, H.; Guo, H.; Kong, J.; Huang, Q.; et al. Fluoxetine improves behavioral performance by suppressing the production of soluble β-amyloid in APP/PS1 mice. Curr. Alzheimer Res. 2014, 11, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.N.; Chao, F.L.; Zhang, Y.; Jiang, L.; Zhang, L.; Fan, J.H.; Wu, Y.X.; Dou, X.Y.; Tang, Y. Fluoxetine delays the cognitive function decline and synaptic changes in a transgenic mouse model of early Alzheimer’s disease. J. Comp. Neurol. 2019, 527, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Bharti, V.; Tan, H.; Deol, J.; Wu, Z.; Wang, J.F. Upregulation of antioxidant thioredoxin by antidepressants fluoxetine and venlafaxine. Psychopharmacology 2020, 237, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Meejuru, G.F.; Somavarapu, A.; Danduga, R.C.S.R.; Nissankara Roa, L.S.; Kola, P.K. Protective effects of duloxetine against chronic immobilisation stress-induced anxiety, depression, cognitive impairment and neurodegeneration in mice. J. Pharm. Pharmacol. 2021, 73, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Ogata, N.; de Souza Dantas, L.M.; Crowell-Davis, S.L. Selective serotonin reuptake inhibitors. In Veterinary Psychopharmacology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2022; pp. 103–128. [Google Scholar] [CrossRef]
- Truchot, L.; Costes, N.; Zimmer, L.; Laurent, B.; Le Bars, D.; Thomas-Antérion, C.; Mercier, B.; Hermier, M.; Vighetto, A.; Krolak-Salmon, P. A distinct [18F]MPPF PET profile in amnestic mild cognitive impairment compared to mild Alzheimer’s disease. Neuroimage 2008, 40, 1251–1256. [Google Scholar] [CrossRef]
- Momma, Y.; Tsuji, M.; Oguchi, T.; Ohashi, H.; Nohara, T.; Ito, N.; Yamamoto, K.; Nagata, M.; Kimura, A.M.; Nakamura, S.; et al. The curcumin derivative GT863 protects cell membranes in cytotoxicity by Aβ oligomers. Int. J. Mol. Sci. 2023, 24, 3089. [Google Scholar] [CrossRef]
- Nohara, T.; Tsuji, M.; Oguchi, T.; Momma, Y.; Ohashi, H.; Nagata, M.; Ito, N.; Yamamoto, K.; Murakami, H.; Kiuchi, Y. Neuroprotective potential of raloxifene via G-protein-coupled estrogen receptors in Aβ-oligomer-induced neuronal injury. Biomedicines 2023, 11, 2135. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2021, 421, 117253. [Google Scholar] [CrossRef] [PubMed]
- Arimon, M.; Takeda, S.; Post, K.L.; Svirsky, S.; Hyman, B.T.; Berezovska, O. Oxidative stress and lipid peroxidation are upstream of amyloid pathology. Neurobiol. Dis. 2015, 84, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Perry, G.; Smith, M.A. Oxidative stress and neurotoxicity. Chem. Res. Toxicol. 2008, 21, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Disabato, B.M.; Restivo, J.L.; Verges, D.K.; Goebel, W.D.; Sathyan, A.; Hayreh, D.; D’Angelo, G.; Benzinger, T.; Yoon, H.; et al. Serotonin signaling is associated with lower amyloid-β levels and plaques in transgenic mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 14968–14973. [Google Scholar] [CrossRef] [PubMed]
- Gelain, D.P.; Antonio Behr, G.; Birnfeld de Oliveira, R.; Trujillo, M. Antioxidant therapies for neurodegenerative diseases: Mechanisms, current trends, and perspectives. Oxid Med. Cell. Longev. 2012, 2012, 895153. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.R.; Liu, R.T. The toxicity and polymorphism of β-amyloid oligomers. Int. J. Mol. Sci. 2020, 21, 4477. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal 2009, 11, 2985–3011. [Google Scholar] [CrossRef] [PubMed]
- Inkielewicz-Stępniak, I. Impact of fluoxetine on liver damage in rats. Pharmacol. Rep. 2011, 63, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Novío, S.; Núñez, M.J.; Amigo, G.; Freire-Garabal, M. Effects of fluoxetine on the oxidative status of peripheral blood leucocytes of restraint-stressed mice. Basic. Clin. Pharmacol. Toxicol. 2011, 109, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Grasso, M.; Fidilio, A.; Torrisi, S.A.; Musso, N.; Geraci, F.; Tropea, M.R.; Privitera, A.; Tascedda, F.; Puzzo, D.; et al. Antioxidant activity of fluoxetine and vortioxetine in a non-transgenic animal model of Alzheimer’s disease. Front. Pharmacol. 2021, 12, 809541. [Google Scholar] [CrossRef] [PubMed]
- Engel, D.F.; de Oliveira, J.; Lieberknecht, V.; Rodrigues, A.L.S.; de Bem, A.F.; Gabilan, N.H. Duloxetine protects human neuroblastoma cells from oxidative stress-induced cell death through Akt/Nrf-2/HO-1 pathway. Neurochem. Res. 2018, 43, 387–396. [Google Scholar] [CrossRef]
- Kolla, N.; Wei, Z.; Richardson, J.S.; Li, X.M. Amitriptyline and fluoxetine protect PC12 cells from cell death induced by hydrogen peroxide. J. Psychiatry Neurosci. 2005, 30, 196–201. [Google Scholar] [PubMed]
- Pu, Y.; Zhao, L.; Xi, Y.; Xia, Y.; Qian, Y. The protective effects of mirtazapine against lipopolysaccharide (LPS)-induced brain vascular hyperpermeability. Bioengineered 2022, 13, 3680–3693. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.J.; Heiser, P.; Hemmeter, U.M.; Krieg, J.C.; Vedder, H. Effects of antidepressants on mRNA levels of antioxidant enzymes in human monocytic U-937 cells. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1567–1573. [Google Scholar] [CrossRef]
- Díaz-Tufinio, C.A.; Palma-Aguirre, J.A.; González-Covarrubias, V. Pharmacogenetic variants associated with fluoxetine pharmacokinetics from a bioequivalence study in healthy subjects. J. Pers. Med. 2023, 13, 1352. [Google Scholar] [CrossRef]
- Knadler, M.P.; Lobo, E.D.; Chappell, J.; Bergstrom, R.F. Duloxetine: Clinical pharmacokinetics and drug interactions. Clin. Pharmacokinet. 2011, 50, 281–294. [Google Scholar] [CrossRef]
- Timmer, C.J.; Sitsen, J.M.A.; Delbressine, L.P.C. Clinical pharmacokinetics of mirtazapine. Clin. Pharmacokinet. 2000, 38, 461–474. [Google Scholar] [CrossRef]
- Gao, W.; Chen, R.; Xie, N.; Tang, D.; Zhou, B.; Wang, D. Duloxetine-induced neural cell death and promoted neurite outgrowth in N2a cells. Neurotox. Res. 2020, 38, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Sarikaya, S.B.; Gulcin, I. Radical scavenging and antioxidant capacity of serotonin. Curr. Bioact. Compd. 2013, 9, 143–152. [Google Scholar] [CrossRef]
- Lobayan, R.M.; Schmit, M.C.P. Conformational and NBO studies of serotonin as a radical scavenger. Changes induced by the OH group. J. Mol. Graph. Model. 2018, 80, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Weng, R.; Shen, S.; Tian, Y.; Burton, C.; Xu, X.; Liu, Y.; Chang, C.; Bai, Y.; Liu, H. Metabolomics approach reveals integrated metabolic network associated with serotonin deficiency. Sci. Rep. 2015, 5, 11864. [Google Scholar] [CrossRef] [PubMed]
- Nagayasu, K.; Kitaichi, M.; Nishitani, N.; Asaoka, N.; Shirakawa, H.; Nakagawa, T.; Kaneko, S. Chronic effects of antidepressants on serotonin release in rat raphe slice cultures: High potency of milnacipran in the augmentation of serotonin release. Int. J. Neuropsychopharmacol. 2013, 16, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Popova, N.K.; Naumenko, V.S. 5-HT1A receptor as a key player in the brain 5-HT system. Rev. Neurosci. 2013, 24, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Albert, P.R.; Vahid-Ansari, F. The 5-HT1A receptor: Signaling to behavior. Biochimie 2019, 161, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Rojas, P.S.; Fiedler, J.L. What do we really know about 5-HT1A receptor signaling in neuronal cells? Front. Cell. Neurosci. 2016, 10, 272. [Google Scholar] [CrossRef]
- Verdurand, M.; Zimmer, L. Hippocampal 5-HT1A receptor expression changes in prodromal stages of Alzheimer’s disease: Beneficial or deleterious? Neuropharmacology 2017, 123, 446–454. [Google Scholar] [CrossRef]
- Mattsson, P.; Cselényi, Z.; Andrée, B.; Borg, J.; Nag, S.; Halldin, C.; Farde, L. Decreased 5-HT1A binding in mild Alzheimer’s disease-A positron emission tomography study. Synapse 2022, 76, e22235. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activities | Measurements | Flx | Dlx | Mir |
|---|---|---|---|---|
| Viability | MTT | + | ++ | ++ |
| Antioxidant effects | ROS | + | ++ | + |
| Mito-ROS | + | ++ | ++ | |
| Phospholipid peroxidation | + | + | + | |
| Activation of antioxidative enzyme activity | HO-1 | − | + | − |
| Mn-SOD | + | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, K.; Tsuji, M.; Oguchi, T.; Momma, Y.; Ohashi, H.; Ito, N.; Nohara, T.; Nakanishi, T.; Ishida, A.; Hosonuma, M.; et al. Comparison of Protective Effects of Antidepressants Mediated by Serotonin Receptor in Aβ-Oligomer-Induced Neurotoxicity. Biomedicines 2024, 12, 1158. https://doi.org/10.3390/biomedicines12061158
Yamamoto K, Tsuji M, Oguchi T, Momma Y, Ohashi H, Ito N, Nohara T, Nakanishi T, Ishida A, Hosonuma M, et al. Comparison of Protective Effects of Antidepressants Mediated by Serotonin Receptor in Aβ-Oligomer-Induced Neurotoxicity. Biomedicines. 2024; 12(6):1158. https://doi.org/10.3390/biomedicines12061158
Chicago/Turabian StyleYamamoto, Ken, Mayumi Tsuji, Tatsunori Oguchi, Yutaro Momma, Hideaki Ohashi, Naohito Ito, Tetsuhito Nohara, Tatsuya Nakanishi, Atsushi Ishida, Masahiro Hosonuma, and et al. 2024. "Comparison of Protective Effects of Antidepressants Mediated by Serotonin Receptor in Aβ-Oligomer-Induced Neurotoxicity" Biomedicines 12, no. 6: 1158. https://doi.org/10.3390/biomedicines12061158
APA StyleYamamoto, K., Tsuji, M., Oguchi, T., Momma, Y., Ohashi, H., Ito, N., Nohara, T., Nakanishi, T., Ishida, A., Hosonuma, M., Nishikawa, T., Murakami, H., & Kiuchi, Y. (2024). Comparison of Protective Effects of Antidepressants Mediated by Serotonin Receptor in Aβ-Oligomer-Induced Neurotoxicity. Biomedicines, 12(6), 1158. https://doi.org/10.3390/biomedicines12061158