
Phillygenin Suppresses Glutamate Exocytosis in Rat Cerebrocortical Nerve Terminals (Synaptosomes) through the Inhibition of Cav2.2 Calcium Channels



Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals and Ethics
2.3. Synaptosome Isolation
2.4. Measurement of Glutamate Release
2.5. Measurement of Synaptosomal Plasma Membrane Potential
2.6. Measurement of Intrasynaptosomal Ca2+ Concentration ([Ca2+]i)
2.7. Western Blotting
2.8. Data Analysis
3. Results
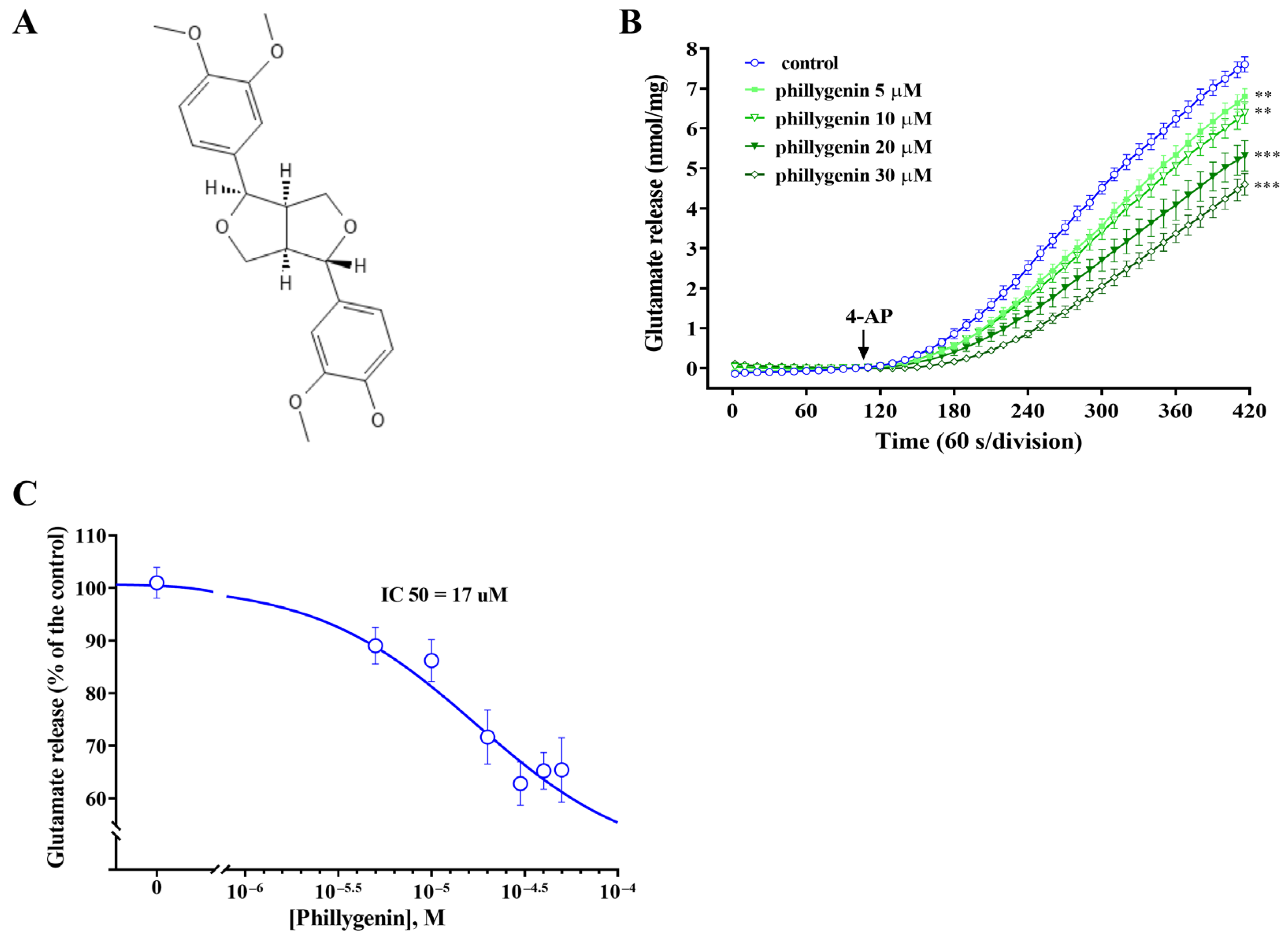
3.1. Phillygenin Inhibits Glutamate Release in Rat Cerebrocortical Synaptosomes
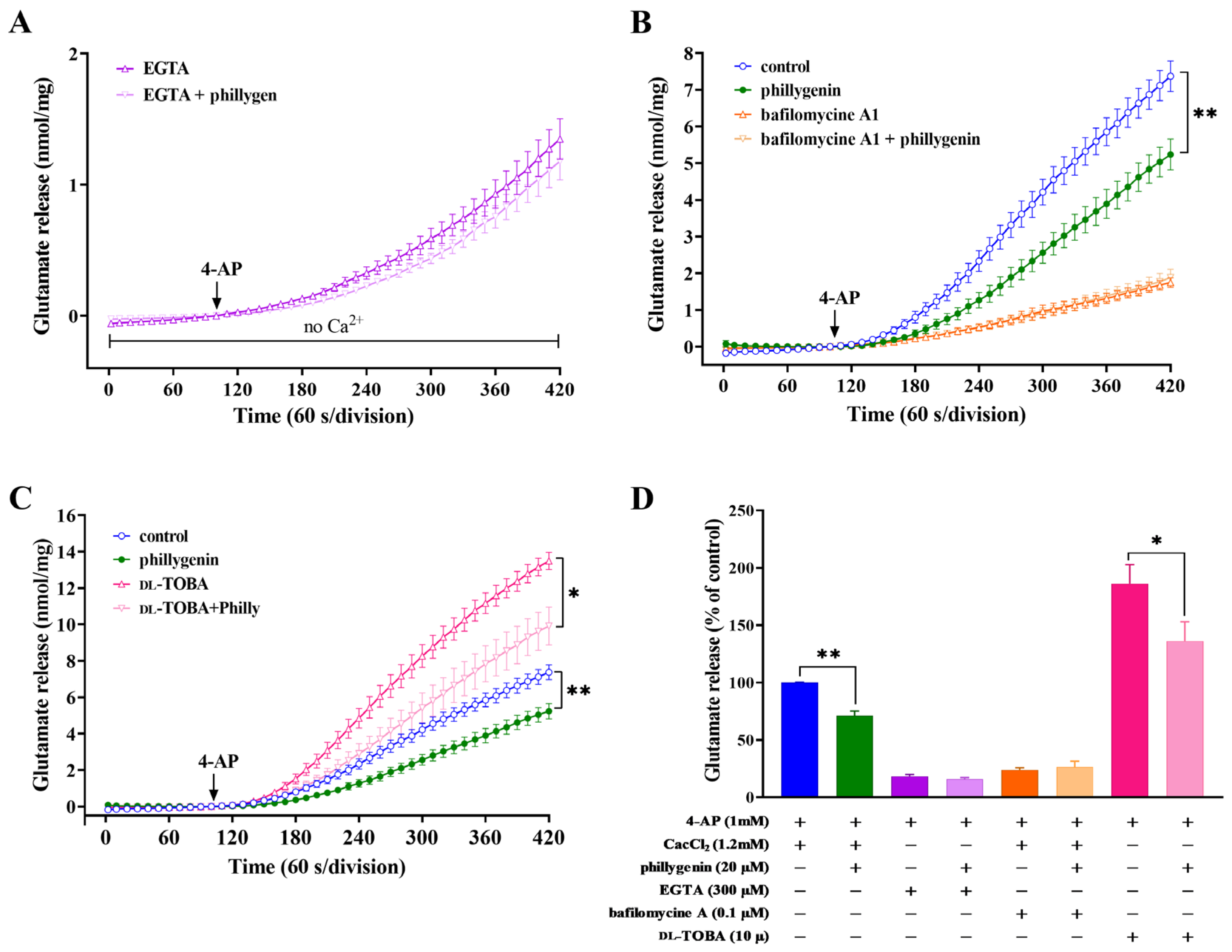
3.2. Effects of Calcium Ion Chelators, Glutamate Transporter Inhibitors or Vesicular Transporter Inhibitors on the Inhibition of 4-AP-Evoked Glutamate Release by Phillygenin
3.3. Effect of Phillygenin on Nerve Terminal Excitability and 4-AP Induced Ca2+ Influx
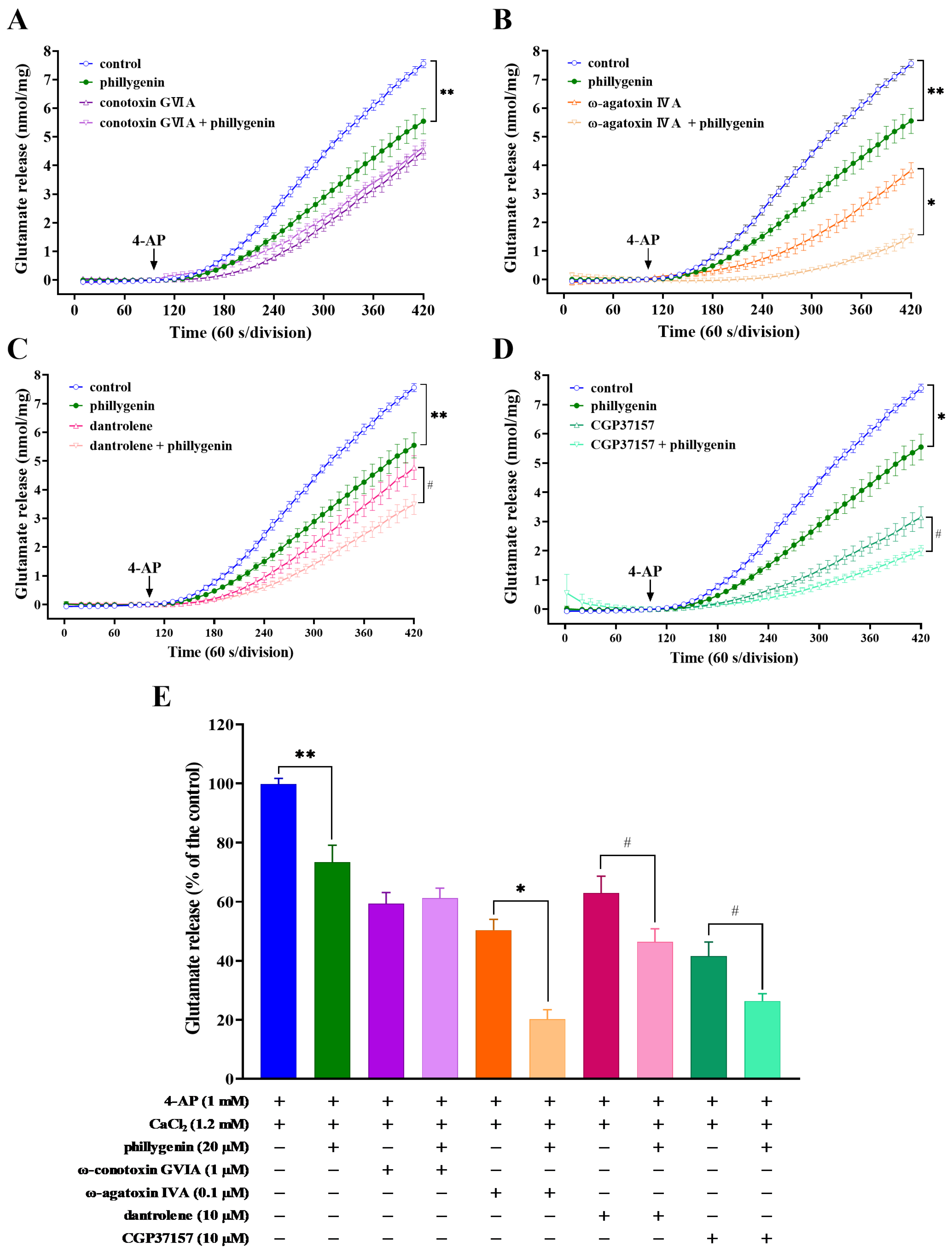
3.4. Effects of Ca2+-Channel Antagonists, Intracellular Ca2+ Release Inhibitors or Mitochondrial Na+/Ca2+ Exchanger Inhibitors on Phillygenin-Mediated Inhibition of 4-AP-Induced Glutamate Release
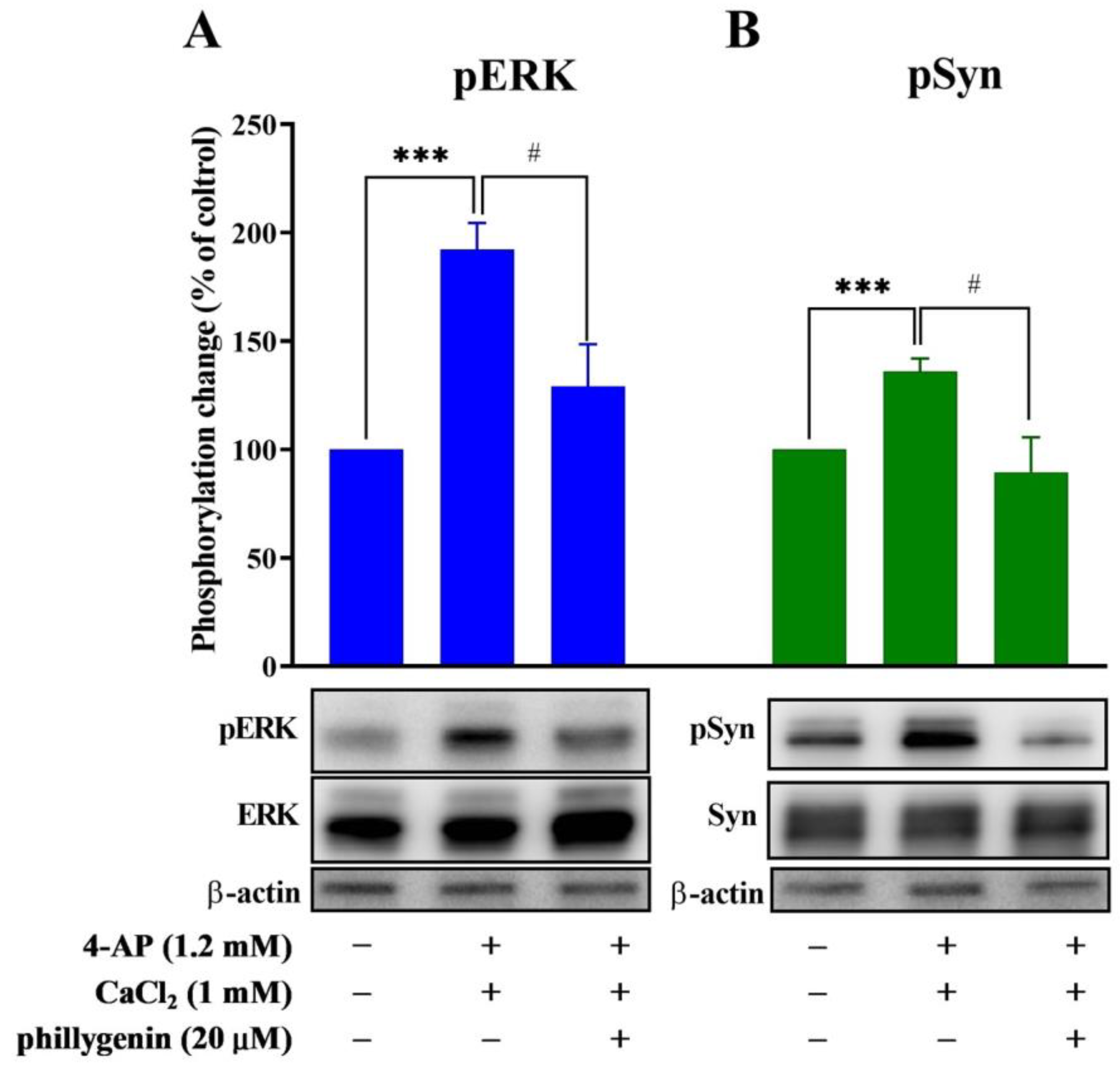
3.5. Phillygenin Inhibits 4-AP-Evoked Glutamate Release through Extracellular Signal-Regulated Kinase Signaling
4. Discussion
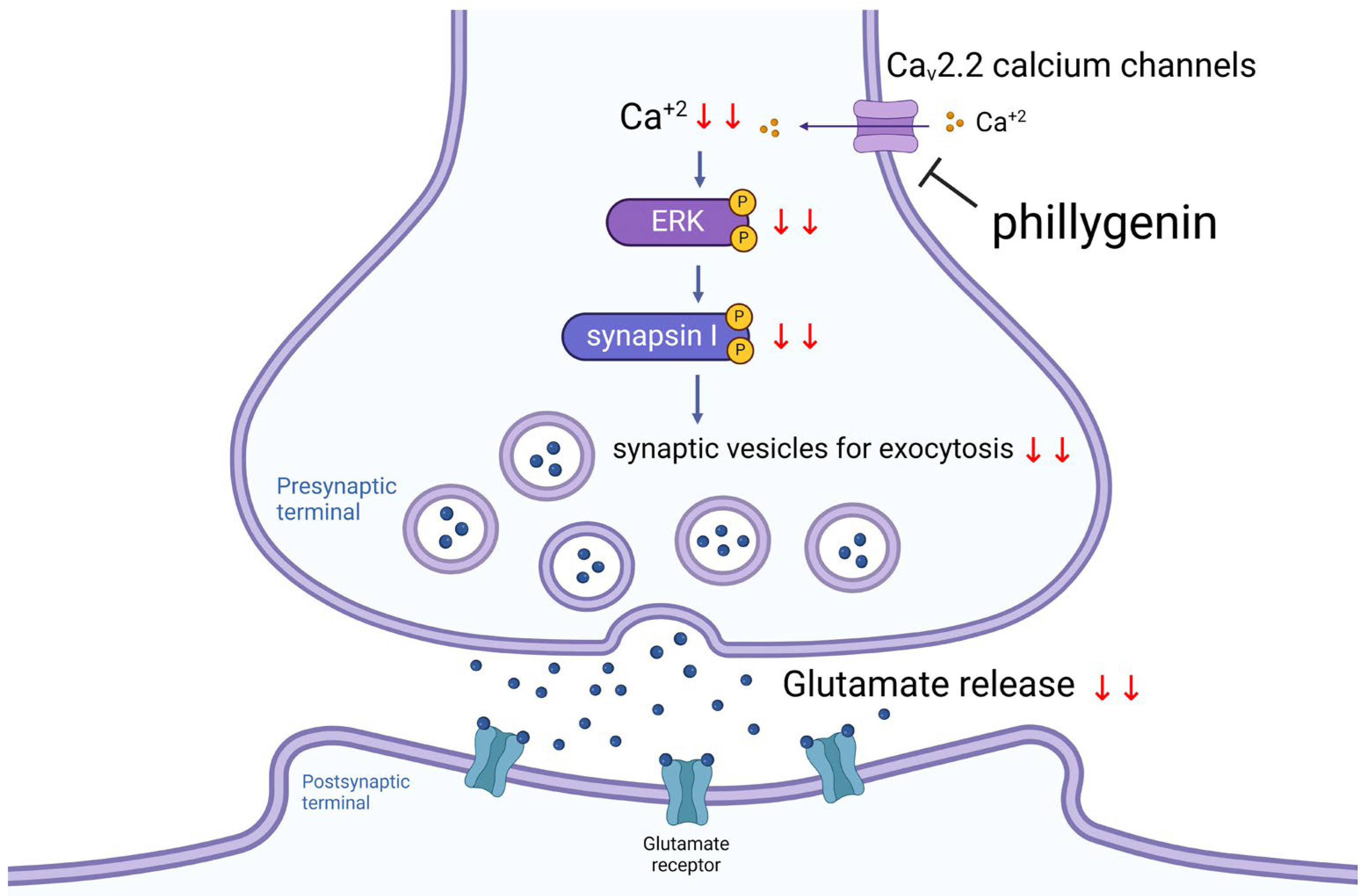
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Platt, S.R. The role of glutamate in central nervous system health and disease—A review. Vet. J. 2007, 173, 278–286. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef]
- Dhaouadi, N.; Vitto, V.A.M.; Pinton, P.; Galluzzi, L.; Marchi, S. Ca2+ signaling and cell death. Cell Calcium 2023, 113, 102759. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Neves, D.; Salazar, I.L.; Almeida, R.D.; Silva, R.M. Molecular mechanisms of ischemia and glutamate excitotoxicity. Life Sci. 2023, 328, 121814. [Google Scholar] [CrossRef] [PubMed]
- Green, J.L.; Dos Santos, W.F.; Fontana, A.C.K. Role of glutamate excitotoxicity and glutamate transporter EAAT2 in epilepsy: Opportunities for novel therapeutics development. Biochem. Pharmacol. 2021, 193, 114786. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Ince, P.G. Glutamate, excitotoxicity and amyotrophic lateral sclerosis. J. Neurol. 1997, 244 (Suppl. 2), S3–S14. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Wali, A.F.; Ahmad, A.; Shakeel, S.; Rasool, S.; Ali, R.; Rashid, S.M.; Madkhali, H.; Ganaie, M.A.; Khan, R. Neuroprotective strategies for neurological disorders by natural products: An update. Curr. Neuropharmacol. 2019, 17, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Mathur, S.; Hoskins, C. Drug development: Lessons from nature. Biomed. Rep. 2017, 6, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xia, Q.; Liu, X.; Liu, W.; Huang, W.; Mei, X.; Luo, J.; Shan, M.; Lin, R.; Zou, D.; et al. Phytochemistry, pharmacology, quality control and future research of Forsythia suspensa (Thunb.) Vahl: A review. J. Ethnopharmacol. 2018, 210, 318–339. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.H.; Li, Y.X.; Peng, C.; Gong, X.H.; Zheng, X.G. Determination of phillygenin in rat plasma by high-performance liquid chromatography and its application to pharmacokinetic studies. Eur. J. Drug Metab. Pharmacokinet. 2013, 38, 201–207. [Google Scholar] [CrossRef]
- Song, W.; Wu, J.; Yu, L.; Peng, Z. Evaluation of the pharmacokinetics and hepatoprotective effects of phillygenin in mouse. Biomed. Res. Int. 2018, 2018, 7964318. [Google Scholar] [CrossRef]
- Du, B.; Zhang, L.; Sun, Y.; Zhang, G.; Yao, J.; Jiang, M.; Pan, L.; Sun, C. Phillygenin exhibits anti-inflammatory activity through modulating multiple cellular behaviors of mouse lymphocytes. Immunopharmacol. Immunotoxicol. 2019, 41, 76–85. [Google Scholar] [CrossRef]
- Xue, H.H.; Li, J.J.; Li, S.F.; Guo, J.; Yan, R.P.; Chen, T.G.; Shi, X.H.; Wang, J.D.; Zhang, L.W. Phillygenin attenuated colon inflammation and improved intestinal mucosal barrier in DSS-induced colitis mice via TLR4/Src mediated MAPK and NF-κB signaling pathways. Int. J. Mol. Sci. 2023, 24, 2238. [Google Scholar] [CrossRef]
- Kang, W.; Wang, J. In vitro antioxidant properties and in vivo lowering blood lipid of Forsythiasuspense leaves. Med. Chem. Res. 2010, 19, 617–628. [Google Scholar] [CrossRef]
- Guo, J.; Yan, W.R.; Tang, J.K.; Jin, X.; Xue, H.H.; Wang, T.; Zhang, L.W.; Sun, Q.Y.; Liang, Z.X. Dietary phillygenin supplementation ameliorates aflatoxin B1-induced oxidative stress, inflammation, and apoptosis in chicken liver. Ecotoxicol. Environ. Saf. 2022, 236, 113481. [Google Scholar] [CrossRef]
- Ding, X.; Lu, D.; Fan, J. A natural product phillygenin suppresses osteosarcoma growth and metastasis by regulating the SHP-1/JAK2/STAT3 signaling. Biosci. Biotechnol. Biochem. 2021, 85, 307–314. [Google Scholar] [CrossRef]
- Li, H.; Chen, M.; Yang, Z.; Wang, Q.; Wang, J.; Jin, D.; Yang, X.; Chen, F.; Zhou, X.; Luo, K. Phillygenin, a MELK Inhibitor, Inhibits Cell Survival and Epithelial-Mesenchymal Transition in Pancreatic Cancer Cells. OncoTargets Ther. 2020, 13, 2833–2842. [Google Scholar] [CrossRef]
- Li, R.J.; Xu, J.Y.; Wang, X.; Liao, L.J.; Wei, X.; Xie, P.; Xu, W.Y.; Xu, Z.Y.; Xie, S.H.; Jiang, Y.Y.; et al. Therapeutic effect of demethylated hydroxylated phillygenin derivative on Helicobacter pylori infection. Front. Microbiol. 2023, 14, 1071603. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, J.; Zhang, K.; Wang, X.; Zhang, K.; Guo, Z.; Han, S.; Wang, L.; Qiu, Z.; Wang, G.; et al. Phillygenin activates PKR/eIF2α pathway and induces stress granule to exert anti-avian infectious bronchitis virus. Int. Immunopharmacol. 2022, 108, 108764. [Google Scholar] [CrossRef]
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef]
- Lalkovičová, M.; Danielisová, V. Neuroprotection and antioxidants. Neural. Regen. Res. 2016, 11, 865–874. [Google Scholar] [CrossRef]
- Lin, Y.; Yang, P. Phillygenin inhibits the inflammation and apoptosis of pulmonary epithelial cells by activating PPARγ signaling via downregulation of MMP8. Mol. Med. Rep. 2021, 24, 775. [Google Scholar] [CrossRef]
- Binvignat, O.; Olloquequi, J. Excitotoxicity as a Target Against Neurodegenerative Processes. Curr. Pharm. Des. 2020, 26, 1251–1262. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Sihra, T.S. Synaptosomes possess an exocytotic pool of glutamate. Nature 1986, 321, 772–773. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Pan, T.L.; Wang, P.W.; Chiu, K.M.; Lee, M.Y.; Wang, S.J. Asiatic acid prevents cognitive deficits by inhibiting calpain activation and preserving synaptic and mitochondrial function in rats with kainic acid-induced seizure. Biomedicines 2021, 9, 284. [Google Scholar] [CrossRef]
- Chiu, K.M.; Lee, M.Y.; Lu, C.W.; Lin, T.Y.; Wang, S.J. Plantainoside D reduces depolarization-evoked glutamate release from rat cerebral cortical synaptosomes. Molecules 2023, 28, 1313. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Chiu, K.M.; Lee, M.Y.; Huang, J.H.; Wang, S.J. Silymarin inhibits glutamate release and prevents against kainic acid-induced excitotoxic injury in rats. Biomedicines 2020, 8, 486. [Google Scholar] [CrossRef]
- Akerman, K.E.; Scott, I.G.; Heikkilä, J.E.; Heinonen, E. Ionic dependence of membrane potential and glutamate receptor-linked responses in synaptoneurosomes as measured with a cyanine dye, DiS-C2-(5). J. Neurochem. 1987, 48, 552–559. [Google Scholar] [CrossRef]
- Magi, S.; Piccirillo, S.; Amoroso, S.; Lariccia, V. Excitatory amino acid transporters (EAATs): Glutamate transport and beyond. Int. J. Mol. Sci. 2019, 20, 5674. [Google Scholar] [CrossRef]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Ladera, C.; Martín, R.; Bartolomé-Martín, D.; Torres, M.; Sánchez-Prieto, J. Partial compensation for N-type Ca2+ channel loss by P/Q-type Ca2+ channels underlines the differential release properties supported by these channels at cerebrocortical nerve terminals. Eur. J. Neurosci. 2009, 29, 1131–1140. [Google Scholar] [CrossRef]
- Chiu, K.M.; Lin, T.Y.; Lee, M.Y.; Lu, C.W.; Wang, S.J. Lappaconitine inhibits glutamate release from rat cerebrocortical nerve terminals by suppressing Ca2+ influx and protein kinase A cascade. Neurotoxicology 2022, 91, 218–227. [Google Scholar] [CrossRef]
- Falcón-Moya, R.; Rodríguez-Moreno, A. Metabotropic actions of kainate receptors modulating glutamate release. Neuropharmacology 2021, 197, 108696. [Google Scholar] [CrossRef]
- Meldrum, B.; Garthwaite, J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. Sci. 1990, 11, 379–387. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Wu, L.G.; Saggau, P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997, 20, 204–212. [Google Scholar] [CrossRef]
- Pongs, O.; Leicher, T.; Berger, M.; Roeper, J.; Bähring, R.; Wray, D.; Giese, K.P.; Silva, A.J.; Storm, J.F. Functional and molecular aspects of voltage-gated K+ channel beta subunits. Ann. N. Y. Acad. Sci. 1999, 868, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Sihra, T.S.; Nichols, R.A. Mechanisms in the regulation of neurotransmitter release from brain nerve terminals: Current hypotheses. Neurochem. Res. 1993, 18, 47–58. [Google Scholar] [CrossRef]
- Südhof, T.C. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 2013, 80, 675–690. [Google Scholar] [CrossRef]
- Attwell, D.; Barbour, B.; Szatkowski, M. Nonvesicular release of neurotransmitter. Neuron 1993, 11, 401–407. [Google Scholar] [CrossRef]
- Nicholls, D.G. Presynaptic modulation of glutamate release. Prog. Brain Res. 1998, 116, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, E.M.; Vivar, C.; Camandola, S. Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 2012, 3, 61. [Google Scholar] [CrossRef]
- Jarvis, S.E.; Zamponi, G.W. Interactions between presynaptic Ca2+ channels, cytoplasmic messengers and proteins of the synaptic vesicle release complex. Trends Pharmacol. Sci. 2001, 22, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Leenders, A.G.; Sheng, Z.H. Modulation of neurotransmitter release by the second messenger-activated protein kinases: Implications for presynaptic plasticity. Pharmacol. Ther. 2005, 105, 69–84. [Google Scholar] [CrossRef]
- Pereira, D.B.; Carvalho, A.P.; Duarte, C.B. Non-specific effects of the MEK inhibitors PD098,059 and U0126 on glutamate release from hippocampal synaptosomes. Neuropharmacology 2002, 42, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Millán, C.; Torres, M.; Sánchez-Prieto, J. Co-activation of PKA and PKC in cerebrocortical nerve terminals synergistically facilitates glutamate release. J. Neurochem. 2003, 87, 1101–1111. [Google Scholar] [CrossRef]
- Chi, P.; Greengard, P.; Ryan, T.A. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 2003, 38, 69–78. [Google Scholar] [CrossRef]
- Guo, J.; Tang, J.K.; Wang, B.F.; Yan, W.R.; Li, T.; Guo, X.J.; Zhang, L.; Wang, T.; Sun, Q.Y.; Zhang, L.W. Phillygenin from Forsythia suspensa leaves exhibits analgesic potential and anti-inflammatory activity in carrageenan-induced paw edema in mice. J. Food Biochem. 2022, 46, e14460. [Google Scholar] [CrossRef]
- Hu, N.; Wang, C.; Dai, X.; Zhou, M.; Gong, L.; Yu, L.; Peng, C.; Li, Y. Phillygenin inhibits LPS-induced activation and inflammation of LX2 cells by TLR4/MyD88/NF-κB signaling pathway. J Ethnopharmacol 2020, 248, 112361. [Google Scholar] [CrossRef]
- Wang, C.; Wu, R.; Zhang, S.; Gong, L.; Fu, K.; Yao, C.; Peng, C.; Li, Y. A comprehensive review on pharmacological, toxicity, and pharmacokinetic properties of phillygenin: Current landscape and future perspectives. Biomed. Pharmacother. 2023, 166, 115410. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Y.; Zhang, Y.; Chen, L.; Xu, X.; Dang, Y.; Ti, X. Phillygenin regulates proliferation and apoptosis of non-small cell lung cancer through by AMPK/ERK/NF-κB axis. Pharmazie 2020, 75, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Piao, X.L.; Zhang, Q.; Wang, D.; Piao, X.S.; Kim, S.W. Protective effects of Forsythia suspensa extract against oxidative stress induced by diquat in rats. Food Chem. Toxicol. 2010, 48, 764–770. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane Potential (Fluorescence Units) | Glutamate Release (nmol mg−1 Protein per 5 min) | [Ca2+]i (nM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Basal | 4-AP | n | Basal | KCl | n | Basal | 4-AP | n | |
| Control | 0.01 ± 0.03 | 25.63 ± 0.86 | 5 | −0.08 ± 0.08 | 5.18 ± 0.4 | 5 | 218.2 ± 10.62 | 327.48 ± 19.29 | 5 |
| Phillygenin | 0.02 ± 0.12 | 27.08 ± 1.47 | 5 | −0.07 ± 0.04 | 2.65 ± 0.63 ** | 5 | 211.38 ± 8.29 | 240.16 ± 11.23 * | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.-Y.; Lin, T.-Y.; Chang, Y.-Y.; Chiu, K.-M.; Wang, S.-J. Phillygenin Suppresses Glutamate Exocytosis in Rat Cerebrocortical Nerve Terminals (Synaptosomes) through the Inhibition of Cav2.2 Calcium Channels. Biomedicines 2024, 12, 495. https://doi.org/10.3390/biomedicines12030495
Lee M-Y, Lin T-Y, Chang Y-Y, Chiu K-M, Wang S-J. Phillygenin Suppresses Glutamate Exocytosis in Rat Cerebrocortical Nerve Terminals (Synaptosomes) through the Inhibition of Cav2.2 Calcium Channels. Biomedicines. 2024; 12(3):495. https://doi.org/10.3390/biomedicines12030495
Chicago/Turabian StyleLee, Ming-Yi, Tzu-Yu Lin, Ya-Ying Chang, Kuan-Ming Chiu, and Su-Jane Wang. 2024. "Phillygenin Suppresses Glutamate Exocytosis in Rat Cerebrocortical Nerve Terminals (Synaptosomes) through the Inhibition of Cav2.2 Calcium Channels" Biomedicines 12, no. 3: 495. https://doi.org/10.3390/biomedicines12030495
APA StyleLee, M.-Y., Lin, T.-Y., Chang, Y.-Y., Chiu, K.-M., & Wang, S.-J. (2024). Phillygenin Suppresses Glutamate Exocytosis in Rat Cerebrocortical Nerve Terminals (Synaptosomes) through the Inhibition of Cav2.2 Calcium Channels. Biomedicines, 12(3), 495. https://doi.org/10.3390/biomedicines12030495