1. Introduction
Chemotherapy, especially together with immunotherapy, has resulted in major advances in cancer treatment [
1,
2]. The development of personalized chemotherapy using inhibitors of specific signal transduction proteins such as specific forms of oncogenic ras-p21 [
3] have likewise resulted in improved cancer treatment modalities, especially in relation to solid tissue tumors such as colorectal cancer [
4] and lung cancer [
5]. These recently developed methods still have off-target effects that include the development of autoimmune states in patients treated with anti-PD1 (programmed cell death protein on the surface of T cells) and CTLA (cytotoxic T-lymphocyte-associated protein 4) agents [
6] and sometimes compromised efficacy when tumors undergo changes such as mutations or use of alternate signal transduction pathways [
1].
This review concerns the recent discovery of a set of peptides, the main prototype of which is PNC-27, containing p53 residues 12–26, from its transactivating HDM-2-protein-binding domain, attached to a transmembrane-penetrating peptide sequence, described below, that kills a wide variety of cancer cells, including solid tissue tumors and hematopoietic cancers, but that has no effects on the viability and growth of normal cells. The mechanism involves the interaction of these peptides with HDM-2 (human-double-minute-binding protein) expressed in the cell membranes of cancer cells but expressed either at low levels of or not at all in the membranes of normal cells. Colocalization of these peptides with membrane-expressed HDM-2 results in the formation of transmembrane pores, leading to the explosive release of the cancer cells’ intracellular contents, resulting in tumor cell necrosis, termed poptosis. These peptides have been tested in vivo against human tumors and have been found to eradicate them. The advantage in the use of these peptides is that their anti-cancer effects do not depend on the inhibition of intracellular mechanisms such as specific signal transduction pathways, specific proteins on these pathways, specific DNA-repair processes, DNA methylation, or the inhibition of multiple drug-resistant (MDR) gene products, giving them the potential to treat many different forms of cancer with the only requirement being that they interact with cancer-cell-membrane-expressed HDM-2.
It should be noted that several other peptides have been developed that have also been found to induce pore formation in cancer cells. Several membrane-active antibiotic peptides such as magainin, which contains positively charged amino acid residues, allowing for an interaction with the negatively charged surfaces of cells, have been found to be membranolytic, preferentially to cancer cells [
7], but can also affect normal cells. Certain cell-penetrating peptides (CPP), that enable the delivery of cargo, such as peptides and poly-nucleic acids into cells, target cancer cells that have been found to contain significant levels of gangliosides in their membranes that are not present in normal cells [
8]. Other peptides have been found to bind preferentially to cancer cell membranes. An example of the use of cancer-cell-targeting CPPs is the use of the peptide derived from the antimicrobial peptide buforin, called BR2 (whose sequence is RAGLQFPVGRLLRRLLR). This peptide was found to enter cancer cells selectively (it did enter normal cells, although to a significantly less degree) and was used to deliver the inactivating anti-ras-p21 antibody (Y13-259) into cancer cells, resulting in tumor cell necrosis. This effect was abolished if the cells were incubated with a ganglioside synthesis inhibitor, strongly suggesting that the ganglioside composition of cancer cells may be different from that of untransformed cells, allowing them to be targeted by agents that bind to membrane-expressed gangliosides [
8].
Using this approach, a series of decoy peptides were designed to block the activation of the AAC-11 anti-apoptotic protein in tumor cells, most notably in Sezary’s leukemia cells [
9,
10,
11]. AAC-11 is thought to activate PAK1 (p21-activated kinase) on its anti-apoptotic pathway. These peptides contained the heptad leucine zipper sequence involved in the activation domain of AAC-11 and were attached on the amino termini to a CPP (penetratin) sequence. These peptides were found to colocalize with PAK1 in the cell membranes of Sezary’s leukemia cells in vitro and in vivo, and they induced cell death via pore formation and apoptosis. The release of damage-associated molecular pattern agents (DAMPs) results in the activation of immune responses to the cancer cells. Thus, besides inducing tumor cell killing, these peptides appear to activate an anti-tumor immune response. These peptides have yet to be tested against other human cancers [
11].
In this current review, we show that specific peptides from the HDM-2-binding domain of p53 attached to a CPP from antennapaedia on their carboxyl terminal ends induce tumor cell necrosis of a wide range of cancers, including solid tissue and hematopoietic cancers, by inducing transmembrane pores, and that they have no effect on the growth and viability of a wide variety of normal cells. These peptides act by binding to the HDM-2 protein, which is expressed significantly in the membranes of cancer but not normal cells. When bound to membrane-attached HDM-2, the peptide-HDM-2 complexes form transmembrane pores that can be directly observed in high-resolution immuno-scanning electron microscopy.
2. Design and Effects of PNC-27 and PNC-28
The most effective of these poptosis-inducing agents is PNC-27, which contains amino acid residues 12–26 of the transactivating domain of p53, which have been found to be involved in the binding of p53 to the H(M)DM-2 protein [
12]. This sequence is attached to a transmembrane-penetrating sequence, also called penetratin, as shown in entry 1 of
Table 1 [
13]. The leader sequence was added to this peptide (and other peptides used in our studies, such as those shown in
Table 1) to induce the crossing of the peptides across the cell membrane since the isolated p53 12–26 peptide had no effect on cancer cells due to its inability to cross the cell membrane [
13].
The second peptide shown in
Table 1 is PNC-28, which is a shorter version of PNC-27 and contains p53 residues 17–26 that contain the p53 residues that contacts HDM-2, and has anti-cancer activity similar to that of PNC-27. The third peptide shown in
Table 1 is a negative control peptide, containing a sequence (×13) from human cytochrome p450 attached to the leader sequence on its carboxyl terminal end. H (human) or M (mouse) DM2 (double minute-binding protein-2) is mainly a nuclear protein that is an E3 ubiquitin ligase that binds to p53 in its amino terminal domain (residues 1–109) and several other proteins that target these proteins for ubiquitination and degradation in the proteosome [
12]. Thus, HDM-2 limits the ability of p53 to stimulate the apoptosis of transformed cells.
PNC-27 was originally designed by our research group to block the binding of p53 to HDM-2 in the nuclei of cancer cells as a “decoy” peptide, i.e., the peptide would cross the cancer cell and nuclear membranes using its penetratin sequence and bind to HDM-2 in place of the full p53 protein, hopefully enhancing p53-induced apoptosis of the cancer cells [
13].
2.1. PNC-27 and 28 Induce Tumor Cell Necrosis of Multiple Different Types of Cancers
In multiple studies, we found that PNC-27 and PNC-28 (
Table 1) induced rapid cancer cell necrosis but did not affect untransformed cells [
13], as illustrated in
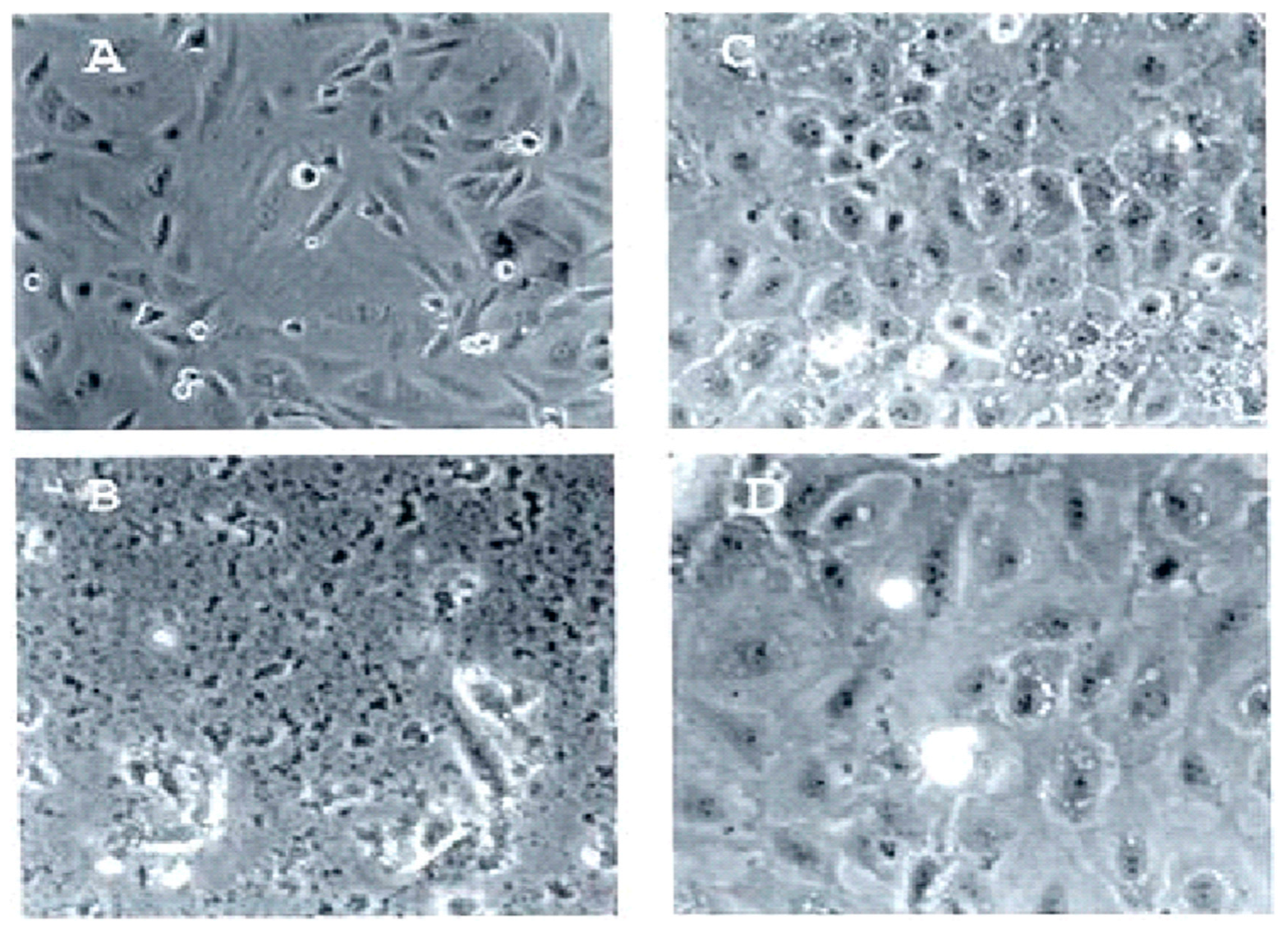
Figure 1, wherein rat pancreatic acinar cancer cells called TUC-3 (the spindle-shaped cancer cells shown in
Figure 1A) and their normal counterpart pancreatic acinar cells called BMRPA1 (shown in
Figure 1B) were incubated with PNC-28 for 24 h. As shown in
Figure 1C, all the TUC-3 cells were killed by PNC-28, while, as shown in
Figure 1D, the BMRPA1 cells remained viable [
13].
In further studies, PNC-27 and PNC-28 have been found to be cytotoxic to a large variety of human and mammalian solid tissue and non-solid tissue cancer cells, including pancreatic [
13], breast [
13], ovarian [
14], colon [
15], cervical (HeLa) [
13], non-small cell lung carcinoma [
13], angiosarcoma [
13], osteogenic sarcoma [
13], acute myelogenous leukemia [
16,
17], and chronic myelogenous leukemia [
18], with IC
50 values that ranged from 6–80 μM, while neither peptide was found to be cytotoxic to untransformed control cells that included human fibroblasts [
13], pancreatic acinar cells [
13], human breast epithelial cells [
13], keratinocytes [
13], human umbilical vein (HUVEC) cells [
14], and rat mononuclear cells [
16] at the highest concentrations used with the corresponding cancer cells. Importantly, PNC-28 was found to have no effect on the abilities of human hematopoietic stem cells from the cord blood of five different donors to differentiate into mature hematopoietic cells in the presence of growth factors [
13], strongly suggesting that neither peptide would suppress bone marrow during cancer treatment. This finding was further confirmed in a recent study [
17], showing that PNC-27 was cytotoxic to human AML stem cells in vitro and in vivo but had no effect on normal human myeloid stem cells.
Typical dose–response curves for the cancer cell cytotoxicity of PNC-27 are shown in [
14,
15,
16] for ovarian cancers, colon cancers, and acute myelogenous leukemias. PNC-27 killed over 90 percent of these tumor cells (approximately 1 × 10
6) in a four-hour period. On the other hand, it can be seen in these figures that the negative control PNC-29 peptide had no effect on any of these cell lines. In addition, included for each cancer cell line, a corresponding normal cell line was likewise incubated with PNC-27 and PNC-29. For SKOV-3 cells, the control cell line was HUVEC cells (umbilical vein endothelial cells) for HCT-116 CCD-18Co colonic fibroblasts, and for U-937, rat mononuclear cells. Neither PNC-27 nor PNC-29 affected these untransformed cells. Importantly, PNC-27 was also found to be cytotoxic to OVCAR-3 cells that are multi-drug-resistant [
14], indicating that cancer cell killing was independent of intracellular processes.
Included among the cancer cells tested with PNC-27 were primary ovarian cancer cells, i.e., cells that were removed from patients who underwent excision of these tumors under an IRB protocol and that were then incubated in culture with PNC-27. Complete killing of these cells by 60 μM PNC-27 (by not by PNC-29) was achieved after 4 h incubation, suggesting the efficacy of this peptide in the treatment of ovarian cancer. In addition, PNC-27 was found to be cytotoxic to cancer stem cells, including colon cancer cell lines such as CTA, CTR, and SW1222 [
15], that are enriched in CD44-positive tumor stem cells. As discussed below, PNC-27 is cytotoxic to human acute myelogenous tumor stem cells [
17] without affecting normal hematopoietic stem cells [
13,
17], as discussed above. Thus, PNC-27 kills primary human tumors, chemoresistant cancers, and tumor stem cells without affecting normal cells.
2.2. PNC-28 Kills TUC-3 Cells In Vivo
As discussed above, PNC-27 and PNC-28 kill murine TUC-3 pancreatic cancer cells, but not their counterpart normal pancreatic acinar BMRPA1 cells, in culture. TUC-3 cells form highly metastatic tumors in nude mice within two weeks after transplantation. We implanted TUC-3 cells in nude mice either intraperitoneally or subdermally and treated these mice with continuous infusion of PNC-28 or negative control PNC-29 over a two-week period using intraperitoneally placed Alzet minipumps. PNC-28 eradicated these tumors after two weeks; no tumor growth was observed over the successive two-week period. In contrast, tumors treated with PNC-29 were found to grow to large (3500 mg) tumors that metastasized over the four-week period. In the mice treated with PNC-28, there was no evidence of undesirable side effects in that the treated mice thrived and gained weight that was statistically insignificantly different from a control group of untreated mice [
13]. As discussed below, PNC-27 has been found to eradicate human-stem-cell-enriched acute myelogenous leukemia in vivo [
17].
2.3. PNC-27 Eradicates Human Acute Myelogenous Leukemia (AML) in Nude Mice
Although most of our studies on PNC-27 and PNC-28 have been focused on solid tissue tumors, both in vitro and in vivo, we also found that these peptides are cytotoxic to hematopoietic cell cancers, including AML [
16] and CML (chronic myelogenous leukemia) [
18], in vitro. Recently, a large in vivo study [
17] was performed at the City of Hope Medical Center in Los Angeles on stem-cell-enriched acute myelogenous leukemia (AML) cells harvested from the bone marrows from nine different patients. These cells were transplanted into the bone marrows of nude mice, which developed AML and high white cell counts. These mice were then treated with daily intraperitoneal injections of PNC-27 over a three-week period. At the end of this period, the white cell counts normalized. Bone marrow cells were then explanted from these mice into the bone marrows of naïve mice whose white cell counts were followed and found to be normal. Both cohorts of mice showed markedly prolonged Kaplan–Meier survival curves compared with those of treated mice with a negative control peptide, PNC-27.
3. PNC-27 and PNC-28 Are Cytotoxic to Cancer Cells by a p53-Independent Mechanism
An unexpected finding obtained for the cell lines studied was that PNC-27 and 28 both induced rapid cell death, which began to occur minutes after incubation of cell lines with either peptide but was not observed in cells undergoing apoptosis. For many tumor cell lines, complete tumor cell death occurred in 4 h. In none of these studies were the usual markers for apoptosis expressed, such as DNA laddering [
13], annexin V labeling of the membrane phospholipid, and caspase expression [
13,
14,
15,
16,
19]. In contrast, the release of LDH occurred almost immediately after the incubation of cell lines, with either peptide indicative of tumor cell death.
The Leader Sequence of PNC-27 and 28 Cause the Mechanism of Cell Killing from Apoptosis to Tumor Cell Necrosis
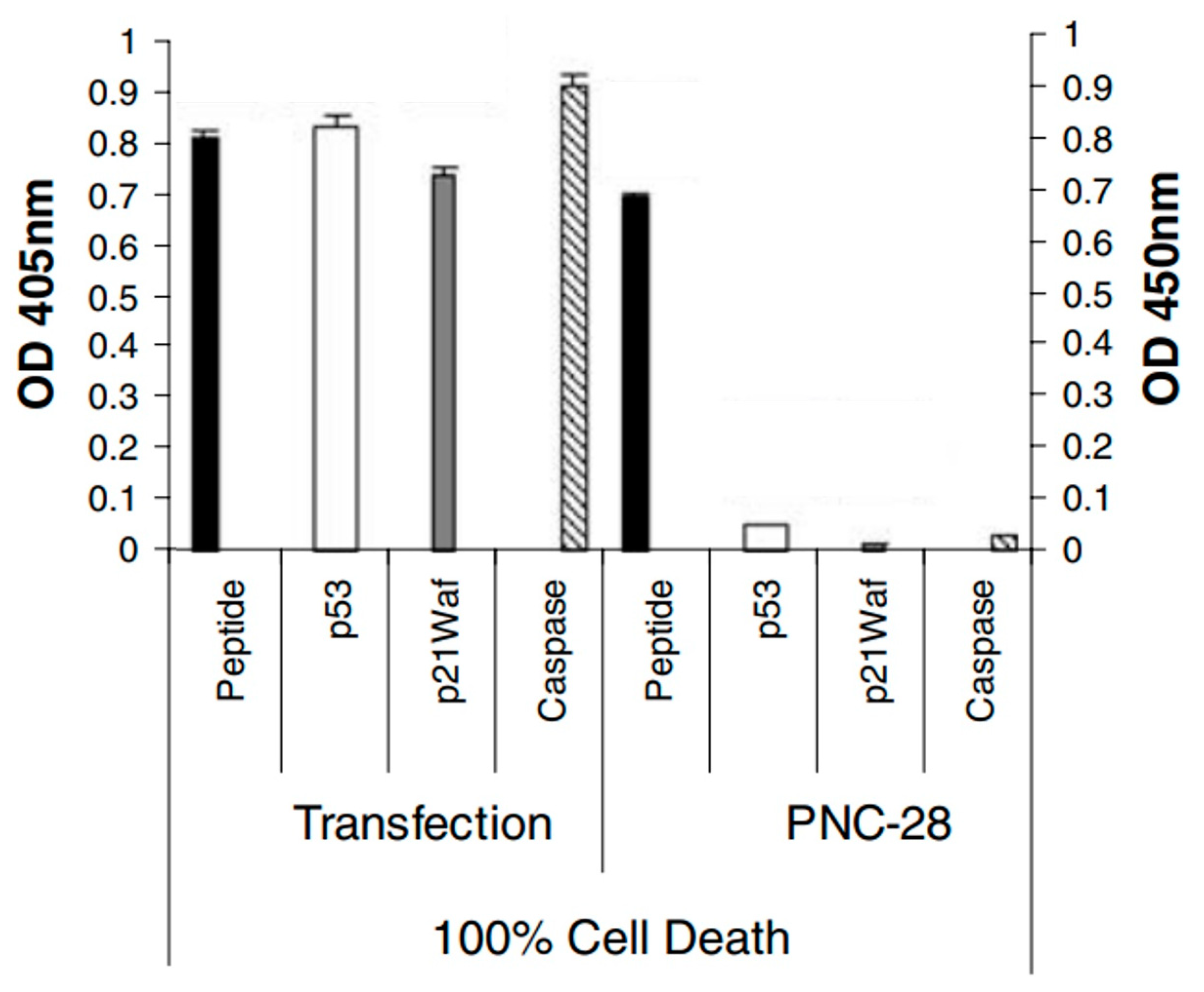
Since PNC-27 and 28 peptides were designed to bind to HDM-2 using the p53 12 (17)-26 sequence in the nucleus, we designed a plasmid [
19] that uniquely encoded this p53 sequence and transfected it into MIA-PaCa-2 pancreatic carcinoma cells. As shown in
Figure 2, these transfected cells expressed high levels of caspase-3 and p21
waf (another marker for apoptosis), as opposed to MIA-PaCa-2 cells that were incubated with PNC-28. In addition, p53 was found to be elevated in the transfected cells but not in the cells treated with PNC-28, as would be expected if the plasmid-expressed p53 peptide blocked HDM-2-induced degradation of the p53 protein. On the other hand, LDH release occurred almost immediately in the cells treated with PNC-28, but not in the transfected cells, pointing to a major difference in the mechanism of cancer cell killing.
Thus, the p53 peptide directly introduced into the cancer cells caused apoptosis, as was expected for the p53 peptide attached to the leader sequence. Evidently, the leader sequence of PNC-27 and -28 changed the mechanism of cancer cell death induced by both peptides, resulting in rapid tumor cell necrosis. Since the activation of p53 would be expected to result in apoptosis of the tumor cells, this novel mechanism apparently does not involve p53 activation.
This conclusion was verified by our finding that several cell lines killed by PNC-27 and PNC-28, such as MDA-MB-453 breast cancer cells [
13] and SAOS2 osteogenic sarcoma cells [
13], are known to have the p53 gene homozygous deleted. Thus, both peptides must be inducing cancer cell death by a non-p53-dependent mechanism, explaining the absence of the expected apoptosis.
4. Both PNC-27 and PNC-28 Induce Transmembrane Pore Formation, Resulting in the Extrusion of Cancer Cell Contents in a New Process Termed “Poptosis”
Since there is a rapid release of LDH indicative of membrane damage, we subjected tumor cells, i.e., breast cancer cells (MDA-MB-468) [
13], pancreatic cancer cells (MIA-PaCa-2) [
13], and melanoma cells (A2058) [
13], to transmission electron microscopy after incubating them for 15 min with PNC-27 or PNC-28. As illustrated in
Figure 3 for MIA-PaCa-2 cells incubated with PNC-28, pores were seen to form across the cell membranes of these cells, which were not seen in the case of untreated cells. As discussed below, no pore formation was observed in the electron microscopy when untransformed cells were incubated with either peptide. Time lapse photographs of cancer cells treated with PNC-27 show that this process is explosive, hence the term “poptosis”. The question arose as to how these PNC-27 and PNC-28 induced such pores.
5. Membrane-Bound HDM-2 Is Expressed Uniquely in Cancer Cells
Western blots of the cell membrane and nuclear fractions of a series of untransformed cells (MCF-10-2A human breast epithelial cells, BMRPA1 rat pancreatic acinar cells, and AG13145 human fibroblast cells) and another series of cancer cells (TUC-3 rat pancreatic acinar cancer cells, MIA-PaCa-2 human pancreatic cancer cells, MCF-7 human breast cancer cells, and A2058 human melanoma cancer cells) showed that all cell lines expressed significant levels of nuclear HDM-2. However, all four cancer cells lines were found to have high levels of expression of HDM-2 in their membrane fractions, while the three untransformed cell lines expressed low or no observable levels of HDM-2 in their membrane fractions [
13]. Similar results have now been confirmed in a variety of different cancers including ovarian cancers (SKOV-3, OVCAR-3, HUVEC negative control) [
14], colon cancers (CTA, CTP, CTR, SW1222, HCT116, CT26, CCD-18Co negative control [
15]), AML (U937, OCI-AML-3, HL60, rat mononuclear cells negative control) [
16], and human-stem-cell-enriched AML cells (normal stem cells negative control) [
17]. Since cancer cells uniquely express HDM-2 in their cell membranes, we investigated whether PNC-27 and -28 interact with membrane-bound HDM-2.
5.1. PNC-27 Colocalizes with HDM-2 in the Cancer Cell Membrane
To determine whether our peptides interacted with cancer-cell-membrane-bound HDM-2, we performed co-localization studies on cancer cells incubated with PNC-27. In these studies, cancer cells were incubated for short periods with PNC-27 and then incubated with two antibody systems: one a green-fluorescent-labeled anti-PNC-27 monoclonal antibody (DO1) and the other a red-fluorescent-labeled polyclonal anti-HDM-2 antibody [
13,
14,
15,
16]. Colocalization was indicated by the presence of merged green and red fluorescence to generate yellow emission.
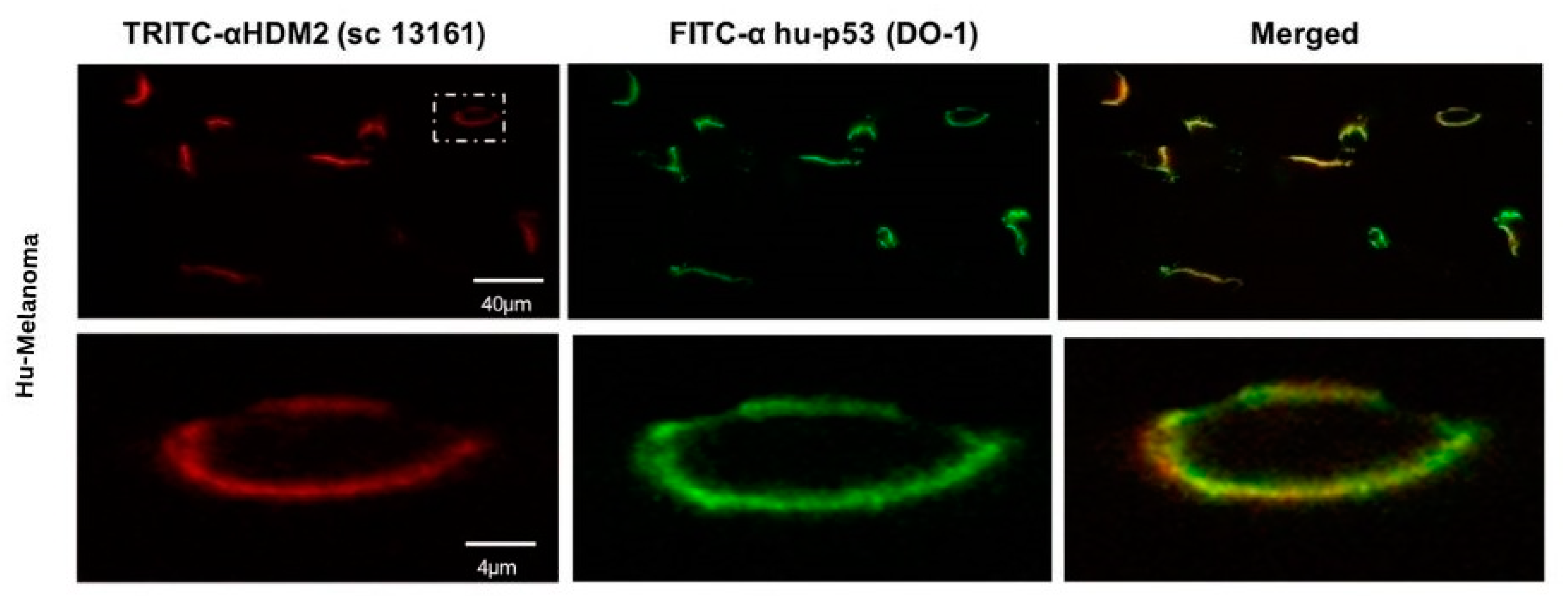
An example is shown in
Figure 4 for A2058 human melanoma cells. In this figure, A2058 human melanoma cells were incubated in the above-described manner with PNC-27 and the two labeled antibody systems. The top panels show low power cells in which PNC-27 labeled green (leftmost panel) and HDM-2 labeled red (middle panel) fluoresced, showing yellow fluorescence as merged images, indicating that both fluorescent probes must be proximate to one another. The two lower sets of panels were high-powered views of two such cells, showing that all the fluorescence is on the cell membrane. The combined green and red fluorescence is seen to give a yellow-colored fluorescence due to the proximity of the probes in the cell membrane to one another, indicating a colocalization of PNC-27 with HDM-2. Identical results have been obtained on numerous other cancer cell lines, including TUC-3, MIA-PaCa-2, MCF-7, SKOV-3, OVCAR-3, HCT-116, CT-26, CTA, CTP, CTR, SW1222, U937, OCI-AML3, and HL60, but not on any normal cells, including MCF-10-2A, BMRPA1, AG13145, HUVEC, CCD-18Co, and rat mononuclear cells [
13,
14,
15,
16]. Importantly, in a major in vivo study of the effect of PNC-27 on human acute myelogenous leukemia cells transplanted into the bone marrows of nude mice, the IC
50 of PNC-27 in killing these tumors in vivo correlated linearly with the extent of co-localization of PNC-27 with membrane-bound HDM-2 [
17]. In addition, we found that the anti-HDM-2 antibody, but not control antibodies, blocks PNC-27 from inducing cancer cell death in a dose-dependent manner, strongly suggesting that it is the PNC-27-HDM2 membrane complexes that ultimately induce cancer cell death [
22].
5.2. Expression of HDM-2 on the Cancer Cell Membrane Is Necessary for PNC-27-Induced Tumor Cell Necrosis
We transfected untransformed MCF-10-2A human breast epithelial cells that remain viable when treated with PNC-27, using plasmids based on the Origene Precision Shuttle Destination Vector, expressing different forms of HDM-2, including full-length HDM-2, full-length HDM-2 with a nuclear localization peptide sequence on its carboxyl terminal end that targets proteins to the cell membrane [
13], and del1-109HDM-2 (missing the p53/PNC-27 binding domain) attached to the nuclear localization peptide. The plasmid used was designed by Origene, called the Precision Shuttle Destination Vector. Western blots of the membrane fractions of the two cell lines expressing the HDM-2 proteins with the membrane localization peptide showed high levels of expression in the membrane fraction. In contrast, Western blots of the membrane fractions of cells transfected with an empty vector or transfected with full-length HDM-2 without the membrane localization peptide revealed the absence of HDM-2. PNC-27 was found to colocalize with the membrane-expressed HDM-2 with the membrane localization peptide on cells that had been transfected, with the plasmid expressing this form of HDM-2 but not in cells transfected with an empty vector or with full-length HDM-2, with no membrane localization peptide [
13].
Each of these transfected cell lines was incubated with PNC-27 to determine if they were killed by this peptide. Only the cells expressing full-length HDM-2 with the membrane localization peptide were killed by PNC-27. These results suggest that the interaction between PNC-27/28 with the p53 binding site (residues 1–109) of membrane-expressed HDM-2 results in tumor cell necrosis. Since cancer cells treated with either peptide have been found to have transmembrane pores (
Figure 3), we surmised that the PNC-27/28-membrane-expressed HDM-2 induced pore formation. Since the isolated p53 12–26 peptide has no effect on cancer cells [
13], the presence of the leader sequence evidently is vital to the process of tumor cell necrosis. We therefore sought to determine how the leader sequence would affect the interaction between PNC-27 and residues 1–109 of the HDM-2-binding site and whether complexes of PNC-27 and HDM-2 are involved in the structure of the EM-observed transmembrane pores.
5.3. Putative Structure for the PNC-27-1-109 HDM-2 Complex
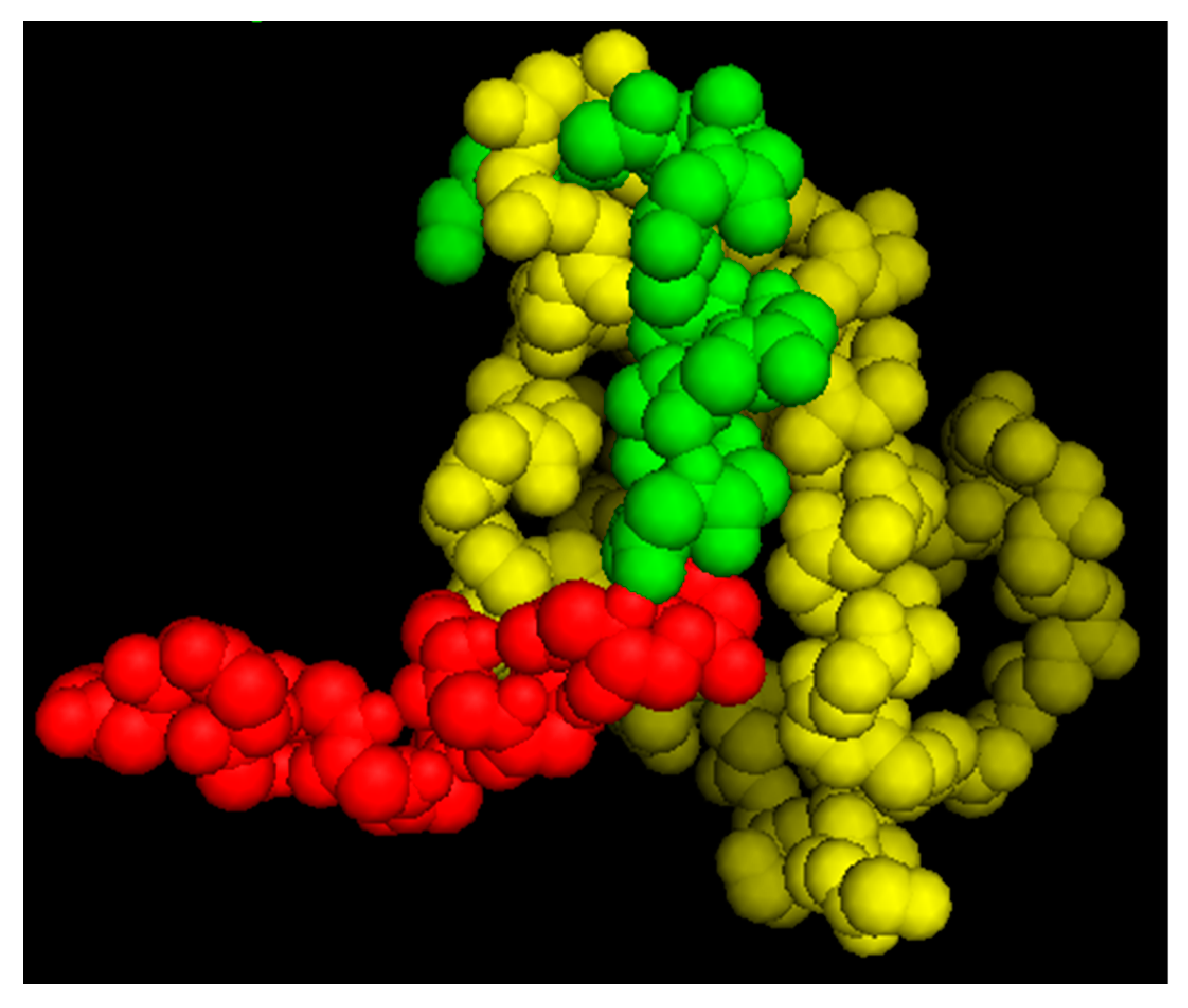
As noted above, we determined the solution structure of PNC-27 by 2D-NMR [
13] and found that p53 residues 17–26 were superimposable [
13] on the X-ray crystal structure of the 15–29 p53 peptide bound to HDM-2 residues 1–109 [
12]. Using this superimposed structure of PNC-27 in the HDM-2 binding site as a starting structure for systematic energy minimizations, we obtained a lowest energy structure, as shown in
Figure 5. The structure of residues 15–26 remained superimposable on the corresponding residues of the 15–29 peptide in the X-ray structure. In addition, there were critical binding residues in PNC-27 and the 15–29 peptide, including Leu 22, Trp 23, and Leu 26 that form part of the hydrophobic face of the peptide and interact closely with HDM2 residues Ile 99, Leu 54, Ile 61, and Met 62. These critical interactions were preserved in both structures (our structure for PNC-27 and the X-ray structure of the p53 15–29 peptide). In the PNC-27 final energy-minimized structure, the leader sequence projected away from the PNC-27-HDM-2 complex, making few contacts with HDM-2, suggesting that it is free to interact with other molecules in the membrane, possibly other PNC-27-HDM-2 complexes resulting in the transmembrane pores. However, in view of the absence of a structure for the whole HDM-2 protein, the orientation of the leader sequence in the PNC-27-HDM-2 complex remains to be determined.
6. Structure of PNC-27-Induced Transmembrane Pores
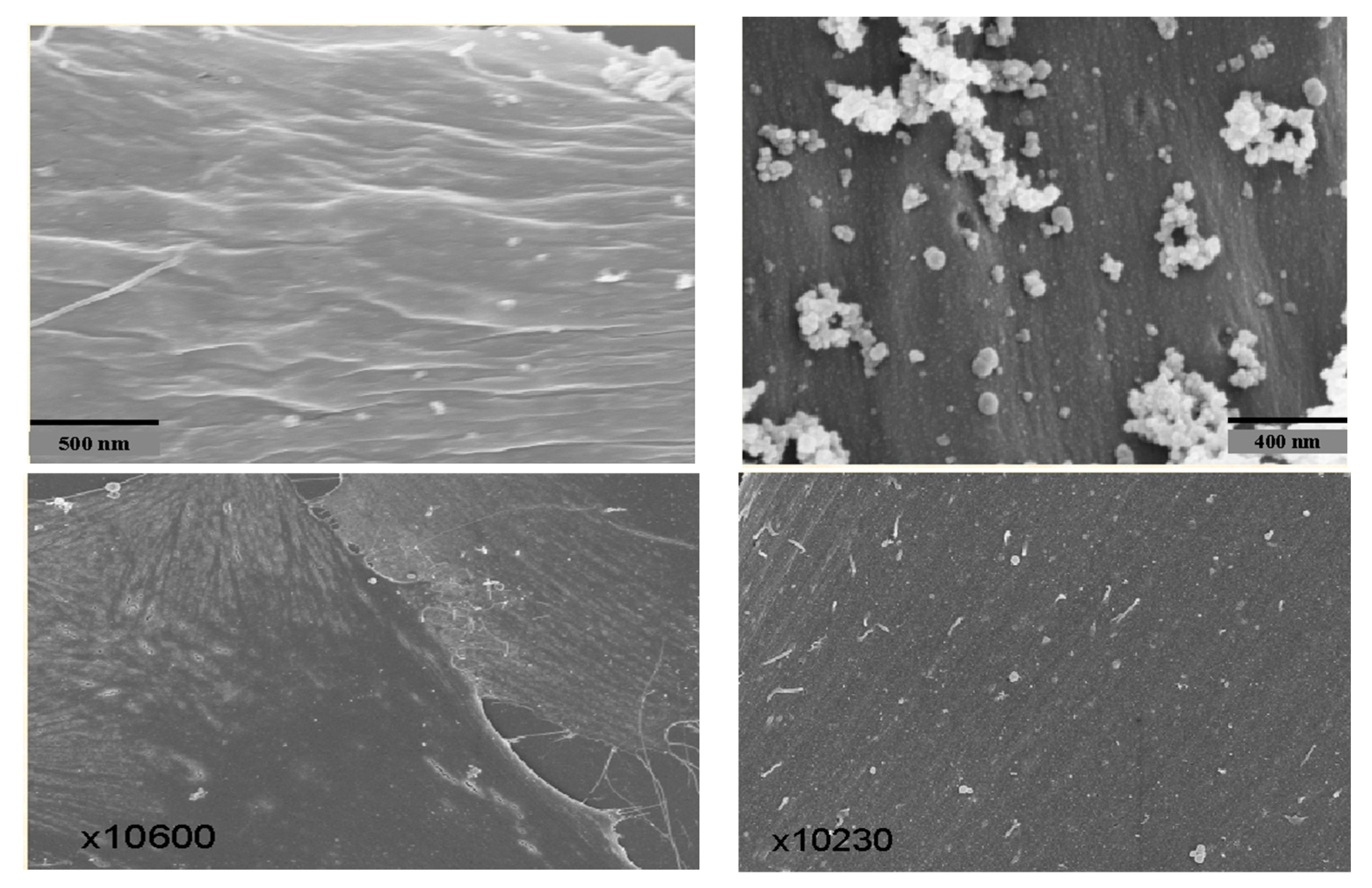
To help determine if the PNC-27-HDM-2 complex is involved in transmembrane pore formation, we performed high-resolution immune scanning electron microscopy (SEM) with high backscatter on cancer cells treated with PNC-27. In these studies, we incubated the cancer cells treated for short periods with PNC-27 and then with gold-labeled secondary antibodies in the anti-PNC-27 and anti-HDM-2 antibody system described for the colocalization experiments. Uniform gold particles of 6µ were used in the anti-PNC-27 antibody system and 15 µ for the anti-HDM-2 antibody system.
Figure 6 (upper left) shows the SEM of the cell membrane of an untreated MIA-PaCa-2 pancreatic cancer cell as a ruffled surface (caused by oncogenic ras-p21). In contrast,
Figure 6 (upper right) shows the membrane surface of a MIA-PaCa-2 cell that was treated with PNC-27 for 15 min. There were multiple sphere-shaped rings around holes representing transmembrane pores.
Figure 6 (lower left) shows the SEM of the membrane of a typical, normal, untreated AG13145 fibroblast, and
Figure 6 (lower right) shows the membrane of the fibroblasts treated with PNC-27 for 15 min. No transmembrane pores were seen. These figures indicate that PNC-27 directly induces transmembrane pores in cancer cells only.
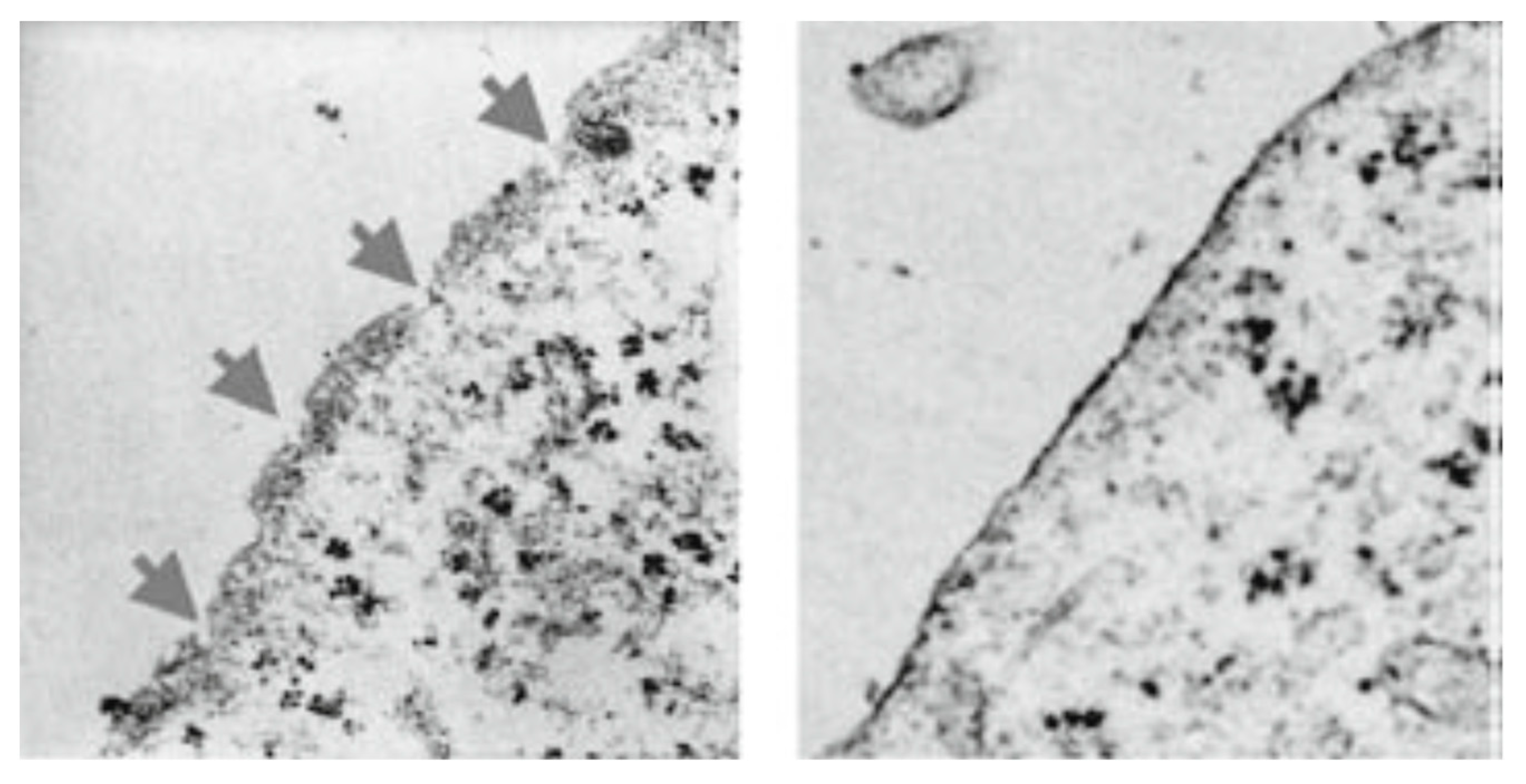
Figure 7 shows the membrane of a MIA-PaCa-2 cell treated with PNC-27 and then incubated with the two gold-labeled antibody systems. As can be seen in this figure, the transmembrane pores were formed from complexes of PNC27, labeled with the yellow arrows pointing to the 6u gold-labeled structures, with HDM-2, labeled with red arrows pointing to the 15 µ gold-labeled structures. Thus, the pores were lined with PNC-27-HDM-2 complexes, a finding that is discussed further below. These complexes appeared to occur as doublets, i.e., two sets of PNC-27–HDM-2 complexes lining a single pore. The average pore size was 37.7 nm. Thus, PNC-27 and -28 complexed with cancer-cell-membrane-bound HDM-2 directly formed transmembrane pores in cancer cells.
PNC-27-induced pore formation shows parallels with other known transmembrane-pore-inducing agents such as streptolysin O, which has been found to induce transmembrane channels by associating with membrane cholesterol, resulting in multimers of streptolysin–cholesterol complexes that line the transmembrane pores [
21,
24]. The formation of these complexes involved two discreet steps: a relatively temperature-independent streptolysin–cholesterol binding event followed by a strongly temperature-dependent multimer-forming process resulting in pore formation [
21,
24].
We found that PNC-27 induces pore formation in two similar steps [
23]. Incubation of PNC-27 with a variety of cancer cells such as MIA-PaCa-2 (human pancreatic cancer) and A2058 (human melanoma) at 17 °C resulted in the absence of LDH release (no cytotoxicity), minimal cytotoxicity at 25 °C but 100 percent LDH release, and cytotoxicity at 37 °C. When these cells were incubated with PNC-27 at 17 °C and were then washed at this temperature, removing unbound PNC-27, and then were incubated at 37 °C, 100 percent cell killing occurred. The dose–response curve for the re-incubated cells was identical to that observed when the cells were incubated at 37 °C only. These findings suggest that binding of PNC-27 to membrane-bound HDM-2 is the temperature-independent step, followed by a strongly temperature-dependent co-migration event in which PNC-27–HDM-2 complexes diffuse in the cell membrane to form the transmembrane pores lined by what appear in
Figure 7 to be dimers of PNC-27–HDM-2 complexes and possibly higher multimeric forms, resulting in poptosis of these cells.
6.1. Dependence of Pore Formation on Membrane-Associated E-Cadherin
In earlier studies [
25], HDM-2 was found in metastatic breast cancer cell lines to occur in the membranes of these cells and interacted with the cell–cell adhering protein E-cadherin in the promotion of contact inhibition, inducing its ubiquitination and proteosomal degradation. In the in vivo studies on the effects of PNC-27 on human AML cells, a further investigation of the possible interactions of PNC-27 with E-cadherin were carried out [
17]. (These studies indicated paradoxically that PNC-27 activated HDM-2-induced ubiquitination of E-cadherin, both in cell-free systems and in AML cells, but not in normal human stem cells.) In addition, human MV4–11 (monomyelocytic) AML cells were transfected with lentivirus-containing E-cadherin silencing (sh) RNA or control shRNA, and they were found to undergo tumor cell necrosis (termed necrobiosis), i.e., rapid release of LDH, pore formation, and consequent cell death, unlike the same cells transfected with a control vector. Interestingly, human AML blast cells treated with inactivating anti-E-cadherin antibody diminished the cytoxicity of PNC-27 to these cells, as did the proteosomal inhibitory drug bortezumib, presumably blocking the degradation of ubiquitinated E-cadherin. These results suggest the possibility that the degradation of membrane-bound E-cadherin is necessary for transmembrane pore formation and that E-cadherin blocks this process, as well as the fact that pore formation is independent of the presence of PNC-27, which acts to enhance E-cadherin degradation. However, these findings may also be explained by a blockade by these agents of PNC-27 from the interaction with its target, HDM-2.
Furthermore, our SEM studies suggest that PNC-27 is required for pore formation. As shown in
Figure 7, PNC-27 forms discrete membrane complexes with HDM-2 lining the pores, clearly implicating PNC-27 directly in pore formation. In addition, if the activation of membrane-bound HDM-2 to E-cadherin degradation is the unique requirement for transmembrane pore formation, one would expect the known HDM-2-binding agents such as the p53 12–26 peptide [
13] and the drug nutlin [
26] to activate this process. As noted above, we found that the p53 12–26 peptide had no effect on cancer cell growth but, as shown in
Figure 2, when expressed via transfected plasmid into cancer cells, interacted with intracellular HDM-2 to induce apoptosis of these cells by preventing it from binding to p53 and targeting it for ubiquitination and proteosomal degradation [
13]. Nutlin, which is an HDM-2-binding drug [
26], likewise induces apoptosis of cancer cells by blocking intracellular HDM-2 from binding to p53. One would expect this agent to interact with membrane-bound HDM-2 and, presumably, would activate degradation of E-cadherin, resulting in pore formation that has not been observed. There is also the known phenomenon of cancer cells [
27], such as lobular breast carcinoma cells, which are membrane E-cadherin-depleted but do not exhibit pore formation and are viable [
28]. Moreover, we found that PNC-27 is cytotoxic to several breast cancer cell lines (MDA-MB-157 and MDA-MB-453) that are known not to express E-cadherin [
13].
6.2. Model for the Transmembrane Pore Formation Induced by PNC-27 and -28
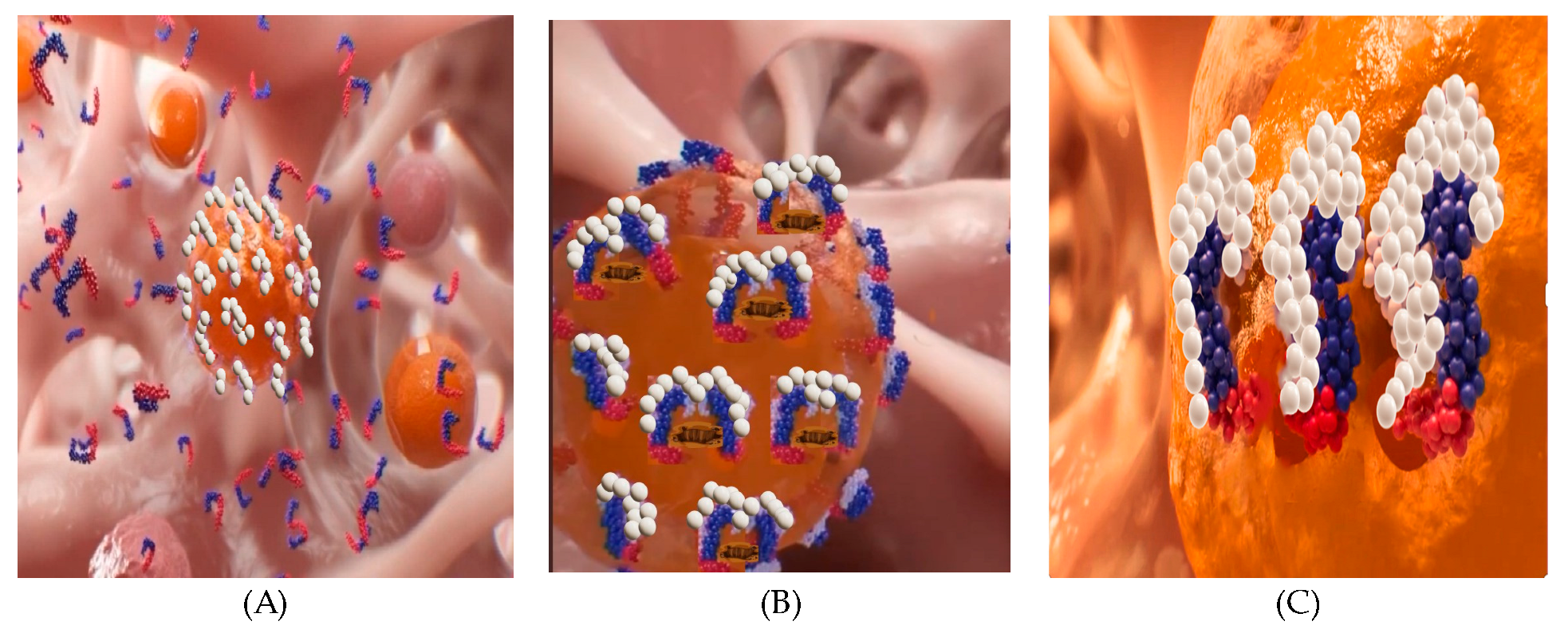
Figure 8 shows a model for the action of PNC-27 (and PNC-28) on cancer cells. In
Figure 8A, PNC-27 shown as a helix (blue p53 residues 12–26)-loop-helix (red leader sequence) is present around the cancer cell membrane, which expresses membrane-bound HDM-2 (white structures). In
Figure 8B, PNC-27 forms dimeric complexes with HDM-2. This results in multiple dimeric complexes present in the membrane, but no pore formation occurs. In
Figure 8C, pairs of dimeric PNC-27-HDM-2 complexes form intramembranously, with each pair lining a transmembrane pore. It is assumed that these dimers line the pores based on the immuno-SEM structure of PNC-27-HDM-2 complexes shown in
Figure 7, although this does not preclude the possibility of higher-order multimer formation.
7. Assessment
PNC-27 and PNC-28 peptides induce cancer cell death by a novel mechanism involving an interaction with membrane-expressed HDM-2 that appears to be unique to cancer cells since a wide variety of normal cells express either no or low levels of this protein in their membranes. Binding of these peptides to HDM-2 results in a two-step process in which the peptides bind to HDM-2 and the complexes that form associate to form transmembrane pores, resulting in an explosive release of intracellular contents (poptosis). Both peptides have been found to eradicate a solid tissue cancer and a number of human acute myelogenous leukemias in vivo, with minimal off-target effects, which is consistent with our finding that these peptides do not affect normal cells in culture.
Our results also call attention to the membranes of cancer cells as possible targets for selective killing. In a separate study [
8] reviewed recently [
3] and referred to in the Introduction section, a specific cell-penetrating peptide called BR2 with the sequence RAGLQFPVGRLLRRLLR selectively enters cancer cells by interacting with gangliosides in the cancer cell membrane. Using BR2 attached to an anti-ras-p21-inactivating antibody to enter colon cancer cells, this construct was found to selectively kill these cells [
8]. Thus, specific membrane components of cancer cell membranes may enable selective cell killing.
Overall, the findings that PNC-27 and PNC-28 are selectively cytotoxic to cancers in vivo with minimal off-target effects due to their binding to HDM-2 expressed uniquely in cancer cell membranes suggest that these agents have strong potential in treating a variety of human cancers.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}