
Advances in Pharmacological Approaches for Managing Hypercholesterolemia: A Comprehensive Overview of Novel Treatments
 ,
,  , ,
, ,  , and
, and
Abstract
1. Introduction
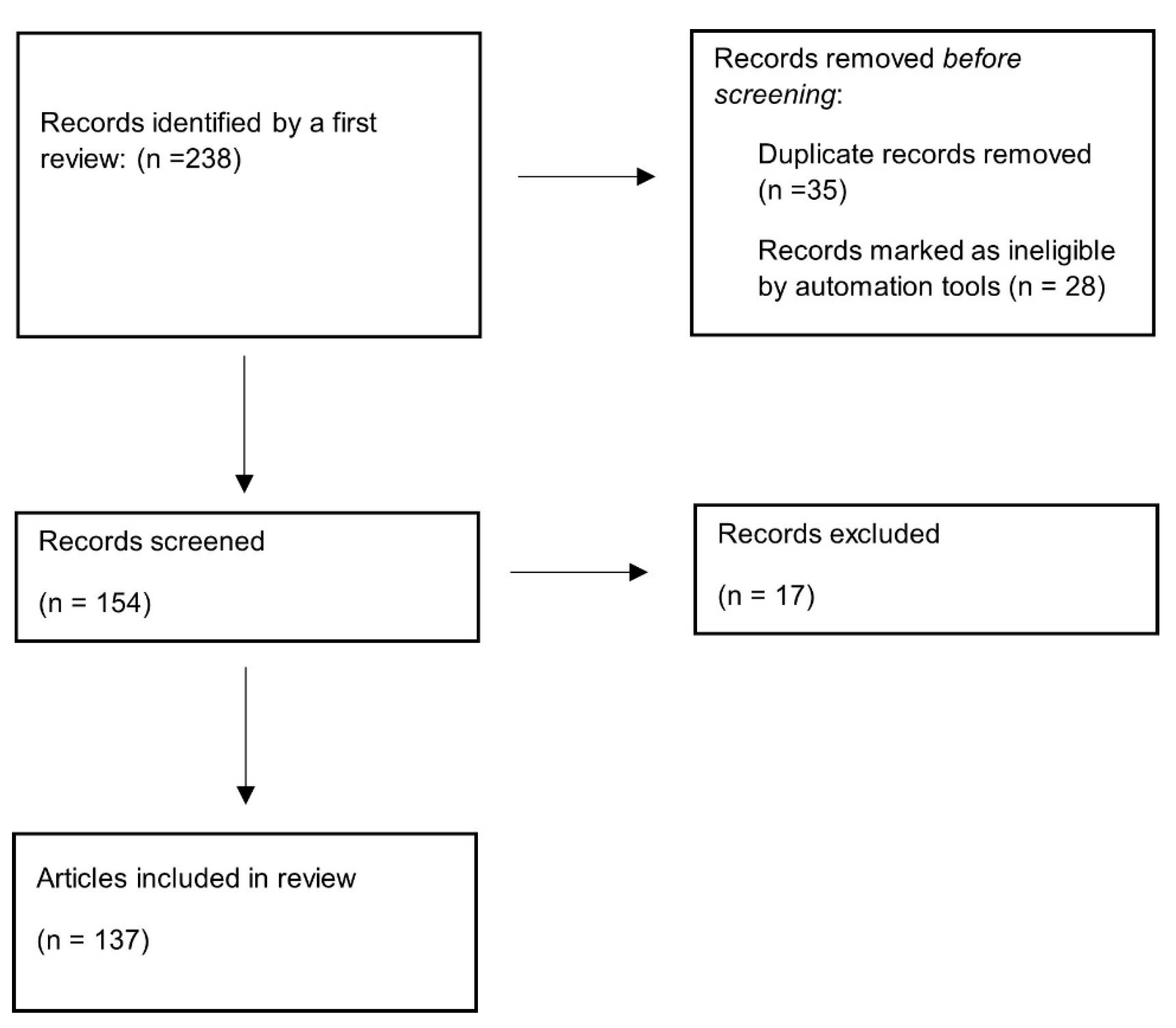
Data Collection
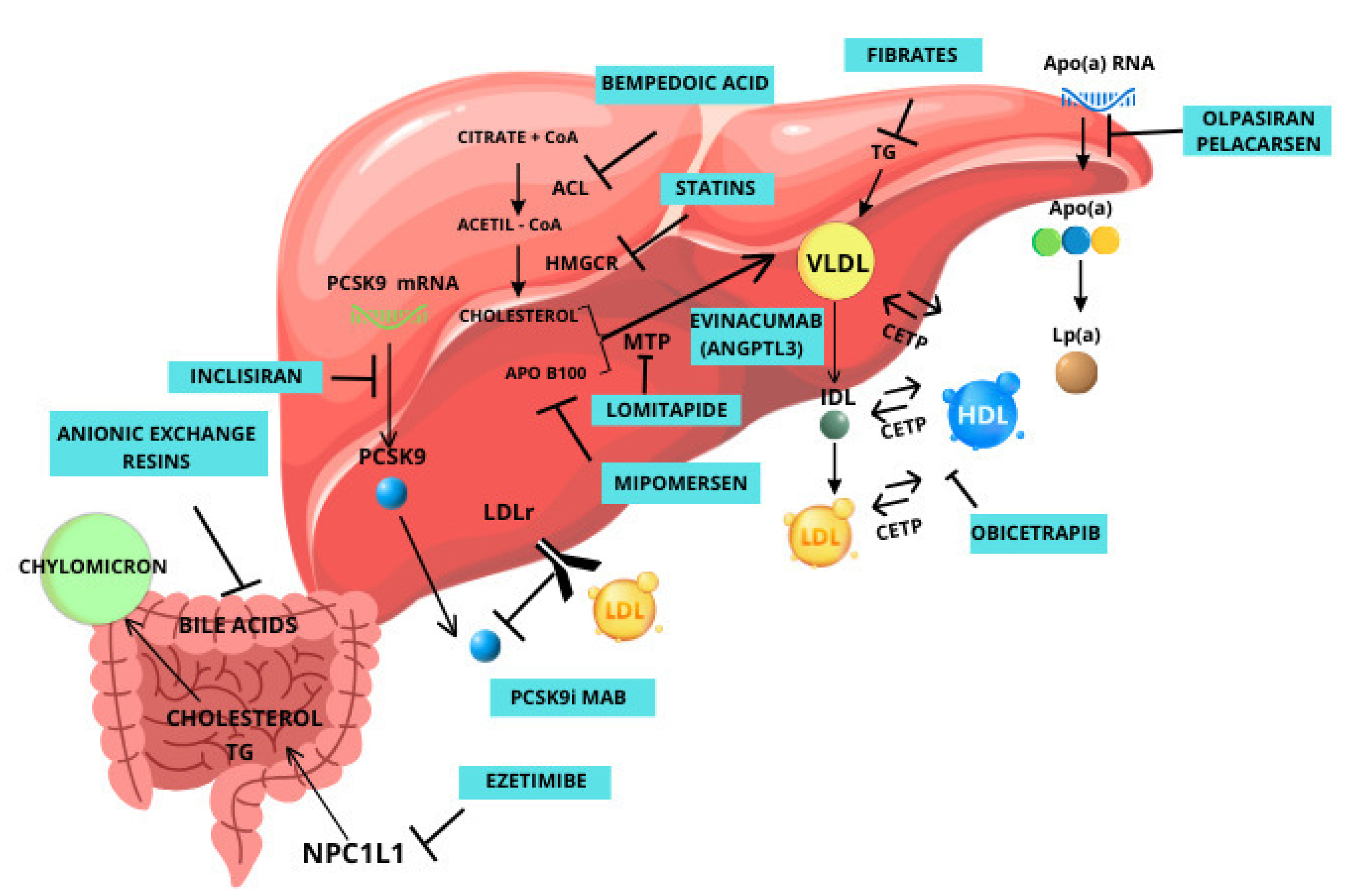
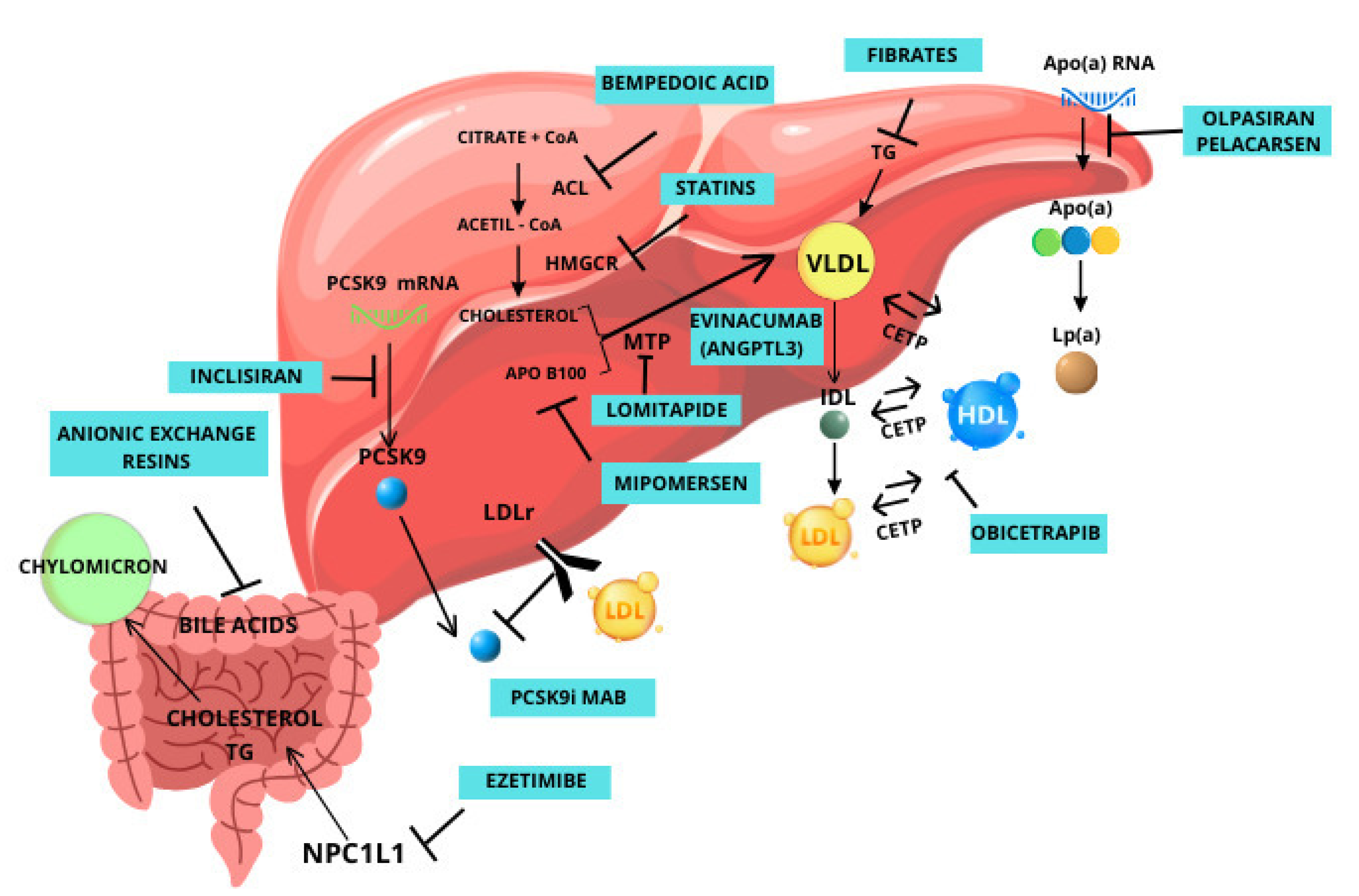
2. Currently Available Drugs
2.1. Mipomersen
2.2. Lomitapide
2.3. Inclisiran
2.4. Bempedoic Acid
2.5. Pelacarsen
3. Novel Therapeutic Options That Are Not on the Market Yet
3.1. Olpasiran
3.2. ANGPTL3
3.3. CETP
3.4. HMG-CoA Reductase Degrader
3.5. ASGR1
3.6. PCSK9 Vaccine
3.7. Additional Emerging Strategies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Libby, P.; Sukhova, G.; Lee, R.T.; Liao, J.K. Molecular biology of atherosclerosis. Int. J. Cardiol. 1997, 62, S23–S29. [Google Scholar] [CrossRef]
- Kumric, M.; Borovac, J.A.; Martinovic, D.; Kurir, T.T.; Bozic, J. Circulating Biomarkers Reflecting Destabilization Mechanisms of Coronary Artery Plaques: Are We Looking for the Impossible? Biomolecules 2021, 11, 881. [Google Scholar] [CrossRef]
- Yurdagul, A.; Finney, A.C.; Woolard, M.D.; Orr, A.W. The arterial microenvironment: The where and why of atherosclerosis. Biochem. J. 2016, 473, 1281–1295. [Google Scholar] [CrossRef]
- Sasso, F.C.; Simeon, V.; Galiero, R.; Caturano, A.; De Nicola, L.; Chiodini, P.; Rinaldi, L.; Salvatore, T.; Lettieri, M.; Nevola, R.; et al. The number of risk factors not at target is associated with cardiovascular risk in a type 2 diabetic population with albuminuria in primary cardiovascular prevention. Post-hoc analysis of the NID-2 trial. Cardiovasc. Diabetol. 2022, 21, 235. [Google Scholar] [CrossRef]
- Caturano, A.; D’Angelo, M.; Mormone, A.; Russo, V.; Mollica, M.P.; Salvatore, T.; Galiero, R.; Rinaldi, L.; Vetrano, E.; Marfella, R.; et al. Oxidative Stress in Type 2 Diabetes: Impacts from Pathogenesis to Lifestyle Modifications. Curr. Issues Mol. Biol. 2023, 45, 6651–6666. [Google Scholar] [CrossRef]
- Bore’n, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; Baigent, C.; et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [PubMed]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin toxicity mechanistic insights and clinical implications. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Loffredo, G.; Rinaldi, L.; Catalini, C.; Gjeloshi, K.; Albanese, G.; Di Martino, A.; et al. Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options. Biomedicines 2022, 10, 2274. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Di Angelantonio, E.; Gao, P.; Pennells, L.; Kaptoge, S.; Caslake, M.; Thompson, A.; Butterworth, A.S.; Sarwar, N.; Wormser, D.; et al. Lipid-related markers and cardiovascular disease prediction. JAMA 2012, 307, 2499–2506. [Google Scholar]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar]
- Hajar, R. Statins: Past and present. Heart Views 2011, 12, 121–127. [Google Scholar] [CrossRef]
- Sirtori, C.R. The pharmacology of statins. Pharm. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef]
- Gentile, S.; Turco, S.; Guarino, G.; Sasso, C.F.; Amodio, M.; Magliano, P.; Salvatore, T.; Corigliano, G.; Agrusta, M.; De Simone, G.; et al. Comparative efficacy study of atorvastatin vs simvastatin, pravastatin, lovastatin and placebo in type 2 diabetic patients with hypercholesterolaemia. Diabetes Obes. Metab. 2000, 2, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Naeem, F.; McKay, G.; Fisher, M. Cardiovascular outcomes trials with non-statin lipid-lowering drugs in diabetes. Br. J. Diabetes 2018, 18, 5. [Google Scholar] [CrossRef]
- Habibe, M.N.; Kellar, J.Z. Niacin Toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Lloyd-Jones, D.M.; Morris, P.B.; Ballantyne, C.M.; Birtcher, K.K.; Daly, D.D., Jr.; De Palma, S.M.; Minissian, M.B.; Orringer, C.E.; Smith, S.C., Jr. ACC expert consensus decision pathway on the role of non-statin therapies for ldl-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: A report of the American college of cardiology task force on clinical expert consensus documents. J. Am. Coll. Cardiol. 2016, 68, 92–125. [Google Scholar] [PubMed]
- Baigent, C.; Landray, M.J.; Reith, C.; Emberson, J.; Wheeler, D.C.; Tomson, C.; Wanner, C.; Krane, V.; Cass, A.; Craig, J.; et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): A randomised placebo-controlled trial. Lancet 2011, 377, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Rossebo, A.B.; Pedersen, T.R.; Boman, K.; Brudi, P.; Chambers, J.B.; Egstrup, K.; Gerdts, E.; Gohlke-Bärwolf, C.; Holme, I.; Kesäniemi, Y.A.; et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N. Engl. J. Med. 2008, 359, 1343–1356. [Google Scholar] [CrossRef] [PubMed]
- Kosoglou, T.; Statkevich, P.; Johnson-Levonas, A.O.; Paolini, J.F.; Bergman, A.J.; Alton, K.B. Ezetimibe: A review of its metabolism, pharmacokinetics and drug interactions. Clin. Pharmacokinet. 2005, 44, 467–494. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.D.; Fazio, S. From Lipids to Inflammation: New Approaches to Reducing Atherosclerotic Risk. Circ. Res. 2016, 118, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [PubMed]
- Qian, Y.W.; Schmidt, R.J.; Zhang, Y.; Chu, S.; Lin, A.; Wang, H.; Wang, X.; Beyer, T.P.; Bensch, W.R.; Li, W.; et al. Secreted PCSK9 downregulates low density lipoprotein receptor through receptor-mediated endocytosis. J. Lipid Res. 2007, 48, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Lagace, T.A.; McNutt, M.C.; Horton, J.D.; Deisenhofer, J. Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. USA 2008, 105, 1820–1825. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Neutel, J.; Cropp, A.; Duggan, W.; Wang, E.Q.; Plowchalk, D.; Sweeney, K.; Kaila, N.; Vincent, J.; Bays, H. Results of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, from a randomized, placebo-controlled, dose-ranging study in statin-treated subjects with hypercholesterolemia. Am. J. Cardiol. 2015, 115, 1212–1221. [Google Scholar] [CrossRef]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014, 129, S1–S45. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Puri, R.; Mehta, V.; Duell, P.B.; Nair, D.; Mohan, J.C.; Yusuf, J.; Dalal, J.J.; Mishra, S.; Kasliwal, R.R.; Agarwal, R.; et al. Proposed low-density lipoprotein cholesterol goals for secondary prevention and familial hypercholesterolemia in India with focus on PCSK9 inhibitor monoclonal antibodies: Expert consensus statement from Lipid Association of India. J. Clin. Lipidol. 2020, 14, e1–e13. [Google Scholar] [CrossRef]
- Goldberg, A.C. Novel therapies and new targets of treatment for familial hypercholesterolemia. J. Clin. Lipidol. 2010, 4, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Goldberg, T. Mipomersen (kynamro): A novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. Pharm. Ther. 2014, 39, 119–122. [Google Scholar]
- Parham, J.S.; Goldberg, A.C. Mipomersen and its use in familial hypercholesterolemia. Expert Opin. Pharmacother. 2019, 20, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Geary, R.S. Clinical pharmacological properties of mipomersen (Kynamro), a second generation antisense inhibitor of apolipoprotein B. Br. J. Clin. Pharmacol. 2013, 76, 269–276. [Google Scholar] [CrossRef]
- McGowan, M.P.; Tardif, J.C.; Ceska, R.; Burgess, L.J.; Soran, H.; Gouni-Berthold, I.; Wagener, G.; Chasan-Taber, S. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS ONE 2012, 7, e49006. [Google Scholar] [CrossRef]
- Chambergo-Michilot, D.; Alur, A.; Kulkarni, S.; Agarwala, A. Mipomersen in Familial Hypercholesterolemia: An Update on Health-Related Quality of Life and Patient-Reported Outcomes. Vasc. Health Risk Manag. 2022, 18, 73–80. [Google Scholar] [CrossRef]
- Astaneh, B.; Makhdami, N.; Astaneh, V.; Guyatt, G. The Effect of Mipomersen in the Management of Patients with Familial Hypercholesterolemia: A Systematic Review and Meta-Analysis of Clinical Trials. J. Cardiovasc. Dev. Dis. 2021, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.A.; Dufour, R.; Gagne, C.; Gaudet, D.; East, C.; Donovan, J.M.; Chin, W.; Tribble, D.L.; McGowan, M. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: Results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation 2012, 126, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, P.S. Impact of new lipid management guidelines on the treatment of extreme and very high-risk patients: AACE/ACE and AHA/ACC guidelines. J. Diabetes 2020, 12, 105–109. [Google Scholar] [CrossRef]
- Jellinger, P.S.; Handelsman, Y.; Rosenblit, P.D.; Bloomgarden, Z.T.; Fonseca, V.A.; Garber, A.J.; Grunberger, G.; Guerin, C.K.; Bell, D.S.H.; Mechanick, J.I.; et al. American association of clinical endocrinologists and american college of endocrinology guidelines for management of dyslipidemia and prevention of cardiovascular disease—Executive summarycomplete Appendix to Guidelines. Endocr. Pract. 2017, 23, 479–497. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Morris, P.B.; Ballantyne, C.M.; Birtcher, K.K.; Daly, D.D.; DePalma, S.M.; Minissian, M.B.; Orringer, C.E.; Smith, S.C. 2017 Focused Update of the 2016 ACC Expert Consensus Decision Pathway on the Role of Non-Statin Therapies for LDL-Cholesterol Lowering in the Management of Atherosclerotic Cardiovascular Disease Risk: A Report of the American College of Cardiology Task Force on Expert Consensus Decision Pathways. J. Am. Coll. Cardiol. 2017, 70, 1785–1822. [Google Scholar] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, e285–e350. [Google Scholar] [CrossRef] [PubMed]
- Samaha, F.F.; McKenney, J.; Bloedon, L.T.; Sasiela, W.J.; Rader, D.J. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Andrus, B.; Lacaille, D. 2013 ACC/AHA guideline on the assessment of cardiovascular risk. J. Am. Coll. Cardiol. 2014, 63, 2886. [Google Scholar] [CrossRef] [PubMed]
- Ajufo, E.; Rader, D.J. New Therapeutic Approaches for Familial Hypercholesterolemia. Annu. Rev. Med. 2018, 69, 113–131. [Google Scholar] [CrossRef]
- Cuchel, M.; Bloedon, L.T.; Szapary, P.O.; Kolansky, D.M.; Wolfe, M.L.; Sarkis, A.; Millar, J.S.; Ikewaki, K.; Siegelman, E.S.; Gregg, R.E.; et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N. Engl. J. Med. 2007, 356, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Meagher, E.A.; du Toit Theron, H.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 2013, 381, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Crismaru, I.; Pantea Stoian, A.; Bratu, O.G.; Gaman, M.A.; Stanescu, A.M.A.; Bacalbasa, N.; Diaconu, C.C. Low-density lipoprotein cholesterol lowering treatment: The current approach. Lipids Health Dis. 2020, 19, 85. [Google Scholar] [CrossRef]
- Soffer, D.; Stoekenbroek, R.; Plakogiannis, R. Small interfering ribonucleic acid for cholesterol lowering—Inclisiran: Inclisiran for cholesterol lowering. J. Clin. Lipidol. 2022, 16, 574–582. [Google Scholar] [CrossRef]
- Alshaer, W.; Zureigat, H.; Al Karaki, A.; Al-Kadash, A.; Gharaibeh, L.; Hatmal, M.M.; Aljabali, A.A.A.; Awidi, A. siRNA: Mechanism of action, challenges, and therapeutic approaches. Eur. J. Pharmacol. 2021, 905, 174178. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.L.; et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Stoekenbroek, R.M.; Kallend, D.; Nishikido, T.; Leiter, L.A.; Landmesser, U.; Wright, R.S.; Wijngaard, P.L.J.; Kastelein, J.J.P. Effect of 1 or 2 doses of inclisiran on low-density lipoprotein cholesterol levels: One-year follow-up of the ORION-1 randomized clinical trial. JAMA Cardiol. 2019, 4, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.J.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Lawrence, D.; Friedman, A.; et al. Inclisiran and cardiovascular events: A patient-level analysis of phase III trials. Eur. Heart J. 2023, 44, 129–138. [Google Scholar] [CrossRef]
- US Food & Drug Administration. Drugs@FDA—Leqvio (Inclisiran). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/214012Orig1s000TOC.cfm (accessed on 27 June 2023).
- Wright, R.S.; Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Friedman, A.; et al. Pooled patient-level analysis of inclisiran trials in patients with familial hypercholesterolemia or atherosclerosis. J. Am. Coll. Cardiol. 2021, 77, 1182–1193. [Google Scholar] [CrossRef]
- Ray, K.K.; Troquay, R.P.T.; Visseren, F.L.J.; Leiter, L.A.; Scott Wright, R.; Vikarunnessa, S.; Talloczy, Z.; Zang, X.; Maheux, P.; Lesogor, A.; et al. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): Results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endocrinol. 2023, 11, 109–119. [Google Scholar] [CrossRef]
- Wright, R.S.; Collins, M.G.; Stoekenbroek, R.M.; Robson, R.; Wijngaard, P.L.J.; Landmesser, U.; Leiter, L.A.; Kastelein, J.J.P.; Ray, K.K.; Kallend, D. Effects of renal impairment on the pharmacokinetics, efficacy, and safety of inclisiran: An analysis of the ORION-7 and ORION-1 studies. Mayo Clin. Proc. 2020, 95, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Vrablik, M.; Seifert, B.; Parkhomenko, A.; Banach, M.; Jóźwiak, J.J.; Kiss, R.G.; Gaita, D.; Rašlová, K.; Zachlederova, M.; Bray, S.; et al. Lipid-lowering therapy use in primary and secondary care in Central and East ern Europe: DA VINCI observational study. Atherosclerosis 2021, 334, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Galli, C.; Franceschini, G. Fraudulent (and non fraudulent) fatty acids for human health. Eur. J. Clin. Investig. 1993, 23, 686. [Google Scholar] [CrossRef]
- Ruscica, M.; Banach, M.; Sahebkar, A.; Corsini, A.; Sirtori, C.R. ETC-1002 (Bempedoic acid) for the management of hyperlipidemia: From preclinical studies to phase 3 trials. Expert Opin. Pharmacother. 2019, 20, 791–803. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Newton, R.S.; Day, E.A.; Ford, R.J.; Lhotak, S.; Austin, R.C.; Birch, C.M.; Smith, B.K.; Filippov, S.; Groot, P.H.E.; et al. Liver-specific ATP citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 2016, 7, 13457. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Filippov, S.; Srivastava, R.A.; Hanselman, J.C.; Bradshaw, C.D.; Hurley, T.R.; Cramer, C.T.; Spahr, M.A.; Brant, A.F.; Houghton, J.L.; et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J. Lipid. Res. 2013, 54, 134–151. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Bays, H.; Catapano, A.L.; Goldberg, A.; Ray, K.K.; Saseen, J.J. Role of bempedoic acid in clinical practice. Cardiovasc. Drugs Ther. 2021, 35, 853–864. [Google Scholar] [CrossRef]
- Amore, B.M.; Sasiela, W.J.; Ries, D.K.; Tresh, P.; Emery, M.G. Pharma cokinetics of bempedoic acid in patients with renal impairment. Clin. Transl. Sci. 2022, 15, 789–798. [Google Scholar] [CrossRef]
- Nilemdo. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/nilemdo (accessed on 19 December 2023).
- Nustendi. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/nustendi#product-information-section] (accessed on 19 December 2023).
- Ballantyne, C.M.; Banach, M.; Mancini, G.B.J.; Lepor, N.E.; Hanselman, J.C.; Zhao, X.; Leiter, L.A. Efficacy and safety of bempedoic acid added to ezetimibe in statin-intolerant patients with hypercholesterolemia: A randomized, placebo controlled study. Atherosclerosis 2018, 277, 195–203. [Google Scholar] [CrossRef]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.C.; Leiter, L.A.; Stroes, E.S.G.; Baum, S.J.; Hanselman, J.C.; Bloedon, L.T.; Lalwani, N.D.; Patel, P.M.; Zhao, X.; Duell, P.B. Effect of bempe doic acid vs placebo added to maximally tolerated statins on low-density lipoprotein cholesterol in patients at high risk for cardiovascular disease: The CLEAR Wisdom Randomized Clini cal Trial. JAMA 2019, 322, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Banach, M.; Mancini, G.B.J.; Gaudet, D.; Bloedon, L.T.; Sterling, L.R.; Kelly, S.; Stroes, E.S.G. Efficacy and safety of bempedoic acid in patients with hypercholesterolemia and statin intolerance. J. Am. Heart Assoc. 2019, 8, e011662. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Duell, P.B.; Gotto, A.M., Jr.; Laufs, U.; Leiter, L.A.; Mancini, G.B.J.; Ray, K.K.; Flaim, J.; Ye, Z.; Catapano, A.L. Association of bempedoic acid administration with atherogenic lipid levels in phase 3 randomized clinical trials of patients with hypercho lesterolemia. JAMA Cardiol. 2020, 5, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Banach, M.; Bays, H.E.; Catapano, A.L.; Laufs, U.; Stroes, E.S.G.; Robinson, P.; Lei, L.; Ray, K.K. Long-term safety and efficacy of bempedoic acid in patients with atherosclerotic cardio vascular disease and/or heterozygous familial hypercholesterolemia (from the CLEAR Harmony Open-Label Extension Study). Am. J. Cardiol. 2022, 174, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Banach, M.; Catapano, A.L.; Duell, P.B.; Gotto, A.M., Jr.; Laufs, U.; Leiter, L.A.; Mancini, G.B.J.; Ray, K.K.; Bloedon, L.T.; et al. Bempedoic acid safety analysis: Pooled data from four phase 3 clinical trials. J. Clin. Lipidol. 2020, 14, 649–659.e646. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, A.; Lupoli, R.; Calcaterra, I.; Poggio, P.; Forte, F.; Spadarella, G.; Ambrosino, P.; Iannuzzo, G.; Di Minno, M.N.D. Efficacy and safety of bempedoic acid in patients with hypercholesterolemia: Systematic review and meta-analysis of randomized controlled trials. J. Am. Heart Assoc. 2020, 9, e016262. [Google Scholar] [CrossRef]
- Dehlin, M.; Jacobsson, L.; Roddy, E. Global epidemiology of gout: Prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 2020, 16, 380–390. [Google Scholar] [CrossRef]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-targeted therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef]
- Berg, K. A new serum type system in man-the Lp system. Acta Pathol. Microbiol. Scand. 1963, 59, 369–382. [Google Scholar] [CrossRef]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef]
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Borén, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur. Heart J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef]
- Varvel, S.; McConnell, J.P.; Tsimikas, S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532 359 patients in the United States. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2239–2245. [Google Scholar] [CrossRef] [PubMed]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bossé, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- van Buuren, F.; Horstkotte, D.; Knabbe, C.; Hinse, D.; Mellwig, K.P. Incidence of elevated lipoprotein (a) levels in a large cohort of patients with cardiovascular disease. Clin. Res. Cardiol. Suppl. 2017, 12, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S. A test in context: Lipoprotein(a): Diagnosis, prognosis, controversies, and emerging therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; López, J.A.G.; Knusel, B.; Gencer, B.; Wang, H.; Wu, Y.; Kassahun, H.; Sabatine, M.S. Study design and rationale for the Olpasiran trials of Cardiovascular Events And lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Study design and rationale for the Olpasiran trials of Cardiovascular Events and lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am. Heart J. 2022, 251, 61–69. [Google Scholar]
- Tsutsumi, K. Lipoprotein lipase and atherosclerosis. Curr. Vasc. Pharmacol. 2003, 1, 11–17. [Google Scholar] [CrossRef]
- Langlois, S.; Deeb, S.; Brunzell, J.D.; Kastelein, J.J.; Hayden, M.R. A major insertion accounts for a significant proportion of mutations underlying human lipoprotein lipase deficiency. Proc. Natl. Acad. Sci. USA 1989, 86, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Abildgaard, S.; Wittrup, H.H.; Steffensen, R.; Jensen, G.; Tybjaerg-Hansen, A. Heterozygous lipoprotein lipase deficiency: Frequency in the general population, effect on plasma lipid levels, and risk of ischemic heart disease. Circulation 1997, 96, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. New insights into angiopoietin-like proteins in lipid metabolism and cardiovascular disease risk. Curr. Opin. Lipidol. 2019, 30, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Koishi, R.; Ando, Y.; Ono, M.; Shimamura, M.; Yasumo, H.; Fujiwara, T.; Horikoshi, H.; Furukawa, H. Angptl3 regulates lipid metabolism in mice. Nat. Genet. 2002, 30, 151–157. [Google Scholar] [CrossRef]
- Adam, R.C.; Mintah, I.J.; Alexa-Braun, C.A.; Shihanian, L.M.; Lee, J.S.; Banerjee, P.; Hamon, S.C.; Kim, H.I.; Cohen, J.C.; Hobbs, H.H.; et al. Angiopoietin-like protein 3 governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. J. Lipid Res. 2020, 61, 1271–1286. [Google Scholar] [CrossRef]
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.P.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.C.; Gipe, D.A.; et al. ELIPSE HoFH Investigators. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 711–720. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Burgess, L.J.; Ebenbichler, C.F.; Baum, S.J.; Stroes, E.S.G.; Ali, S.; Khilla, N.; Hamlin, R.; Pordy, R.; Dong, Y.; et al. Evinacumab in Patients with Refractory Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 2307–2319. [Google Scholar] [CrossRef]
- Gaudet, D.; Karwatowska-Prokopczuk, E.; Baum, S.J.; Hurh, E.; Kingsbury, J.; Bartlett, V.J.; Figueroa, A.L.; Piscitelli, P.; Singleton, W.; Witztum, J.L.; et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur. Heart J. 2020, 41, 3936–3945. [Google Scholar] [CrossRef]
- Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-ionis-announce-discontinuation-vupanorsen (accessed on 19 December 2023).
- Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05217667 (accessed on 19 December 2023).
- Hesler, C.B.; Swenson, T.L.; Tall, A.R. Purification and characterization of a human plasma cholesteryl ester transfer protein. J. Biol. Chem. 1987, 262, 2275–2282. [Google Scholar] [CrossRef] [PubMed]
- Inazu, A.; Brow, M.L.; Hesler, C.B.; Agellon, L.B.; Koizumi, J.; Takata, K.; Maruhama, Y.; Mabuchi, H.; Tall, A.R. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N. Engl. J. Med. 1990, 323, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Rader, D. Trials and Tribulations of CETP Inhibitors. Circ. Res. 2018, 122, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Pafundi, P.C.; Gelso, A.; Bono, V.; Costagliola, C.; Marfella, R.; Sardu, C.; Rinaldi, L.; Galiero, R.; Acierno, C.; et al. High HDL cholesterol: A risk factor for diabetic retinopathy? Findings from NO BLIND study. Diabetes Res. Clin. Pract. 2019, 150, 236–244. [Google Scholar] [CrossRef]
- Hovingh, G.K.; Kastelein, J.J.; Van Deventer, S.J.; Round, P.; Ford, J.; Saleheen, D.; Rader, D.J.; Brewer, H.B.; Barter, P.J. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 2015, 386, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Ditmarsch, M.; Kastelein, J.J.; Rigby, S.P.; Kling, D.; Curcio, D.L.; Alp, N.J.; Davidson, M.H. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: A randomized phase 2 trial. Nat. Med. 2022, 28, 1672–1678. [Google Scholar] [CrossRef]
- Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05202509 (accessed on 19 December 2023).
- Available online: https://classic.clinicaltrials.gov/ct2/show/NCT01252953 (accessed on 19 December 2023).
- Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; Sammons, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J. Lipid Res. 1980, 21, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Reihnér, E.; Rudling, M.; Ståhlberg, D.; Berglund, L.; Ewerth, S.; Björkhem, I.; Einarsson, K.; Angelin, B. Influence of pravastatin, a specific inhibitor of HMG-CoA reductase, on hepatic metabolism of cholesterol. N. Engl. J. Med. 1990, 323, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.Y.; Li, H.; Tang, J.J.; Wang, J.; Luo, J.; Liu, B.; Wang, J.K.; Shi, X.J.; Cui, H.W.; Tang, J.; et al. Discovery of a potent HMG-CoA reductase degrader that eliminates statin-induced reductase accumulation and lowers cholesterol. Nat. Commun. 2018, 9, 5138. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, G.; Morell, A.G. The role of surface carbohydrates in the hepatic recognition and transport of circulating glycoproteins. Adv. Enzymol. Relat. Areas Mol. Biol. 1974, 41, 99–128. [Google Scholar] [PubMed]
- Ashwell, G.; Harford, J. Carbohydrate-specific receptors of the liver. Annu. Rev. Biochem. 1982, 51, 531–554. [Google Scholar] [CrossRef] [PubMed]
- Weigel, P.H.; Yik, J.H. Glycans as endocytosis signals: The cases of the asialoglycoprotein and hyaluronan/chondroitin sulfate receptors. Biochim. Biophys. Acta 2002, 1572, 341–363. [Google Scholar] [CrossRef]
- Hoober, J.K. ASGR1 and Its Enigmatic Relative, CLEC10A. Int. J. Mol. Sci. 2020, 21, 4818. [Google Scholar] [CrossRef]
- Nioi, P.; Sigurdsson, A.; Thorleifsson, G.; Helgason, H.; Agustsdottir, A.B.; Norddahl, G.L.; Helgadottir, A.; Magnusdottir, A.; Jonasdottir, A.; Gretarsdottir, S.; et al. Variant ASGR1 Associated with a Reduced Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 374, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Li, L.L.; Hu, A.; Deng, G.; Wei, J.; Li, Y.F.; Liu, Y.B.; Lu, X.Y.; Qiu, Z.P.; Shi, X.J.; et al. Inhibition of ASGR1 decreases lipid levels by promoting cholesterol excretion. Nature 2022, 608, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Shi, X.; Li, Y.; Xia, B.; Zhou, J.; Du, M.; Xing, X.; Bai, L.; Liu, E.; Alvarez, F.; et al. Deficiency of ASGR1 in pigs recapitulates reduced risk factor for cardiovascular disease in humans. PLoS Genet. 2021, 17, e1009891. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Xu, S.; Weng, J. ASGR1: An emerging therapeutic target in hypercholesterolemia. Signal Transduct. Target. Ther. 2023, 23, 43. [Google Scholar] [CrossRef] [PubMed]
- Janiszewski, M.; Sohn, W.; Su, C.; Hsu, Y.H.; Finger, E.; Kaufman, A. A randomized, placebo-controlled, double-blind, ascending single-dose, phase 1 study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of amg 529, a novel anti-asgr1 monoclonal antibody, in healthy subjects. J. Am. Coll. Cardiol. 2019, 73, 1755. [Google Scholar] [CrossRef]
- Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03170193 (accessed on 22 December 2023).
- Pan, Y.; Zhou, Y.; Wu, H.; Chen, X.; Hu, X.; Zhang, H.; Zhou, Z.; Qiu, Z.; Liao, Y. A Therapeutic Peptide Vaccine Against PCSK9. Sci. Rep. 2017, 7, 12534. [Google Scholar] [CrossRef]
- Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05762276 (accessed on 22 December 2023).
- Dodart, J.; Boyd, J.; Chirinos-Rojas, C.; Lu, H.; Wang, S.; Ding, S.; Thibodeaux, B.; Vroom, M.; Sahni, J.; Ramos, M.M.; et al. Vxx-401, an investigational pcsk9 vaccine for the prevention of cardiovascular disease. J. Am. Coll. Cardiol. 2023, 81, 1636. [Google Scholar] [CrossRef]
- Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05261126 (accessed on 22 December 2023).
- Ballantyne, C.M.; Banka, P.; Mendez, G.; Garcia, R.; Rosenstock, J.; Rodgers, A.; Mendizabal, G.; Mitchel, Y.; Catapano, A.L. Phase 2b Randomized Trial of the Oral PCSK9 Inhibitor MK-0616. J. Am. Coll. Cardiol. 2023, 81, 1553–1564. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-Course CRISPR Base-Editing Therapy Targeting PCSK9 in Nonhuman Primate and Mouse Models. Circulation 2023, 147, 242–253. [Google Scholar] [CrossRef]
- GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 22 December 2023).
- Sasso, F.C.; Pafundi, P.C.; Gelso, A.; Bono, V.; Costagliola, C.; Marfella, R.; Sardu, C.; Rinaldi, L.; Galiero, R.; Acierno, C.; et al. Relationship between albuminuric CKD and diabetic retinopathy in a real-world setting of type 2 diabetes: Findings from No blind study. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Di Francia, R.; Rinaldi, L.; Cillo, M.; Varriale, E.; Facchini, G.; D’Aniello, C.; Marotta, G.; Berretta, M. Antioxidant diet and genotyping as tools for the prevention of liver disease. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 5155–5163. [Google Scholar] [PubMed]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J. Lipid. Res. 2016, 57, 1953–1975. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Name | Target | Phase | LDL-C Reduction | Adverse Reactions | Mechanism | Clinical Use or Perspectives |
|---|---|---|---|---|---|---|
| Statins [12,13,14,15,16] | HMGCR | Approved | 20% to 50% | Rhabdomyolysis | Competitively inhibits HMG-CoA reductase | Primary and secondary prevention of ASCVD Severe hypercholesterolemia |
| Ezetimibe [20,21,22] | NPC1L1 | Approved | 23% | Stomach pain, mucle pain and cramps, asthenia | Inhibits the intestinal absorption of cholesterol by blocking NPC1L1 | Add-on to statin therapy or alone for ASCVD and for severe hypercholesterolemia |
| Mipomersen [35,36,37,38] | ApoB100 mRNA | Approved, FDA only | 26% | Injection-site reactions, flu-like symptoms, ALT elevations | Anti-sense oligonucleotide that prevents the production of apolipoprotein B | HoFH treatment |
| Lomitapide [41,42,43,44,45,46,47,48,49,50] | MTP | Approved | 40% to 50% | Liver toxicity, GI adverse reactions | Blocks MTP protein | HoFH treatment |
| PCSK9i antibody [27,28,29] | PCSK9 | Approved | 47% | Nasopharyngitis, upper respiratory tract infection, back pain, joint pain, flu-like symptoms, injection-site reactions | Inhibits PCSK9, preventing its interaction with cholesterol receptors | HoFH treatment and prevention of ASCVD in association with statins or alone. |
| Bempedoic acid [62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79] | ACL | Approved | 17% to 21% | Hyperuricaemia, pain in arms or legs, anaemia | Blocks adenosine triphosphate citrate lyase | Add-on to statin for HeFH and prevention of ASCVD |
| Inclisiran [51,52,53,54,55,56,57,58,59,60,61] | PCSK9 mRNA | Approved | 50% | Pain and rash at the injection site. | Reduces the production of PCSK9 through gene silencing | Add-on to statin for HeFH and prevention of ASCVD |
| Evinacumab [101,102] | ANGPTL3 | Approved | 47% | Nasopharyngitis, influenza-like illness, dizziness, rhinorrhea, nausea. | Stops ANGPTL3 from blocking vascular lipases that break down fats | HoFH treatment |
| Pelacarsen [80,81,82,83,84,85,86,87,88] | LPA mRNA | Phase 3 | 26% | Flu-like syndrome, liver impairment, kidney damage, thrombocytopenia | Blocks translation of mRNA of the LPA gene | Prevention of ASCVD |
| Olpasiran [90] | Lp(a) | Phase 3 | Lp(a) < 90% | Injection-site pain | Small-interfering RNA that prevents the assembly of Lp(a) | Reduction in Lp(a) |
| Obicetrapib [110] | CETP | Phase 3 | 45% | Nausea, urinary tract infection, headache | Inhibits CETP, which catalyzes the transfer of cholesteryl esters from HDL to LDL and VLDL | Reduction in LDL and apoB and increase in HDL |
| HMARO-ANG3 [99] | ANGPTL3 mRNA | Phase 2 | 44–48% | Headache, respiratory tract infections, local injection-site reactions. | RNAi, which inhibits ANGPTL3 | Treatment of dyslipidemias, familiar hypercholesterolemia, and hypertriglyceridemia. |
| MK-0616 [128,129] | PCSK9 | Phase 2 | 60.9% | Arthralgia, diarrhea, nausea, dyspepsia | Oral PCSK9 inhibitor | Treatment of hypercholesterolemia |
| PCSK9 vaccine (VXX-401) [126,127] | PCSK9 | Phase 1 | 30–50% * | No damage detected * | Induces immune response against PCSK9, blocking it | Treatment of hypercholesterolemia by inducing antibodies against PCSK9 |
| VERVE-101 [131] | PCSK9 gene | Phase 1 | 69% * | Elevations in liver function tests * | Inhibits PCSK9 through a CRISPR base-editing technique | Treatment of HeFH, hypercholesterolemia, and ASCVD. |
| ASGR1i [121,122,123,124] | ASGR1 | One phase 1 study in 2017 | Potential liver toxicity | Increases cholesterol efflux to bile | Treatment of hypercholesterolemia and ASCVD. | |
| HMGCR degrader [114] | HMGCR | No human trial | No damage detected° | Reduces statin-induced HMGCR accumulation | Reduction in cholesterol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mormone, A.; Tortorella, G.; Esposito, F.; Caturano, A.; Marrone, A.; Cozzolino, D.; Galiero, R.; Marfella, R.; Sasso, F.C.; Rinaldi, L. Advances in Pharmacological Approaches for Managing Hypercholesterolemia: A Comprehensive Overview of Novel Treatments. Biomedicines 2024, 12, 432. https://doi.org/10.3390/biomedicines12020432
Mormone A, Tortorella G, Esposito F, Caturano A, Marrone A, Cozzolino D, Galiero R, Marfella R, Sasso FC, Rinaldi L. Advances in Pharmacological Approaches for Managing Hypercholesterolemia: A Comprehensive Overview of Novel Treatments. Biomedicines. 2024; 12(2):432. https://doi.org/10.3390/biomedicines12020432
Chicago/Turabian StyleMormone, Andrea, Giovanni Tortorella, Francesca Esposito, Alfredo Caturano, Aldo Marrone, Domenico Cozzolino, Raffaele Galiero, Raffaele Marfella, Ferdinando Carlo Sasso, and Luca Rinaldi. 2024. "Advances in Pharmacological Approaches for Managing Hypercholesterolemia: A Comprehensive Overview of Novel Treatments" Biomedicines 12, no. 2: 432. https://doi.org/10.3390/biomedicines12020432
APA StyleMormone, A., Tortorella, G., Esposito, F., Caturano, A., Marrone, A., Cozzolino, D., Galiero, R., Marfella, R., Sasso, F. C., & Rinaldi, L. (2024). Advances in Pharmacological Approaches for Managing Hypercholesterolemia: A Comprehensive Overview of Novel Treatments. Biomedicines, 12(2), 432. https://doi.org/10.3390/biomedicines12020432