
Novel Multi-Antioxidant Approach for Ischemic Stroke Therapy Targeting the Role of Oxidative Stress
 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Ischemic Stroke
3. Oxidative Stress
- Mitochondrial ROS generation;
- Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme;
- Xanthine oxidase (XO) enzyme.
3.1. Mitochondrial ROS Generation
3.2. NADPH Oxidase
3.3. Xanthine Oxidase
3.4. Reactive Nitrogen Species
3.5. Excitotoxicity
3.6. Lipid Peroxidation
- Initiation: A free radical, often a hydroxyl radical, subtracts a hydrogen atom from a PUFA, creating a PUFA radical, which can occur through enzymatic and non-enzymatic reactions. In non-enzymatic reactions, Fe is a critical component that triggers the Fenton and Haber–Weiss reactions to generate hydroxyl radicals, anion superoxide, and hydrogen peroxide. In enzymatic reactions, ROS generation is mediated by several enzymes, such as lipoxygenases (LOXs), cyclooxygenases (COXs), and NOX, among others.
- Propagation: PUFA radical reacts with molecular oxygen to form a lipid peroxyl radical. This peroxyl radical can, in turn, react with another PUFA, propagating the chain reaction.
- Termination: The chain reaction is terminated when two radicals react with each other, often forming non-reactive products.
3.7. Hypoxia-Inducible Factor 1 (HIF-1)
3.8. NF-κB
- Canonical pathway (classical): Increased ROS production and pro-inflammatory cytokines stimulate Toll-like receptors (TLRs), tumour necrosis factor (TNF) receptors, and interleukin (IL)-1 receptor, among others. This stimulation triggers the activation of the IκB kinase β (IKKβ) complex, which phosphorylates IκBs, marking them for degradation by proteasomes. Subsequently, NF-κB is released, translocating into the nucleus and initiating gene transcription.
- Non-canonical pathway: This pathway involves a different member of the NF-κB family, p100, which produces the active subunit p52. Specific cytokine family members activate this pathway through the IκB kinase α (IKKα) complex.
3.9. Nrf2
3.10. Caspases
4. Inflammation
4.1. Janus Kinase 2/Signal Transducer and Activator of Transcription 3
4.2. Microglia Activation
5. Blood–Brain Barrier Disruption
- ECs feature specialised transport systems for selective transcytosis, and tight junctions limit paracellular transport [64].
- Pericytes contribute to BBB maturation and stabilisation, possessing contractile properties that influence blood flow [64].
- Astrocytes, enveloping over 99% of the BBB, provide structural support, regulate blood flow and electrolyte homeostasis, and influence tight junction expression and function. They release factors such as NO and VEGF, impacting vasodilation and oedema. Astrocyte–endothelial cell interactions induce specific phenotypes crucial for maintaining BBB homeostasis, especially during neuroinflammation following IS [37,65].
6. Antioxidant Bioactive Molecules against Ischemic Stroke
6.1. Polyphenols
6.2. Carotenoids
6.3. Vitamins
6.4. Hormones
6.5. Others
- Salvianolic acid B (Sal B), a hydrophilic caffeic acid derived from Salvia miltiorrhiza, has been widely studied due to its antioxidative, anti-inflammatory, and neuroprotective properties, probably mediated by blocking the TLR4, p-p38 MAPK, p-JNK, IL-1β, and NF-κB pathways [83].
- Rhein, an anthraquinone, exerts neuroprotective effects by regulating the NRF2/SLC7A11/GPX4 pathway, inhibiting ferroptosis during IRI following a stroke in murine models [84].
- Crebabine, an alkaloid with neuroprotective effects, was shown to be effective in a murine model of stroke, reducing cerebral damage by suppressing NADPH and NOX2 activity and through the inhibition of the NF-κB and MAPK pathways [87].
- Glycosides, derived from the Buyang Huanwu Decoction, exert a neuroprotective effect in murine stroke models by reducing pyroptosis by regulating the Nrf2 pathway [88].
- The Krüppel-like factor 4 (KLF4) is a transcription factor related to several cell processes, such as cell proliferation and apoptosis. In murine models, its administration as a recombinant human KLF4 protein has been shown to effectively reduce cerebral IRI’s brain damage by inhibiting cellular oxidative stress through the Nrf2/Trx1 pathway [89].
- Cerebrolysin is a mixture of neuropeptides that, through the inhibition of the TLRs/NF-kB/cytokines pathways and the activation of the Keap1/Nrf2 pathway, has shown to be neuroprotective in murine models of cerebral IRI [90].

{kind=link}
{kind=link}
| Family | Drug | Results | Type of Model | Ref. |
|---|---|---|---|---|
| Polyphenols | Resveratrol | Nrf2: Upregulate HO-1 and SOD [91] NF-κB: Downregulate TLR4 [92] Sirt1: Downregulate caspase-3 activity [93] Upregulate JAK, ERK, and STAT [94] Upregulate ERK and CREB [95] Upregulate of BDNF/TrkB signalling pathway [96] | Rat models of cerebral ischemia/reperfusion injury summarised through a meta-analysis [68] | [68] |
| Curcumin | Downregulate NLRP3 inflammasome | Rat models of cerebral ischemia/reperfusion injury | [69] | |
| Quercetin | Nrf2: Upregulate HO-1 Downregulate autophagy Upregulate PI3K/AKT/mTOR pathway | Rat models of cerebral ischemia/reperfusion injury | [70] | |
| Demethylnobiletin (polymethoxy-flavanone) | Nrf2: Upregulate HO-1 | Rat models of cerebral ischemia/reperfusion injury | [66] | |
| Carotenes | Beta-carotene | NF-κB: Downregulate caspase-3, and Bax Upregulate Bcl-2 expression | Rat models of cerebral ischemia/reperfusion injury | [55] |
| Astaxanthin | Upregulate expression of SOD1 and -2 | Gerbil models of cerebral ischemia/reperfusion injury | [72] | |
| Vitamins | Vitamin D | Nrf2: Upregulate HO-1 Downregulate NLRP3-mediated pyroptosis | Rat models of cerebral ischemia/reperfusion injury | [40] |
| Folic acid | Downregulate neurotoxicity by downregulation of NMDAR expression | Rat models of cerebral ischemia/reperfusion injury | [73] | |
| ATRA | Downregulate the JNK/P38 MAPK pathway | Rat models of cerebral ischemia/reperfusion injury | [67] | |
| Hormones | Melatonin | Downregulate the HMGB1: modulates pyroptosis and necrosis Modulation of the TLR4/NF-κB signalling pathway: Upregulate anti-inflammatory mediators MAPK regulation: Downregulate apoptosis | Obese rat models of cerebral ischemia/reperfusion injury | [74] |
| Oestrogen Progesterone | Decrease neurotoxicity by modulating glutamate transporter expression and inducing glutamate re-uptake | Rat models of cerebral ischemia/reperfusion injury | [76] | |
| Erythropoietin | Upregulate STAT | Rat models of cerebral ischemia/reperfusion injury | [77] | |
| Other | Coenzyme Q10 | NF-κB: Downregulate p65, TNF-α, and IL-6 Downregulate caspase-3 apoptosis | Rat models of cerebral ischemia/reperfusion injury | [97] |
| Acetyl-L-carnitine | Suppress excitotoxicity NF-κB: Downregulate p65, TNF-α, and IL-6 Downregulate caspase-3 apoptosis | Rat models of cerebral ischemia/reperfusion injury | [97] | |
| Salvianolic acid B (Sal B) | Downregulate TLR4, NF-κB, and IL-1β | Mice models of cerebral ischemia/reperfusion injury | [83] | |
| Rhein | Nrf2/SLC7A11/GPX4 axis: Inhibit ferroptosis | Rat models of cerebral ischemia/reperfusion injury | [84] | |
| Osmundacetone | Nrf2: Upregulate HO-1 and NQO1 Downregulate caspase-3 pathway | Rat models of cerebral ischemia/reperfusion injury | [85] | |
| Ruscogenin | Nrf2 pathway | Mice models of cerebral ischemia/reperfusion injury | [86] | |
| Crebanine | Downregulate oxidative stress and neuroinflammation mediated by NOX2 in microglia | Rat models of cerebral ischemia/reperfusion injury | [87] | |
| Glycosides | Nrf2: Downregulate pyroptosis | Rat models of cerebral ischemia/reperfusion injury | [88] | |
| KLF4 | Nrf2: Trx1 pathway | Rat models of cerebral ischemia/reperfusion injury | [89] | |
| Cerebrolysin | Downregulate TLR/NF-κB/cytokines Upregulate the Keap1/Nrf2/antioxidant signalling pathway | Mice models of cerebral ischemia/reperfusion injury | [90] |
7. Multi-Antioxidant Therapy for the Improvement of Clinical Outcomes
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACSL4 | Acyl-CoA synthetase long-chain family member 4 |
| Akt | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| ARE | Antioxidant response elements |
| Arg1 | Arginase 1 |
| ATP | Adenosine triphosphate |
| ATRA | All-trans retinoic acid |
| ATX | Astaxanthin |
| BBB | Blood–brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| BNIP3 | BCL2 interacting protein 3 |
| CoQ10 | Coenzyme Q10 |
| COX | Cyclooxygenases |
| CREB | cAMP response element-binding protein |
| DAMPs | Damage-associated molecular patterns |
| E6AP | E6-associated protein |
| ECs | Endothelial cells |
| EDV | Edaravone |
| eNOS | Endothelial NOS |
| ERK | Extracellular signal-regulated kinase |
| ETC | Electron transport chain |
| FHC1 | Ferritin heavy chain 1 |
| GPX | Glutathione peroxidase |
| HIF | Hypoxia-inducible factor |
| HMGB1 | High-mobility group box protein 1 |
| HNE | 4-Hydroxy-2-nonenal |
| HO-1 | Heme oxygenase 1 |
| IFN | Interferon |
| IKKα | IκB kinase α |
| IKKβ | IκB kinase β |
| IL | Interleukin |
| iNOS | Induced NOS |
| IRF | Interferon regulatory factors |
| IRI | Ischemia-reperfusion injury |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LDL | Low-density lipoprotein |
| LOX | Lipoxygenases |
| Maf | Musculoaponeurotic fibrosarcoma |
| MAPK | Mitogen-activated protein kinase |
| MDA | Malondialdehyde |
| MMP | Matrix metalloproteinase |
| MMSE | Mini mental state examination |
| mRS | Modified Rankin scale |
| mTOR | Mammalian target of rapamycin |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NCOA4 | Nuclear receptor coactivator 4 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NIHSS | National Institutes of Health Stroke Scale |
| NLRP3 | NLR family pyrin domain containing 3 |
| NMDAR | N-methyl-D-aspartate receptor |
| nNOS | Nitric oxide synthase |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OEA | Oleoylethanolamide |
| OS | Oxidative stress |
| PI3K | Phosphatidylinositol-3-kinase |
| PRRs | Pattern recognition receptors |
| Prx | Peroxiredoxin |
| PUFA | Polyunsaturated fatty acids |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| Sirt1 | Sirtuin 1 |
| SLC7A11 | Solute carrier family 7 member 11 |
| SOD | Superoxide dismutase |
| STAT | Signal transducer and activator of transcription |
| TAC | Total antioxidant capacity |
| TIGAR | TP53-induced glycolysis and apoptosis regulator |
| TLR | Toll-like receptor |
| TNF | Tumour necrosis factor |
| TP53 | Tumour protein p53 |
| TrkB | Tyrosine receptor kinase B |
| VEGF | Vascular endothelial growth factor |
| XO | Xanthine oxidase |
| βCAR | β-carotene |
References
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abedi, V.; et al. Global, Regional, and National Burden of Stroke and Its Risk Factors, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic Stroke. Nat. Rev. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Urzúa, S.; Briones-Valdivieso, C.; Chichiarelli, S.; Saso, L.; Rodrigo, R. Potential Role of Natural Antioxidants in Countering Reperfusion Injury in Acute Myocardial Infarction and Ischemic Stroke. Antioxidants 2023, 12, 1760. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.; Menon, B.K.; van Zwam, W.H.; Dippel, D.W.J.; Mitchell, P.J.; Demchuk, A.M.; Dávalos, A.; Majoie, C.B.L.M.; van der Lugt, A.; de Miquel, M.A.; et al. Endovascular Thrombectomy after Large-Vessel Ischaemic Stroke: A Meta-Analysis of Individual Patient Data from Five Randomised Trials. Lancet 2016, 387, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Briyal, S.; Ranjan, A.K.; Gulati, A. Oxidative Stress: A Target to Treat Alzheimer’s Disease and Stroke. Neurochem. Int. 2023, 165, 105509. [Google Scholar] [CrossRef] [PubMed]
- Mathias, K.; Machado, R.S.; Stork, S.; dos Santos, D.; Joaquim, L.; Generoso, J.; Danielski, L.G.; Barichello, T.; Prophiro, J.S.; Petronilho, F. Blood-Brain Barrier Permeability in the Ischemic Stroke: An Update. Microvasc. Res. 2024, 151, 104621. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Wen, Y.; Yang, S.; Duan, Y.; Liu, Z. Research Progress and Perspectives of N-Methyl-D-Aspartate Receptor in Myocardial and Cerebral Ischemia-Reperfusion Injury: A Review. Medicine 2023, 102, e35490. [Google Scholar] [CrossRef]
- Salatin, S.; Farhoudi, M.; Farjami, A.; Maleki Dizaj, S.; Sharifi, S.; Shahi, S. Nanoparticle Formulations of Antioxidants for the Management of Oxidative Stress in Stroke: A Review. Biomedicines 2023, 11, 3010. [Google Scholar] [CrossRef]
- Li, Y.; Schappell, L.E.; Polizu, C.; DiPersio, J.; Tsirka, S.E.; Halterman, M.W.; Nadkarni, N.A. Evolving Clinical–Translational Investigations of Cerebroprotection in Ischemic Stroke. J. Clin. Med. 2023, 12, 6715. [Google Scholar] [CrossRef]
- Islam, F.; Roy, S.; Zehravi, M.; Paul, S.; Sutradhar, H.; Yaidikar, L.; Kumar, B.R.; Dogiparthi, L.K.; Prema, S.; Nainu, F.; et al. Polyphenols Targeting MAP Kinase Signaling Pathway in Neurological Diseases: Understanding Molecular Mechanisms and Therapeutic Targets. Mol. Neurobiol. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Lu, W.; Wen, J. H2S-RhoA/ROCK Pathway and Glial Cells in Axonal Remyelination after Ischemic Stroke. Mol. Neurobiol. 2023, 60, 5493–5504. [Google Scholar] [CrossRef]
- Lin, W.; Zhao, X.-Y.; Cheng, J.-W.; Li, L.-T.; Jiang, Q.; Zhang, Y.-X.; Han, F. Signaling Pathways in Brain Ischemia: Mechanisms and Therapeutic Implications. Pharmacol. Ther. 2023, 251, 108541. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-S.; Chen, X.; Li, W.-T.; Shen, J.-G. Targeting RNS/Caveolin-1/MMP Signaling Cascades to Protect against Cerebral Ischemia-Reperfusion Injuries: Potential Application for Drug Discovery. Acta Pharmacol. Sin. 2018, 39, 669–682. [Google Scholar] [CrossRef]
- Muralikrishna Adibhatla, R.; Hatcher, J.F. Phospholipase A2, Reactive Oxygen Species, and Lipid Peroxidation in Cerebral Ischemia. Free Radic. Biol. Med. 2006, 40, 376–387. [Google Scholar] [CrossRef]
- Kahles, T.; Luedike, P.; Endres, M.; Galla, H.-J.; Steinmetz, H.; Busse, R.; Neumann-Haefelin, T.; Brandes, R.P. NADPH Oxidase Plays a Central Role in Blood-Brain Barrier Damage in Experimental Stroke. Stroke 2007, 38, 3000–3006. [Google Scholar] [CrossRef]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2017, 10, 301. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.E.; Hare, J.M. Xanthine Oxidoreductase and Cardiovascular Disease: Molecular Mechanisms and Pathophysiological Implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef]
- Yu, H.; Chen, X.; Guo, X.; Chen, D.; Jiang, L.; Qi, Y.; Shao, J.; Tao, L.; Hang, J.; Lu, G.; et al. The Clinical Value of Serum Xanthine Oxidase Levels in Patients with Acute Ischemic Stroke. Redox Biol. 2023, 60, 102623. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.; Lu, B. Reverse Electron Transport at Mitochondrial Complex I in Ischemic Stroke, Aging, and Age-Related Diseases. Antioxidants 2023, 12, 895. [Google Scholar] [CrossRef]
- Tsao, C.-C.; Baumann, J.; Huang, S.-F.; Kindler, D.; Schroeter, A.; Kachappilly, N.; Gassmann, M.; Rudin, M.; Ogunshola, O.O. Pericyte Hypoxia-Inducible Factor-1 (HIF-1) Drives Blood-Brain Barrier Disruption and Impacts Acute Ischemic Stroke Outcome. Angiogenesis 2021, 24, 823–842. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Liu, C.; Wang, G.; Han, W.; Tian, Q.; Li, M. Ferroptosis: A Potential Therapeutic Target for Stroke. Neural Regen. Res. 2024, 19, 988–997. [Google Scholar] [CrossRef]
- Kamal, F.Z.; Lefter, R.; Jaber, H.; Balmus, I.-M.; Ciobica, A.; Iordache, A.-C. The Role of Potential Oxidative Biomarkers in the Prognosis of Acute Ischemic Stroke and the Exploration of Antioxidants as Possible Preventive and Treatment Options. Int. J. Mol. Sci. 2023, 24, 6389. [Google Scholar] [CrossRef]
- Montuschi, P.; Barnes, P.J.; Roberts, L.J. Isoprostanes: Markers and Mediators of Oxidative Stress. FASEB J. 2004, 18, 1791–1800. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 Exacerbates Ischemic Stroke by Promoting Ferroptosis-Induced Brain Injury and Neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef]
- Fang, Y.; Chen, X.; Tan, Q.; Zhou, H.; Xu, J.; Gu, Q. Inhibiting Ferroptosis through Disrupting the NCOA4–FTH1 Interaction: A New Mechanism of Action. ACS Cent. Sci. 2021, 7, 980–989. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Koyama, R.; Shichita, T. Glial Roles in Sterile Inflammation after Ischemic Stroke. Neurosci. Res. 2023, 187, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, W.; Geng, P.; Du, W.; Guo, C.; Wang, Q.; Zheng, G.-Q.; Jin, X. Role of Crosstalk between Glial Cells and Immune Cells in Blood-Brain Barrier Damage and Protection after Acute Ischemic Stroke. Aging Dis. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Song, T.; Zhang, Y.; Zhu, L.; Zhang, Y.; Song, J. The Role of JAK/STAT Signaling Pathway in Cerebral Ischemia-Reperfusion Injury and the Therapeutic Effect of Traditional Chinese Medicine: A Narrative Review. Medicine 2023, 102, e35890. [Google Scholar] [CrossRef]
- Du, X.; Amin, N.; Xu, L.; Botchway, B.O.A.; Zhang, B.; Fang, M. Pharmacological Intervention of Curcumin via the NLRP3 Inflammasome in Ischemic Stroke. Front. Pharmacol. 2023, 14, 1249644. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation following Ischemic Stroke. Front. Cell. Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.-D.D.; Hayakawa, K.; Seo, J.H.; Nguyen, M.-N.; Som, A.T.; Lee, B.J.; Guo, S.; Kim, K.-W.; Lo, E.H.; Arai, K. Crosstalk between Oligodendrocytes and Cerebral Endothelium Contributes to Vascular Remodeling after White Matter Injury. Glia 2012, 60, 875–881. [Google Scholar] [CrossRef]
- Wan, Y.; Jin, H.-J.; Zhu, Y.-Y.; Fang, Z.; Mao, L.; He, Q.; Xia, Y.-P.; Li, M.; Li, Y.; Chen, X.; et al. MicroRNA-149-5p Regulates Blood–Brain Barrier Permeability after Transient Middle Cerebral Artery Occlusion in Rats by Targeting S1PR2 of Pericytes. FASEB J. 2018, 32, 3133–3148. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Chang, M.-S.; Koh, S.-H.; Choi, Y.K. Repair Mechanisms of the Neurovascular Unit after Ischemic Stroke with a Focus on VEGF. Int. J. Mol. Sci. 2021, 22, 8543. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Xue, S.; Zhou, X.; Yang, Z.-H.; Si, X.-K.; Sun, X. Stroke-Induced Damage on the Blood–Brain Barrier. Front. Neurol. 2023, 14, 1248970. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Wang, S.; Kroemer, G.; Penninger, J.M.; Uversky, V.N.; Pratico, D.; Henninger, N.; Reiter, R.J.; Bruno, A.; Joshipura, K.; et al. Targeting Autophagy in Ischemic Stroke: From Molecular Mechanisms to Clinical Therapeutics. Pharmacol. Ther. 2021, 225, 107848. [Google Scholar] [CrossRef]
- Mao, R.; Zong, N.; Hu, Y.; Chen, Y.; Xu, Y. Neuronal Death Mechanisms and Therapeutic Strategy in Ischemic Stroke. Neurosci. Bull. 2022, 38, 1229–1247. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Ma, H.; Chen, M.; Bai, J. Vitamin D Alleviates Neuronal Injury in Cerebral Ischemia-Reperfusion via Enhancing the Nrf2/HO-1 Antioxidant Pathway to Counteract NLRP3-Mediated Pyroptosis. J. Neuropathol. Exp. Neurol. 2023, 82, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, F.; Duo, K.; Liu, Y.; Yu, J.; Wu, Q.; Cai, Z. The Role of Necroptosis in Cerebral Ischemic Stroke. Mol. Neurobiol. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of Mitochondrial ROS in the Brain: From Physiology to Neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Yang, H.; Qi, C.; Su, F.; Shan, W.; Guo, A.; Wu, J.; Wang, Y.; You, H.; Wang, Q. Cerebral Ischemia/Reperfusion Injury and Pharmacologic Preconditioning as a Means to Reduce Stroke-Induced Inflammation and Damage. Neurochem. Res. 2022, 47, 3598–3614. [Google Scholar] [CrossRef]
- Wang, C.-K.; Ahmed, M.M.; Jiang, Q.; Lu, N.-N.; Tan, C.; Gao, Y.-P.; Mahmood, Q.; Chen, D.-Y.; Fukunaga, K.; Li, M.; et al. Melatonin Ameliorates Hypoglycemic Stress-Induced Brain Endothelial Tight Junction Injury by Inhibiting Protein Nitration of TP53-Induced Glycolysis and Apoptosis Regulator. J. Pineal Res. 2017, 63, e12440. [Google Scholar] [CrossRef]
- Xu, S.-Y.; Ni, S.-M.; Zeng, C.-L.; Peng, Y.-J. Role of Ferroptosis in Glial Cells after Ischemic Stroke. Front. Biosci. 2023, 28, 208. [Google Scholar] [CrossRef]
- He, J.; Liu, J.; Huang, Y.; Tang, X.; Xiao, H.; Hu, Z. Oxidative Stress, Inflammation, and Autophagy: Potential Targets of Mesenchymal Stem Cells-Based Therapies in Ischemic Stroke. Front. Neurosci. 2021, 15, 641157. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.X.; Gelb, A.W. Free Radicals, Antioxidants, and Neurologic Injury: Possible Relationship to Cerebral Protection by Anesthetics. J. Neurosurg. Anesthesiol. 2002, 14, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tao, L.; Wu, H. Effects of Hypoxia-Inducible Factor 1 (HIF-1) Signaling Pathway on Acute Ischemic Stroke. Comput. Math. Methods Med. 2022, 2022, 1860925. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.K. Cerebral Ischemic Stroke Cellular Fate and Therapeutic Opportunities. Front. Biosci. 2019, 24, 435–450. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-ҚB Interplay in Cerebrovascular and Neurodegenerative Disorders: Molecular Mechanisms and Possible Therapeutic Approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-ΚB-Mediated Neuroinflammation in Parkinson’s Disease and Potential Therapeutic Effect of Polyphenols. Neurotox. Res. 2020, 37, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Chen, Z.; Wen, J. The Role of RhoA/ROCK Pathway in the Ischemic Stroke-Induced Neuroinflammation. Biomed. Pharmacother. 2023, 165, 115141. [Google Scholar] [CrossRef]
- Li, R.; Zhou, Y.; Zhang, S.; Li, J.; Zheng, Y.; Fan, X. The Natural (Poly)Phenols as Modulators of Microglia Polarization via TLR4/NF-ΚB Pathway Exert Anti-Inflammatory Activity in Ischemic Stroke. Eur. J. Pharmacol. 2022, 914, 174660. [Google Scholar] [CrossRef] [PubMed]
- Althurwi, H.N.; Abdel-Rahman, R.F.; Soliman, G.A.; Ogaly, H.A.; Alkholifi, F.K.; Abd-Elsalam, R.M.; Alqasoumi, S.I.; Abdel-Kader, M.S. Protective Effect of Beta-Carotene against Myeloperoxidase-Mediated Oxidative Stress and Inflammation in Rat Ischemic Brain Injury. Antioxidants 2022, 11, 2344. [Google Scholar] [CrossRef] [PubMed]
- Fadoul, G.; Ikonomovic, M.; Zhang, F.; Yang, T. The Cell-Specific Roles of Nrf2 in Acute and Chronic Phases of Ischemic Stroke. CNS Neurosci. Ther. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Pan, Z.; Ma, G.; Kong, L.; Du, G. Hypoxia-Inducible Factor-1: Regulatory Mechanisms and Drug Development in Stroke. Pharmacol. Res. 2021, 170, 105742. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Zhang, Y.; Li, C.; Luo, L.; Hua, Y.; Hu, J.; Bai, Y. Repetitive Transcranial Magnetic Stimulation of the Brain after Ischemic Stroke: Mechanisms from Animal Models. Cell. Mol. Neurobiol. 2023, 43, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Uzdensky, A.B. Apoptosis Regulation in the Penumbra after Ischemic Stroke: Expression of pro- and Antiapoptotic Proteins. Apoptosis 2019, 24, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Bu, F.; Min, J.-W.; Munshi, Y.; Lai, Y.-J.; Qi, L.; Urayama, A.; McCullough, L.D.; Li, J. Activation of Endothelial Ras-Related C3 Botulinum Toxin Substrate 1 (Rac1) Improves Post-Stroke Recovery and Angiogenesis via Activating Pak1 in Mice. Exp. Neurol. 2019, 322, 113059. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Min, X.; Chai, Y.; Zhang, J.; Chen, X. NLRP1 Inflammasomes: A Potential Target for the Treatment of Several Types of Brain Injury. Front. Immunol. 2022, 13, 863774. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Yin, B.; Ye, Y.; Dekhel, O.Y.A.T.; Xiong, X.; Jian, Z.; Gu, L. The Bidirectional Role of the JAK2/STAT3 Signaling Pathway and Related Mechanisms in Cerebral Ischemia-Reperfusion Injury. Exp. Neurol. 2021, 341, 113690. [Google Scholar] [CrossRef]
- Raible, D.J.; Frey, L.C.; Brooks-Kayal, A.R. Effects of JAK2-STAT3 Signaling after Cerebral Insults. JAKSTAT 2014, 3, e29510. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, J.; Li, M.; Cao, H.; Zhu, Q.; Yang, D. PAK1 Contributes to Cerebral Ischemia/Reperfusion Injury by Regulating the Blood-Brain Barrier Integrity. iScience 2023, 26, 107333. [Google Scholar] [CrossRef]
- Zeng, M.; Peng, M.; Liang, J.; Sun, H. The Role of Gut Microbiota in Blood–Brain Barrier Disruption after Stroke. Mol. Neurobiol. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Huang, D.; Awad, A.C.A.; Tang, C.; Chen, Y. Demethylnobiletin Ameliorates Cerebral Ischemia-Reperfusion Injury in Rats through Nrf2/HO-1 Signaling Pathway. Environ. Toxicol. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Li, M.; Tian, X.; An, R.; Yang, M.; Zhang, Q.; Xiang, F.; Liu, H.; Wang, Y.; Xu, L.; Dong, Z. All-Trans Retinoic Acid Ameliorates the Early Experimental Cerebral Ischemia–Reperfusion Injury in Rats by Inhibiting the Loss of the Blood–Brain Barrier via the JNK/P38MAPK Signaling Pathway. Neurochem. Res. 2018, 43, 1283–1296. [Google Scholar] [CrossRef]
- Xue, R.; Gao, S.; Zhang, Y.; Cui, X.; Mo, W.; Xu, J.; Yao, M. A Meta-Analysis of Resveratrol Protects against Cerebral Ischemia/Reperfusion Injury: Evidence from Rats Studies and Insight into Molecular Mechanisms. Front. Pharmacol. 2022, 13, 988836. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.; Su, W.; Gao, F.; Ding, Z.; Yang, S.; Ye, L.; Chen, X.; Tian, G.; Xi, J.; Liu, Z. Curcumin Ameliorates White Matter Injury after Ischemic Stroke by Inhibiting Microglia/Macrophage Pyroptosis through NF-ΚB Suppression and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell. Longev. 2021, 2021, 1552127. [Google Scholar] [CrossRef] [PubMed]
- Alattar, A.; Alshaman, R.; Althobaiti, Y.S.; Soliman, G.M.; Ali, H.S.; Khubrni, W.S.; Koh, P.O.; Rehman, N.U.; Shah, F.A. Quercetin Alleviated Inflammasome-Mediated Pyroptosis and Modulated the MTOR/P70S6/P6/EIF4E/4EBP1 Pathway in Ischemic Stroke. Pharmaceuticals 2023, 16, 1182. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, W.; Yu, B.; Liang, H.; Mao, S.; Hu, X.; Feng, Y.; Xu, J.; Chu, L. Quercetin Improves Cerebral Ischemia/Reperfusion Injury by Promoting Microglia/Macrophages M2 Polarization via Regulating PI3K/Akt/NF-ΚB Signaling Pathway. Biomed. Pharmacother. 2023, 168, 115653. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, T.-K.; Kim, D.W.; Ahn, J.H.; Lee, C.-H.; Kim, J.-D.; Shin, M.C.; Cho, J.H.; Lee, J.-C.; Won, M.-H.; et al. Astaxanthin Confers a Significant Attenuation of Hippocampal Neuronal Loss Induced by Severe Ischemia-Reperfusion Injury in Gerbils by Reducing Oxidative Stress. Mar. Drugs 2022, 20, 267. [Google Scholar] [CrossRef]
- Liang, X.; Shi, L.; Wang, M.; Zhang, L.; Gong, Z.; Luo, S.; Wang, X.; Zhang, Q.; Zhang, X. Folic Acid Ameliorates Synaptic Impairment following Cerebral Ischemia/Reperfusion Injury via Inhibiting Excessive Activation of NMDA Receptors. J. Nutr. Biochem. 2023, 112, 109209. [Google Scholar] [CrossRef]
- Yawoot, N.; Sengking, J.; Govitrapong, P.; Tocharus, C.; Tocharus, J. Melatonin Modulates the Aggravation of Pyroptosis, Necroptosis, and Neuroinflammation following Cerebral Ischemia and Reperfusion Injury in Obese Rats. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166785. [Google Scholar] [CrossRef]
- Mehrpooya, M.; Mazdeh, M.; Rahmani, E.; Khazaie, M.; Ahmadimoghaddam, D. Melatonin Supplementation May Benefit Patients with Acute Ischemic Stroke Not Eligible for Reperfusion Therapies: Results of a Pilot Study. J. Clin. Neurosci. 2022, 106, 66–75. [Google Scholar] [CrossRef]
- Nematipour, S.; Vahidinia, Z.; Nejati, M.; Naderian, H.; Beyer, C.; Azami Tameh, A. Estrogen and Progesterone Attenuate Glutamate Neurotoxicity via Regulation of EAAT3 and GLT-1 in a Rat Model of Ischemic Stroke. Iran. J. Basic Med. Sci. 2020, 23, 1346–1352. [Google Scholar] [CrossRef]
- Ma, J.-Y.; Jiang, C.-J.; Wang, Z.-J.; Zhao, Y.-J.; Zhang, Z.-Y.; Tao, J.-J. Erythropoietin Reduces Apoptosis of Brain Tissue Cells in Rats after Cerebral Ischemia/Reperfusion Injury: A Characteristic Analysis Using Magnetic Resonance Imaging. Neural Regen. Res. 2016, 11, 1450. [Google Scholar] [CrossRef]
- Ramezani, M.; Sahraei, Z.; Simani, L.; Heydari, K.; Shahidi, F. Coenzyme Q10 Supplementation in Acute Ischemic Stroke: Is It Beneficial in Short-Term Administration? Nutr. Neurosci. 2020, 23, 640–645. [Google Scholar] [CrossRef]
- Mazdeh, M.; Abolfathi, P.; Sabetghadam, M.; Mohammadi, Y.; Mehrpooya, M. Clinical Evidence of Acetyl-L-Carnitine Efficacy in the Treatment of Acute Ischemic Stroke: A Pilot Clinical Trial. Oxid. Med. Cell. Longev. 2022, 2022, 2493053. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, J.; Zheng, J.; Zhang, X.; Shao, J.; Zhao, L.; Hao, J. Anti-Inflammatory and Antioxidant Effects of Acetyl-L-Carnitine on Atherosclerotic Rats. Med. Sci. Monit. 2020, 26, e920250-1–e920250-11. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Park, C.G.; Park, M.; Lee, S.H.; Park, H.R.; Lim, J.; Paek, S.H.; Choy, Y.B. Intrastriatal Administration of Coenzyme Q10 Enhances Neuroprotection in a Parkinson’s Disease Rat Model. Sci. Rep. 2020, 10, 9572. [Google Scholar] [CrossRef] [PubMed]
- Simani, L.; Ryan, F.; Hashemifard, S.; Hooshmandi, E.; Madahi, M.; Sahraei, Z.; Rezaei, O.; Heydari, K.; Ramezani, M. Serum Coenzyme Q10 Is Associated with Clinical Neurological Outcomes in Acute Stroke Patients. J. Mol. Neurosci. 2018, 66, 53–58. [Google Scholar] [CrossRef]
- Zheng, X.-F.; Zhang, X.-J.; Dong, L.-P.; Zhao, J.-R.; Zhang, C.; Chen, R. Neuroprotective Mechanism of Salvianolic Acid B against Cerebral Ischemia-Reperfusion Injury in Mice through Downregulation of TLR4, P-p38MAPK, P-JNK, NF-κB, and I-L1β. Immun. Inflamm. Dis. 2023, 11, e1030. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, T.-A.; Zhang, W.-Y.; Huang, S.-R.; Hu, Y.; Sun, J. Rhein Attenuates Cerebral Ischemia-Reperfusion Injury via Inhibition of Ferroptosis through NRF2/SLC7A11/GPX4 Pathway. Exp. Neurol. 2023, 369, 114541. [Google Scholar] [CrossRef]
- Li, B.; Yu, W.; Yang, L. Osmundacetone Alleviates Cerebral Ischemia–Reperfusion Injury in Rats. Biol. Pharm. Bull. 2023, 46, 1527–1534. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, Y.; Sheng, M.; Chen, X.; Wu, Q.; Kou, J.; Chen, G. Ruscogenin Timing Administration Mitigates Cerebral Ischemia-Reperfusion Injury through Regulating Circadian Genes and Activating Nrf2 Pathway. Phytomedicine 2023, 120, 155028. [Google Scholar] [CrossRef]
- Yang, Y.; Hao, T.; Yao, X.; Che, Y.; Liu, Y.; Fang, M.; Wang, Y.; Zhou, D.; Chai, H.; Li, N.; et al. Crebanine Ameliorates Ischemia-Reperfusion Brain Damage by Inhibiting Oxidative Stress and Neuroinflammation Mediated by NADPH Oxidase 2 in Microglia. Phytomedicine 2023, 120, 155044. [Google Scholar] [CrossRef]
- She, Y.; Shao, L.; Jiao, K.; Sun, R.; Lang, T.; Long, H.; Tang, Y.; Zhang, W.; Ding, C.; Deng, C. Glycosides of Buyang Huanwu Decoction Inhibits Pyroptosis Associated with Cerebral Ischemia-Reperfusion through Nrf2-Mediated Antioxidant Signaling Pathway Both in Vivo and in Vitro. Phytomedicine 2023, 120, 155001. [Google Scholar] [CrossRef]
- Huang, T.; Yin, J.; Ren, S.; Zhang, X. Protective Effects of KLF4 on Blood–Brain Barrier and Oxidative Stress after Cerebral Ischemia–Reperfusion in Rats through the Nrf2/Trx1 Pathway. Cytokine 2023, 169, 156288. [Google Scholar] [CrossRef]
- Marghani, B.H.; Rezk, S.; Ateya, A.I.; Alotaibi, B.S.; Othman, B.H.; Sayed, S.M.; Alshehri, M.A.; Shukry, M.; Mansour, M.M. The Effect of Cerebrolysin in an Animal Model of Forebrain Ischemic-Reperfusion Injury: New Insights into the Activation of the Keap1/Nrf2/Antioxidant Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 12080. [Google Scholar] [CrossRef]
- Ren, J.; Fan, C.; Chen, N.; Huang, J.; Yang, Q. Resveratrol Pretreatment Attenuates Cerebral Ischemic Injury by Upregulating Expression of Transcription Factor Nrf2 and HO-1 in Rats. Neurochem. Res. 2011, 36, 2352–2362. [Google Scholar] [CrossRef]
- Lei, J.; Tu, X.; Wang, Y.; Tu, D.; Shi, S. Resveratrol Downregulates the TLR4 Signaling Pathway to Reduce Brain Damage in a Rat Model of Focal Cerebral Ischemia. Exp. Ther. Med. 2019, 17, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- Teertam, S.K.; Jha, S.; Prakash babu, P. Up-Regulation of Sirt1/MiR-149-5p Signaling May Play a Role in Resveratrol Induced Protection against Ischemia via P53 in Rat Brain. J. Clin. Neurosci. 2020, 72, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Zhao, Y.; Song, G.; She, K. Resveratrol Protects Hippocampal Neurons against Cerebral Ischemia-Reperfusion Injury via Modulating JAK/ERK/STAT Signaling Pathway in Rats. J. Neuroimmunol. 2018, 315, 9–14. [Google Scholar] [CrossRef]
- Li, Z.; Fang, F.; Wang, Y.; Wang, L. Resveratrol Protects CA1 Neurons against Focal Cerebral Ischemic Reperfusion-Induced Damage via the ERK-CREB Signaling Pathway in Rats. Pharmacol. Biochem. Behav. 2016, 146–147, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Zhu, C.; Li, L. Rehabilitation Training and Resveratrol Improve the Recovery of Neurological and Motor Function in Rats after Cerebral Ischemic Injury through the Sirt1 Signaling Pathway. Biomed Res. Int. 2016, 2016, 1732163. [Google Scholar] [CrossRef] [PubMed]
- Alhusaini, A.; Sarawi, W.; Mattar, D.; Abo-Hamad, A.; Almogren, R.; Alhumaidan, S.; Alsultan, E.; Alsaif, S.; Hasan, I.; Hassanein, E.; et al. Acetyl-L-Carnitine and/or Liposomal Co-Enzyme Q10 Prevent Propionic Acid-Induced Neurotoxicity by Modulating Oxidative Tissue Injury, Inflammation, and ALDH1A1-RA-RARα Signaling in Rats. Biomed. Pharmacother. 2022, 153, 113360. [Google Scholar] [CrossRef] [PubMed]
- Gudarzi, S.; Jafari, M.; Pirzad Jahromi, G.; Eshrati, R.; Asadollahi, M.; Nikdokht, P. Evaluation of Modulatory Effects of Saffron (Crocus sativus L.) Aqueous Extract on Oxidative Stress in Ischemic Stroke Patients: A Randomized Clinical Trial. Nutr. Neurosci. 2022, 25, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Qian, Z.; Chen, J.; Chen, M.; Zhong, W.; Shen, C.; Hu, Z.; Li, R. Effects of Edaravone Dexborneol on Neurological Function and Serum Inflammatory Factor Levels in Patients with Acute Anterior Circulation Large Vessel Occlusion Stroke. Transl. Neurosci. 2023, 14, 20220312. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, A.; Meng, X.; Yalkun, G.; Xu, A.; Gao, Z.; Chen, H.; Ji, Y.; Xu, J.; Geng, D.; et al. Edaravone Dexborneol versus Edaravone Alone for the Treatment of Acute Ischemic Stroke. Stroke 2021, 52, 772–780. [Google Scholar] [CrossRef]
- Sabahi, M.; Ahmadi, S.A.; Kazemi, A.; Mehrpooya, M.; Khazaei, M.; Ranjbar, A.; Mowla, A. The Effect of Oleoylethanolamide (OEA) Add-on Treatment on Inflammatory, Oxidative Stress, Lipid, and Biochemical Parameters in the Acute Ischemic Stroke Patients: Randomized Double-Blind Placebo-Controlled Study. Oxid. Med. Cell. Longev. 2022, 2022, 5721167. [Google Scholar] [CrossRef]
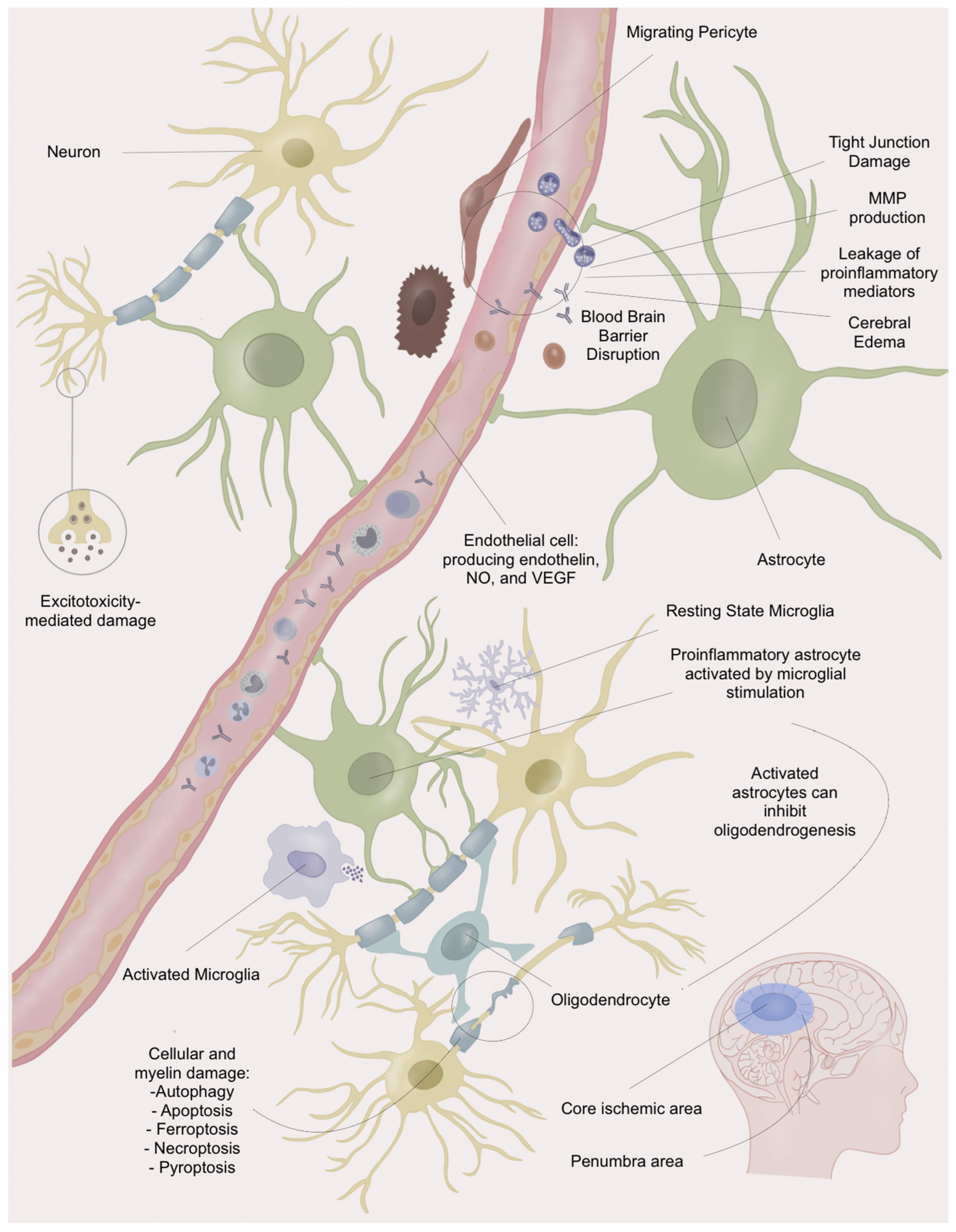

| Pathogenic Processes of Ischemic Stroke | Molecular Mechanisms | Ref. |
|---|---|---|
| Oxidative and Nitrosative Stress | Reactive oxygen species (ROS) and reactive nitrogen species (RNS) induce mitochondrial dysfunction, lipid peroxidation, and disruption of the blood–brain barrier, suppress intrinsic antioxidant effects, and induce DNA damage and cell death | [13,14] |
| Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase catalyses the transfer of electrons from cytosolic NADPH O2 at the extracellular side of the cell membrane culminating with superoxide formation | [15,16] | |
| Xanthine oxidase catalyses the conversion of hypoxanthine and xanthine to uric acid, producing ROS as by-products | [17,18] | |
| Electron leakage occurs at various points along the respiratory chain on the mitochondria, particularly at complex I and complex III, and is responsible for ROS generation | [19] | |
| Differential expression of neuronal nitric oxide synthase (nNOS) induces nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS) with the subsequent nitric oxide production | [12] | |
| Nitric oxide reacts with superoxide to produce peroxynitrite | [5] | |
| Peroxynitrite induces direct nitrosative damage/tyrosine nitration of Keap1 (preventing Nrf2 from being activated with its antioxidant activity) and TP53-induced glycolysis and apoptosis regulator (TIGAR) with a subsequent impaired generation of NADPH | [12] | |
| While hypoxia-inducible factor-1 (HIF-1) activation during ischemia is generally regarded as protective by enhancing VEFG generation and stimulating angiogenesis, its role becomes more nuanced during reperfusion by promoting blood–brain barrier disruption mainly through pericytes | [20] | |
| Nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor upregulates multiple antioxidant response elements (AREs), conferring cytoprotective factors to the cell | [21] | |
| Lipid Peroxidation | Fe2+ produces ROS and promotes lipid peroxidation | [22] |
| Malondialdehyde (reactive aldehydes) can form adducts with cellular proteins and nucleic acids, contributing to cellular dysfunction | [23] | |
| 4-Hydroxynonenal is primarily produced during the peroxidation of omega 6 polyunsaturated fatty acids | [23] | |
| Low-density lipoprotein (LDL) molecules could become highly oxidised to trigger macrophages activation and apoptosis | [23] | |
| ROS could participate in forming F2-isoprostanes through arachidonic acid oxidation | [24] | |
| Acyl-CoA synthetase long-chain family member 4 (ACSL4) facilitates the incorporation of polyunsaturated fatty acids into phospholipids, contributing to lipid peroxidation | [25] | |
| Nuclear receptor coactivator 4 (NCOA4) facilitates the selective autophagic degradation of ferritin, releasing iron and promoting the Fenton reaction, contributing to lipid peroxidation | [26] | |
| Excitotoxicity | Glutamate activates the NMDAR, inducing a large increase in intracellular Ca2+ concentration, directly stimulating ROS/RNS production | [5,27] |
| Inflammation | Microglia display a pro-inflammatory (M1) subtype, producing multiple pro-inflammatory cytokines such as the TNFα, interferon-gamma (IFN-γ) IL-1β, IL-6, and IL-12 | [28,29] |
| Microglia display an anti-inflammatory (M2) subtype, expressing anti-inflammatory cytokines, including IL-10, transforming growth factor beta (TGF-β), insulin-like growth factor 1 (IGF-1), and arginase 1 (Arg1), thus participating in tissue repair and inflammation resolution | ||
| Janus kinase 2 (JAK2)/signal transducer and activator of transcription (STAT3) is the main signalling pathway that is responsible for activating microglia into a pro-inflammatory subtype | [30] | |
| NLR family pyrin domain containing 3 (NLRP3) promotes an inflammatory response and triggers neuronal pyroptosis after ischemic stroke | [31] | |
| Blood–Brain Barrier (BBB) Disruption | Matrix metalloproteinase (MMPs) plays a role in cleaving tight junctions and degrading the extracellular matrix | [32] |
| Oligodendrocytes produce large amounts of MMP-9 as a response to inflammation and oxidative stress | [33] | |
| Pericytes migrate away from the vasculature, thereby contributing to increased BBB permeability | [34] | |
| Astrocytes release vascular endothelial growth factor (VEGF), glial cell-derived neurotrophic factor, MMP, glutamate, and NO | [35,36] | |
| Endothelial cells induce alteration of Ca2+ metabolism, phospholipase-A2 activation, and production of monocyte chemoattractant protein-1 | [37] | |
| Cell Death | Autophagy: Activation by AMP-activated protein kinase (AMPK), activation by phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt), inhibition by mammalian target of rapamycin (mTOR), activation by hypoxia-inducible factor (HIF)-1α/BCL2 interacting protein 3 (BNIP3), inhibition by the sequestration of Beclin1 by Bcl2, activation by p53, and inhibition by TIGAR | [38] |
| Apoptosis: Both the intrinsic (mitochondrial) and extrinsic (death receptors) pathways are involved | [39] | |
| Ferroptosis: Iron-dependent accumulation of lipid peroxides and glutathione peroxidase 4 (GPX4) inactivation | [22] | |
| Pyroptosis: The NLRP3 inflammasome is activated by ROS generated during IRI, thus triggering pyroptosis in brain cells. | [40] | |
| Necroptosis: Mediated by receptor-interacting serine/threonine protein kinase-1 and -3 and mixed lineage kinase domain-like protein | [41] |
| Drug | Dose Frequency Length | Controlled Randomisation Blind | N (Total) N (Intervention) N (Control) | Efficacy Assessment | Main Results | Adverse Effects |
|---|---|---|---|---|---|---|
| Melatonin [75] | 20 mg | Placebo | 65 | NIHSS and mRS | Higher reduction at 30 and 90 days in median NIHSS and mRS scores compared to placebo | No serious adverse events were present |
| 1 per day | Yes | 32 | ||||
| 5 days | Double | 33 | ||||
| Acetyl-L-Carnitine [79] | 1000 mg | Placebo | 69 | NIHSS and mRS | Higher reduction at 90 days in NIHSS and mRS scores compared to placebo | No differences among groups |
| 3 per day | Yes | 34 | ||||
| 3 days | Double | 35 | ||||
| Edaravone dexborneol [100] | 37.5 mg | Edaravone (alone) | 1194 | NIHSS and mRS | Higher reduction at 90 days in mRS score compared to placebo | No differences among groups |
| 2 per day | Yes | 599 | ||||
| 14 days | Double | 595 | ||||
| Saffron [98] | 200 mg | Standard treatment | 40 | NIHSS | Higher reduction at 4 days in NIHSS scores compared to placebo | No data available |
| 2 per day | Yes | 20 | ||||
| 4 days | N/I | 20 | ||||
| Coenzyme Q10 [78] | 100 mg | Placebo | 44 | NIHSS | Higher reduction at 30 days in NIHSS scores compared to placebo | No data available |
| 3 per day | Yes | 21 | mRS | |||
| 4 weeks | Double | 23 | MMSE | |||
| Oleoylethanolamide [101] | 300 or 600 mg | Placebo | 60 | NIHSS mRS | OEA improves the inflammatory parameters, OS balance, and lipids levels | No differences among groups |
| 1 per day | Yes | 40 | ||||
| 3 days | Double | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Briones-Valdivieso, C.; Briones, F.; Orellana-Urzúa, S.; Chichiarelli, S.; Saso, L.; Rodrigo, R. Novel Multi-Antioxidant Approach for Ischemic Stroke Therapy Targeting the Role of Oxidative Stress. Biomedicines 2024, 12, 501. https://doi.org/10.3390/biomedicines12030501
Briones-Valdivieso C, Briones F, Orellana-Urzúa S, Chichiarelli S, Saso L, Rodrigo R. Novel Multi-Antioxidant Approach for Ischemic Stroke Therapy Targeting the Role of Oxidative Stress. Biomedicines. 2024; 12(3):501. https://doi.org/10.3390/biomedicines12030501
Chicago/Turabian StyleBriones-Valdivieso, Camilo, Felipe Briones, Sofía Orellana-Urzúa, Silvia Chichiarelli, Luciano Saso, and Ramón Rodrigo. 2024. "Novel Multi-Antioxidant Approach for Ischemic Stroke Therapy Targeting the Role of Oxidative Stress" Biomedicines 12, no. 3: 501. https://doi.org/10.3390/biomedicines12030501
APA StyleBriones-Valdivieso, C., Briones, F., Orellana-Urzúa, S., Chichiarelli, S., Saso, L., & Rodrigo, R. (2024). Novel Multi-Antioxidant Approach for Ischemic Stroke Therapy Targeting the Role of Oxidative Stress. Biomedicines, 12(3), 501. https://doi.org/10.3390/biomedicines12030501