
Astemizole, a Second-Generation Histamine H1-Receptor Antagonist, Did Not Attenuate the Aggregation Process of α-Synuclein In Vitro
 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. In-Situ ThT Aggregation Assay with Kinetic Analysis
2.3. Congo Red Binding Assay and Cell Viablity Study
2.4. Transmission Electron Microscopy (TEM)
2.5. Statistical Analysis
3. Results
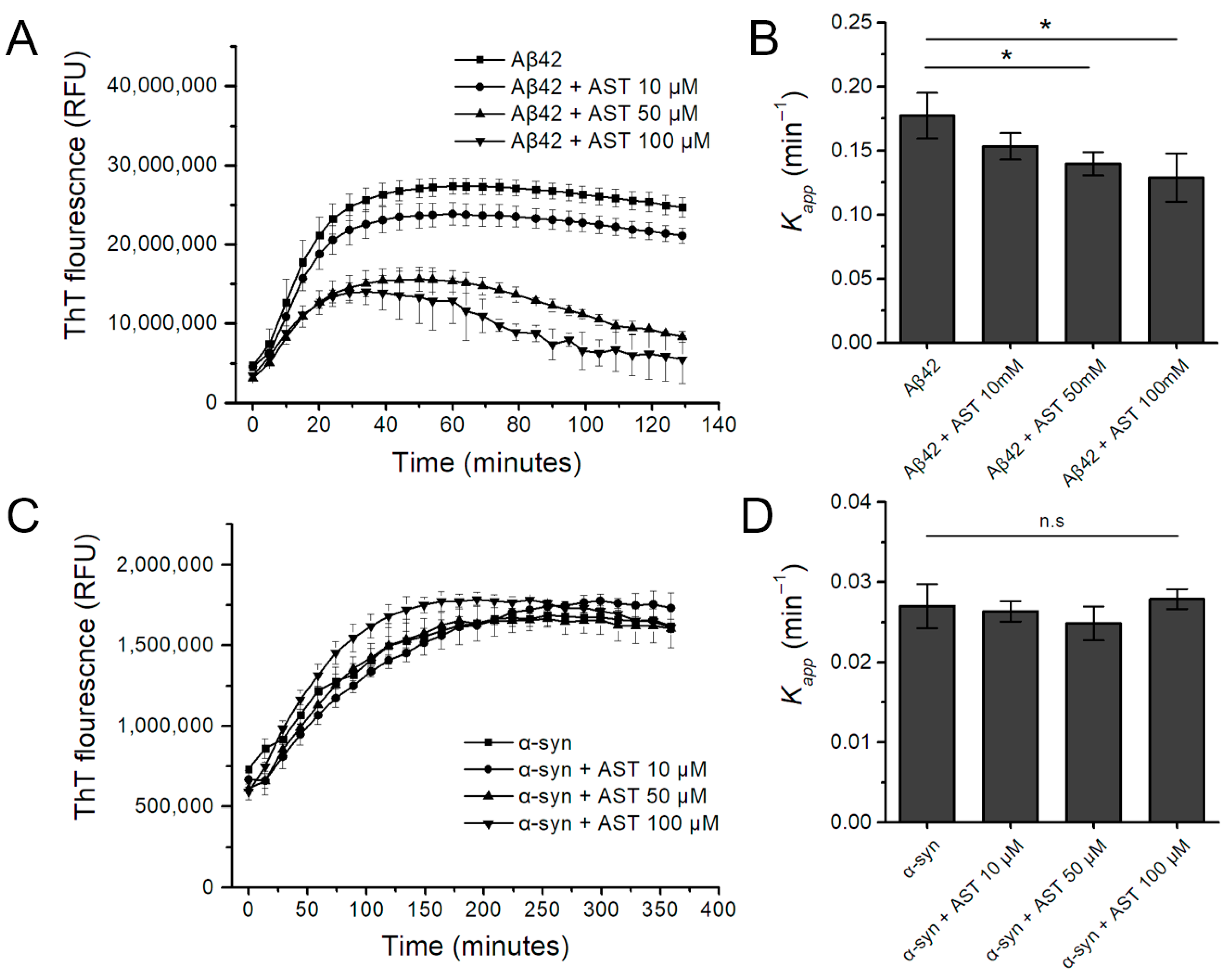
3.1. The Effect of Astemizole on Aβ42 and α-Syn Fibrillation Kinetics in the Real-Time ThT Assay
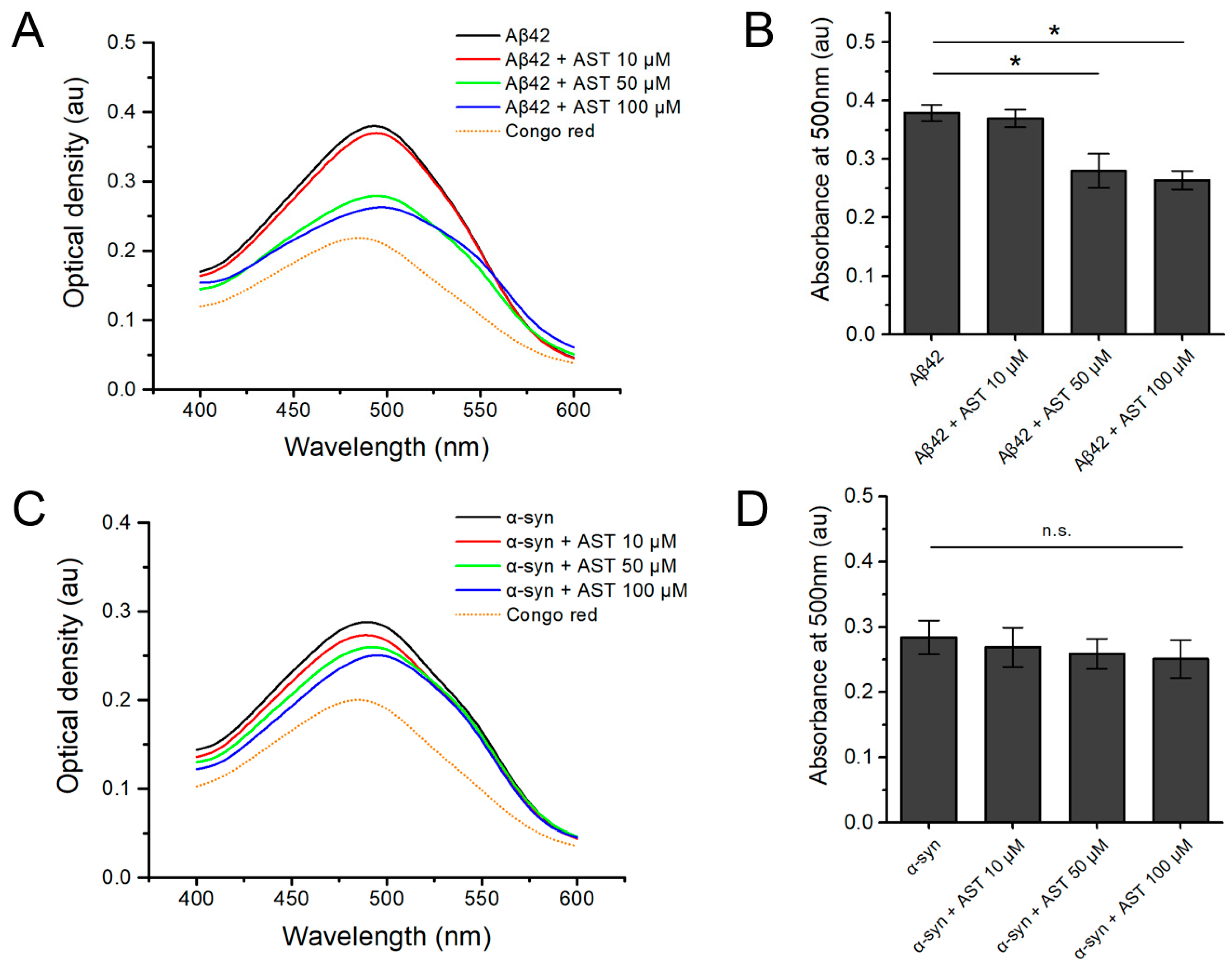
3.2. The Congo Red Binding Assay of Aβ42 and α-Syn Aggregates with or without Astemizole
3.3. The Effect of Astemizole on the Aggregation and Cell Viability Assay
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef]
- Steiner, J.A.; Quansah, E.; Brundin, P. The concept of alpha-synuclein as a prion-like protein: Ten years after. Cell Tissue Res. 2018, 373, 161–173. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat. Rev. Neurosci. 2016, 17, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Baerends, E.; Soud, K.; Folke, J.; Pedersen, A.K.; Henmar, S.; Konrad, L.; Lycas, M.D.; Mori, Y.; Pakkenberg, B.; Woldbye, D.P.D.; et al. Modeling the early stages of Alzheimer’s disease by administering intracerebroventricular injections of human native Aβ oligomers to rats. Acta Neuropathol. Commun. 2022, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Riquelme, A.; Lau, H.H.C.; Stuart, E.; Goczi, A.N.; Wang, Z.; Schmitt-Ulms, G.; Watts, J.C. Prion-like propagation of β-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol. Commun. 2018, 6, 26. [Google Scholar] [CrossRef]
- Olsson, T.T.; Klementieva, O.; Gouras, G.K. Prion-like seeding and nucleation of intracellular amyloid-β. Neurobiol. Dis. 2018, 113, 1–10. [Google Scholar] [CrossRef]
- Henrich, M.T.; Geibl, F.F.; Lakshminarasimhan, H.; Stegmann, A.; Giasson, B.I.; Mao, X.; Dawson, V.L.; Dawson, T.M.; Oertel, W.H.; Surmeier, D.J. Determinants of seeding and spreading of α-synuclein pathology in the brain. Sci. Adv. 2020, 6, eabc2487. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ulusoy, A. Animal models of brain-first and body-first Parkinson’s disease. Neurobiol. Dis. 2022, 163, 105599. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Lakkaraju, A.K.K. Cell Biology of Prions and Prionoids: A Status Report. Trends Cell Biol. 2016, 26, 40–51. [Google Scholar] [CrossRef]
- Chong, C.R.; Chen, X.; Shi, L.; Liu, J.O.; Sullivan, D.J. A clinical drug library screen identifies astemizole as an antimalarial agent. Nat. Chem. Biol. 2006, 2, 415–416. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Karapetyan, Y.E.; Sferrazza, G.F.; Zhou, M.; Ottenberg, G.; Spicer, T.; Chase, P.; Fallahi, M.; Hodder, P.; Weissmann, C.; Lasmézas, C.I. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc. Natl. Acad. Sci. USA 2013, 110, 7044–7049. [Google Scholar] [CrossRef]
- Kocisko, D.A.; Baron, G.S.; Rubenstein, R.; Chen, J.; Kuizon, S.; Caughey, B. New Inhibitors of Scrapie-Associated Prion Protein Formation in a Library of 2,000 Drugs and Natural Products. J. Virol. 2003, 77, 10288–10294. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Lakkaraju, A.K.K.; Frontzek, K. Toward Therapy of Human Prion Diseases. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 331–351. [Google Scholar] [CrossRef] [PubMed]
- Rojo, L.E.; Alzate-Morales, J.; Saavedra, I.N.; Davies, P.; Maccioni, R.B. Selective interaction of lansoprazole and astemizole with tau polymers: Potential new clinical use in diagnosis of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.G.; Lin, A.Q.; Huang, S.Y.; Huo, D.; Cong, C.H. Identification of potential drugs for Parkinson’s disease based on a sub-pathway method. Int. J. Neurosci. 2016, 126, 318–325. [Google Scholar] [CrossRef]
- Styczyńska-Soczka, K.; Zechini, L.; Zografos, L. Validating the Predicted Effect of Astemizole and Ketoconazole Using a Drosophila Model of Parkinson’s Disease. Assay Drug Dev. Technol. 2017, 15, 106–112. [Google Scholar] [CrossRef]
- Tahaei Gilan, S.S.; Yahya Rayat, D.; Mustafa, T.A.; Aziz, F.M.; Shahpasand, K.; Akhtari, K.; Salihi, A.; Abou-Zied, O.K.; Falahati, M. α-synuclein interaction with zero-valent iron nanoparticles accelerates structural rearrangement into amyloid-susceptible structure with increased cytotoxic tendency. Int. J. Nanomed. 2019, 14, 4637–4648. [Google Scholar] [CrossRef]
- Narkiewicz, J.; Giachin, G.; Legname, G. In vitro aggregation assays for the characterization of α-synuclein prion-like properties. Prion 2014, 8, 19–32. [Google Scholar] [CrossRef]
- Gadhave, K.; Bhardwaj, T.; Uversky, V.N.; Vendruscolo, M.; Giri, R. The signal peptide of the amyloid precursor protein forms amyloid-like aggregates and enhances Aβ42 aggregation. Cell Rep. Phys. Sci. 2021, 2, 100599. [Google Scholar] [CrossRef]
- Sanz, F.J.; Solana-Manrique, C.; Muñoz-Soriano, V.; Calap-Quintana, P.; Moltó, M.D.; Paricio, N. Identification of potential therapeutic compounds for Parkinson’s disease using Drosophila and human cell models. Free Radic. Biol. Med. 2017, 108, 683–691. [Google Scholar] [CrossRef]
- Hernández-Parra, H.; Cortés, H.; Avalos-Fuentes, J.A.; Del Prado-Audelo, M.; Florán, B.; Leyva-Gómez, G.; Sharifi-Rad, J.; Cho, W.C. Repositioning of drugs for Parkinson’s disease and pharmaceutical nanotechnology tools for their optimization. J. Nanobiotechnology 2022, 20, 413. [Google Scholar] [CrossRef]
- Tian, J.; Vandermosten, L.; Peigneur, S.; Moreels, L.; Rozenski, J.; Tytgat, J.; Herdewijn, P.; Van den Steen, P.E.; De Jonghe, S. Astemizole analogues with reduced hERG inhibition as potent antimalarial compounds. Bioorg. Med. Chem. 2017, 25, 6332–6344. [Google Scholar] [CrossRef] [PubMed]
- Olasińska-Wiśniewska, A.; Olasiński, J.; Grajek, S. Cardiovascular safety of antihistamines. Postep. Dermatol. Alergol. 2014, 31, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, M.; Edwards, I.R. Risks of non-sedating antihistamines. Lancet 1997, 349, 1322. [Google Scholar] [CrossRef] [PubMed]
- Rajan, R.; Ahmed, S.; Sharma, N.; Kumar, N.; Debas, A.; Matsumura, K. Review of the current state of protein aggregation inhibition from a materials chemistry perspective: Special focus on polymeric materials. Mater. Adv. 2021, 2, 1139–1176. [Google Scholar] [CrossRef]
- Almeida, Z.L.; Brito, R.M.M. Amyloid Disassembly: What Can We Learn from Chaperones? Biomedicines 2022, 10, 3276. [Google Scholar] [CrossRef]
- Wentink, A.; Nussbaum-Krammer, C.; Bukau, B. Modulation of Amyloid States by Molecular Chaperones. Cold Spring Harb. Perspect. Biol. 2019, 11, a033969. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.M.; Darling, A.L.; Uversky, V.N.; Blair, L.J. Small Heat Shock Proteins, Big Impact on Protein Aggregation in Neurodegenerative Disease. Front. Pharmacol. 2019, 10, 1047. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, M.; Anwar, F.; Saleem, U.; Shahzadi, I.; Ahmad, B.; Mir, A.; Ismail, T. Parkinsonism Attenuation by Antihistamines via Downregulating the Oxidative Stress, Histamine, and Inflammation. ACS Omega 2022, 7, 14772–14783. [Google Scholar] [CrossRef]
- Nuutinen, S.; Panula, P. Histamine in neurotransmission and brain diseases. Adv. Exp. Med. Biol. 2010, 709, 95–107. [Google Scholar]
- Nelson, M.P.; Shacka, J.J. Autophagy Modulation in Disease Therapy: Where Do We Stand? Curr. Pathobiol. Rep. 2013, 1, 239–245. [Google Scholar] [CrossRef] [PubMed]
- López-Pérez, Ó.; Badiola, J.J.; Bolea, R.; Ferrer, I.; Llorens, F.; Martín-Burriel, I. An Update on Autophagy in Prion Diseases. Front. Bioeng. Biotechnol. 2020, 8, 975. [Google Scholar] [CrossRef] [PubMed]
- Bamia, A.; Sinane, M.; Naït-Saïdi, R.; Dhiab, J.; Keruzoré, M.; Nguyen, P.H.; Bertho, A.; Soubigou, F.; Halliez, S.; Blondel, M.; et al. Anti-prion Drugs Targeting the Protein Folding Activity of the Ribosome Reduce PABPN1 Aggregation. Neurotherapeutics 2021, 18, 1137–1150. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.I.; Lee, H.; Kim, D.J.; Park, E.S.; Lee, K.Y.; Yang, H.-J. Astemizole, a Second-Generation Histamine H1-Receptor Antagonist, Did Not Attenuate the Aggregation Process of α-Synuclein In Vitro. Biomedicines 2024, 12, 611. https://doi.org/10.3390/biomedicines12030611
Choi JI, Lee H, Kim DJ, Park ES, Lee KY, Yang H-J. Astemizole, a Second-Generation Histamine H1-Receptor Antagonist, Did Not Attenuate the Aggregation Process of α-Synuclein In Vitro. Biomedicines. 2024; 12(3):611. https://doi.org/10.3390/biomedicines12030611
Chicago/Turabian StyleChoi, Jung Il, Hyunjo Lee, Dong Jun Kim, Eun Suk Park, Kyung Yeon Lee, and Hui-Jun Yang. 2024. "Astemizole, a Second-Generation Histamine H1-Receptor Antagonist, Did Not Attenuate the Aggregation Process of α-Synuclein In Vitro" Biomedicines 12, no. 3: 611. https://doi.org/10.3390/biomedicines12030611
APA StyleChoi, J. I., Lee, H., Kim, D. J., Park, E. S., Lee, K. Y., & Yang, H.-J. (2024). Astemizole, a Second-Generation Histamine H1-Receptor Antagonist, Did Not Attenuate the Aggregation Process of α-Synuclein In Vitro. Biomedicines, 12(3), 611. https://doi.org/10.3390/biomedicines12030611