
Intermediate Repeat Expansion in the ATXN2 Gene as a Risk Factor in the ALS and FTD Spanish Population
 ,
,  , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Population
2.2. Genetic Analysis
Determination of CAG Repeat Expansion Length in ATXN2 and C9orf72
- Standard PCR for ATXN2
- Repeat-primed PCR for ATXN2
2.3. Data Analysis
2.4. Ethics Approval
3. Results
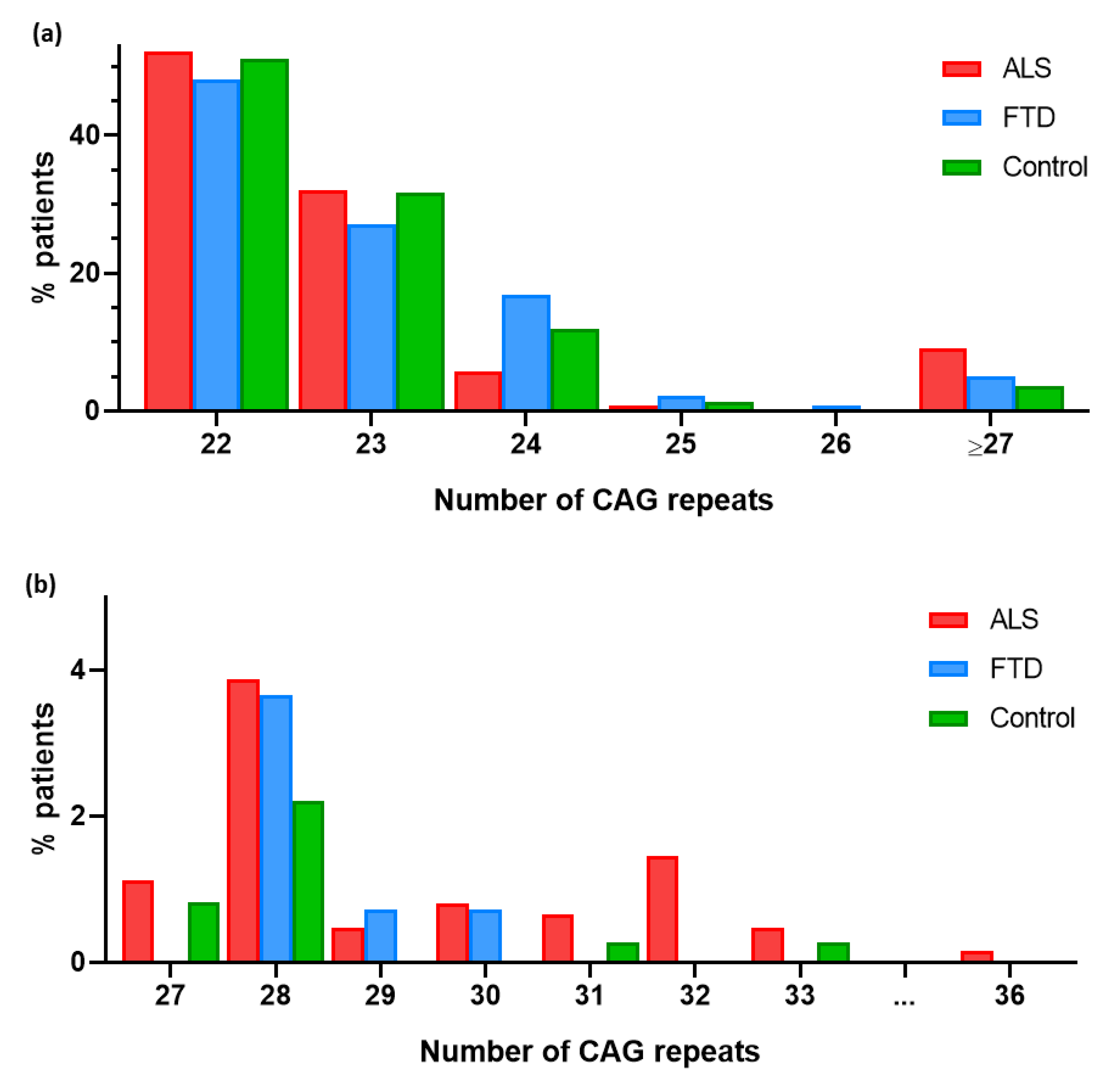
3.1. RE in ATXN2
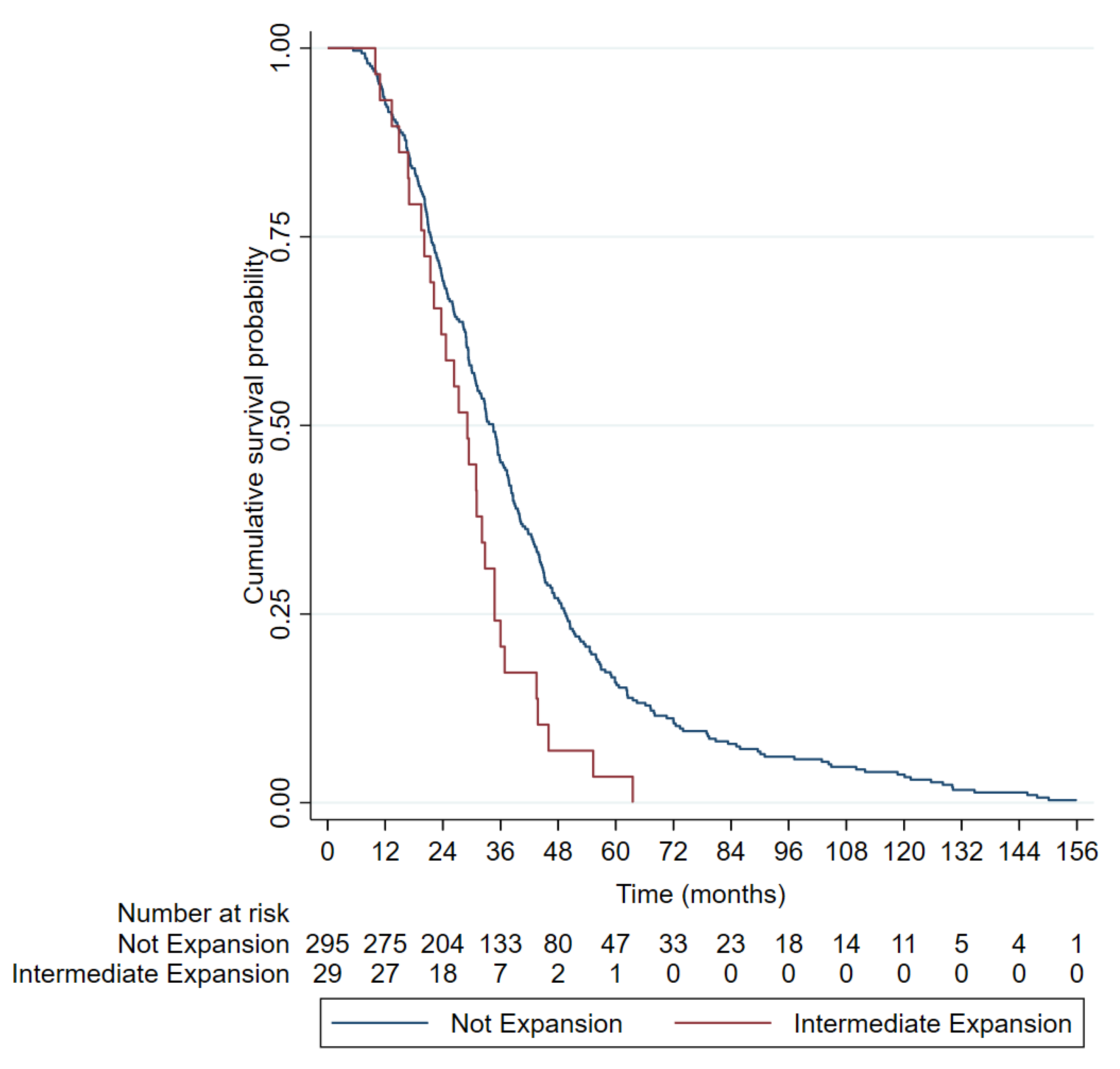
3.2. RE in ATXN2 as a Risk Factor for ALS
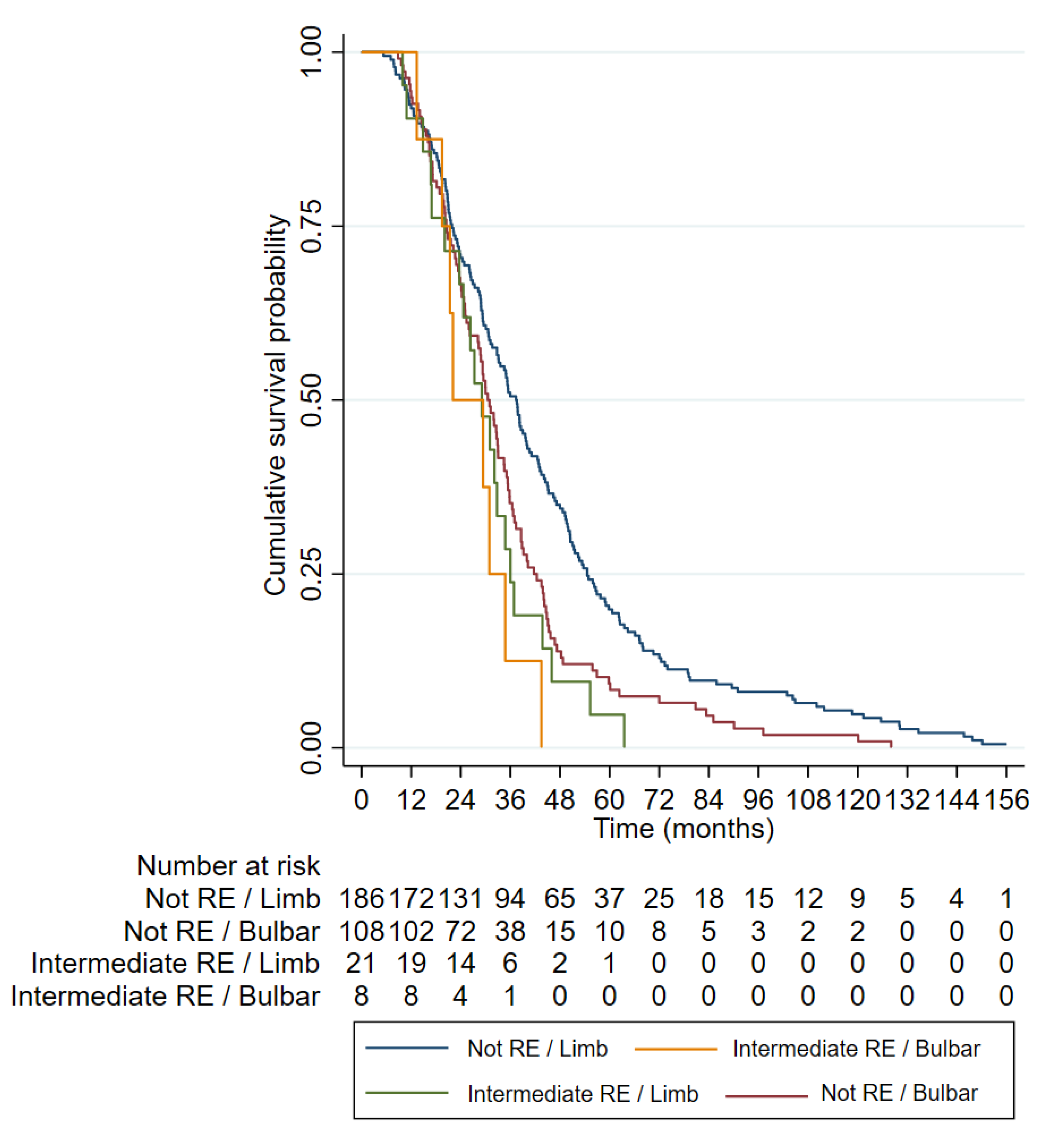
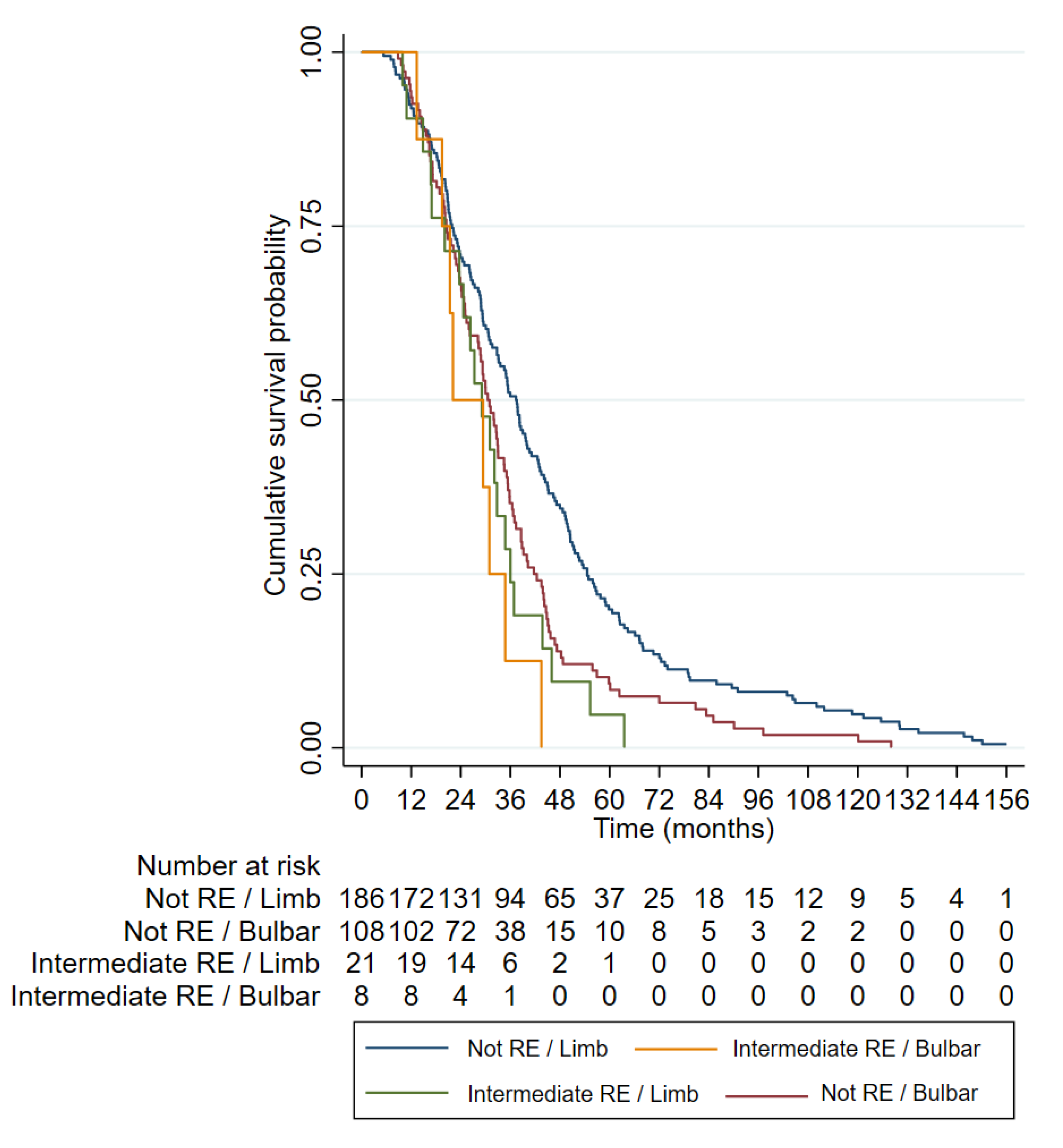
3.3. Clinical Characteristics of ALS Patients with RE in ATXN2
3.4. RE in ATXN2 as a Risk Factor for FTD
3.5. Concomitant RE in ATXN2 and Expansions in C9ORF72
4. Discussion
4.1. RE in ATXN2 Gene in ALS Patients
4.2. Association of Intermediate CAG Repeat Expansion in ATXN2 Gene with FTD
4.3. Concomitant RE in ATXN2 and Expansions in C9orf72
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals Consortium. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar]
- Marangi, G.; Traynor, B.J. Genitic cause of amyotrophic lateral sclerosis: New genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 2015, 1607, 75–93. [Google Scholar]
- Chiò, A.; Mora, G.; Restagno, G.; Brunetti, M.; Ossola, I.; Fuda, G. UNC13A influences survival in Italian ALS patients: A population-based study. Bone 2013, 34. [Google Scholar] [CrossRef]
- Gowland, A.; Opie-Martin, S.; Scott, K.M.; Jones, A.R.; Mehta, P.R.; Batts, C.J.; Ellis, C.M.; Leigh, P.N.; Shaw, C.E.; Sreedharan, J.; et al. Predicting the future of ALS: The impact of demographic change and potential new treatments on the prevalence of ALS in the United Kingdom, 2020–2116. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 264–274. [Google Scholar]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; Van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar]
- Akçimen, F.; Lopez, E.R.; Landers, J.E.; Nath, A.; Chiò, A.; Chia, R.; Traynor, B.J. Amyotrophic lateral sclerosis: Translating genetic discoveries into therapies. Nat. Rev. Genet. 2023, 24, 642–658. [Google Scholar]
- Byrne, S.; Heverin, M.; Elamin, M.; Bede, P.; Lynch, C.; Kenna, K.; MacLaughlin, R.; Walsh, C.; Al Chalabi, A.; Hardiman, O. Aggregation of neurologic and neuropsychiatric disease in ALS kindreds: A population based case controlled cohort study of Familial and Sporadic ALS. Ann. Neurol. 2013, 74, 699–708. [Google Scholar]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Neurosci. 2018, 14, 248–264. [Google Scholar]
- Fang, F.; Ingre, C.; Roos, P.M.; Kamel, F.; Piehl, F. Risk factors for amyotrophic lateral sclerosis. Clin. Epidemiol. 2015, 7, 181–193. [Google Scholar]
- Neumann, M. Frontotemporal lobar degeneration and amyotrophic lateral sclerosis: Molecular similarities and differences. Rev. Neurol. 2013, 169, 793–798. [Google Scholar]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar]
- Imbert, G.; Saudou, F.; Yvert, G.; Devys, D.; Trottier, Y.; Garnier, J.M.; Weber, C.; Mandel, J.L.; Cancel, G.; Abbas, N.; et al. Cloning of the gene for spinooerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 1996, 14, 285–291. [Google Scholar]
- Sanpei, K.; Takano, H.; Igarashi, S.; Sato, T.; Oyake, M.; Sasaki, H.; Wakisaka, A.; Tashiro, K.; Ishida, Y.; Ikeuchi, T.; et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat. Genet. 1996, 14, 277–284. [Google Scholar]
- Lastres-Becker, I.; Rüb, U.; Auburger, G. Spinocerebellar ataxia 2 (SCA2). Cerebellum 2008, 7, 115–124. [Google Scholar]
- Magana, J.J.; Velázquez-Pérez, L.; Cisneros, B. Spinocerebellar ataxia type 2: Clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol. Neurobiol. 2013, 47, 90–104. [Google Scholar]
- Sequeiros, J.; Seneca, S.; Martindale, J. Consensus and controversies in best practices for molecular genetic testing of spinocerebellar ataxias. Eur. J. Hum. Genet. 2010, 18, 1188–1195. [Google Scholar]
- Fan, H.-C.; Ho, L.-I.; Chi, C.-S.; Chen, S.-J.; Peng, G.-S.; Chan, T.-M.; Lin, S.-Z.; Harn, H.-J. Polyglutamine (PolyQ) diseases: Genetics to treatments. Cell Transplant. 2014, 23, 441–458. [Google Scholar]
- Lee, T.; Li, Y.R.; Ingre, C.; Weber, M.; Grehl, T.; Gredal, O.; de Carvalho, M.; Meyer, T.; Tysnes, O.-B.; Auburger, G.; et al. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum. Mol. Genet. 2011, 20, 1697–1700. [Google Scholar]
- Van Langenhove, T.; van der Zee, J.; Engelborghs, S.; Vandenberghe, R.; Santens, P.; Broeck, M.V.D.; Mattheijssens, M.; Peeters, K.; Nuytten, D.; Cras, P.; et al. Ataxin-2 polyQ expansions in FTLD-ALS spectrum disorders in Flanders-Belgian cohorts. Neurobiol. Aging 2012, 33, 1004.e17–1004.e20. [Google Scholar]
- Neuenschwander, A.G.; Thai, K.K.; Figueroa, K.P.; Pulst, S.M. Amyotrophic Lateral Sclerosis Risk for Spinocerebellar Ataxia Type 2 ATXN2 CAG Repeat Alleles: A Meta-analysis. JAMA Neurol. 2014, 71, 1529–1534. [Google Scholar]
- Lahut, S.; Ömür, Ö.; Uyan, Ö.; Agim, Z.S.; Özoğuz, A.; Parman, Y.; Deymeer, F.; Oflazer, P.; Koç, F.; Özçelik, H.; et al. ATXN2 and its neighbouring gene SH2B3 are associated with increased ALS risk in the Turkish population. PLoS ONE 2012, 7, e42956. [Google Scholar]
- Wang, M.-D.; Gomes, J.; Cashman, N.R.; Little, J.; Krewski, D. Intermediate CAG repeat expansion in the ATXN2 gene is a unique genetic risk factor for ALS—A systematic review and meta-analysis of observational studies. PLoS ONE 2014, 9, e105534. [Google Scholar]
- Borghero, G.; Pugliatti, M.; Marrosu, F.; Marrosu, M.G.; Murru, M.R.; Floris, G.; Cannas, A.; Parish, L.D.; Cau, T.B.; Loi, D.; et al. ATXN2 is a modifier of phenotype in ALS patients of Sardinian ancestry. Neurobiol. Aging 2015, 36, 2906.e1–2906.e5. [Google Scholar]
- Rosas, I.; Martinez, C.; Clarimon, J.; Lleo, A.; Illan-Gala, I.; Dols-Icardo, O.; Borroni, B.; Almeida, M.R.; van der Zee, J.; Van Broeckhoven, C.; et al. Role for ATXN1, ATXN2, and HTT intermediate repeats in frontotemporal dementia and Alzheimer’s disease. Neurobiol. Aging 2020, 87, 139.e1–139.e7. [Google Scholar]
- Rubino, E.; Mancini, C.; Boschi, S.; Ferrero, P.; Ferrone, M.; Bianca, S.; Zucca, M.; Orsi, L.; Pinessi, L.; Govone, F.; et al. ATXN2 intermediate repeat expansions influence the clinical phenotype in frontotemporal dementia. Neurobiol. Aging 2019, 73, 231.e7–231.e9. [Google Scholar]
- Fournier, C.; Network, N.-C.N.; Anquetil, V.; Camuzat, A.; Stirati-Buron, S.; Sazdovitch, V.; Molina-Porcel, L.; Turbant, S.; Rinaldi, D.; Sánchez-Valle, R.; et al. Interrupted CAG expansions in ATXN2 gene expand the genetic spectrum of frontotemporal dementias. Acta Neuropathol. Commun. 2018, 6, 41. [Google Scholar]
- Chio, A.; Moglia, C.; Canosa, A.; Manera, U.; Grassano, M.; Vasta, R.; Palumbo, F.; Gallone, S.; Brunetti, M.; Barberis, M.; et al. Exploring the phenotype of Italian patients with ALS with intermediate ATXN2 polyQ repeats. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1216–1220. [Google Scholar]
- Glass, J.D.; Dewan, R.; Ding, J.; Gibbs, J.R.; Dalgard, C.; Keagle, P.J.; Shankaracharya; García-Redondo, A.; Traynor, B.J.; Chia, R.; et al. ATXN2 intermediate expansions in amyotrophic lateral sclerosis. Brain 2022, 145, 2671–2676. [Google Scholar]
- Sproviero, W.; Shatunov, A.; Stahl, D.; Shoai, M.; van Rheenen, W.; Jones, A.R.; Al-Sarraj, S.; Andersen, P.M.; Bonini, N.M.; Conforti, F.L.; et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol. Aging 2017, 51, 178.e1–178.e9. [Google Scholar]
- Shefner, J.M.; Al-Chalabi, A.; Baker, M.R.; Cui, L.-Y.; de Carvalho, M.; Eisen, A.; Grosskreutz, J.; Hardiman, O.; Henderson, R.; Matamala, J.M.; et al. A proposal for new diagnostic criteria for ALS. Clin. Neurophysiol. 2020, 131, 1975–1978. [Google Scholar]
- Lomen-Hoerth, C.; Anderson, T.; Miller, B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002, 59, 1077–1079. [Google Scholar]
- Padovani, A.; Agosti, C.; Premi, E.; Bellelli, G.; Borroni, B. Extrapyramidal symptoms in Frontotemporal Dementia: Prevalence and clinical correlations. Neurosci. Lett. 2007, 422, 39–42. [Google Scholar]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in non-coding region of C9ORF72 causes chromosome 9p-linked frontotemporal dementia and amyotrophic lateral sclerosis. Neuron 2011, 72, 245–256. [Google Scholar]
- Cleary, E.M.; Pal, S.; Azam, T.; Moore, D.J.; Swingler, R.; Gorrie, G.; Stephenson, L.; Colville, S.; Chandran, S.; Porteous, M.; et al. Improved PCR based methods for detecting C9orf72 hexanucleotide repeat expansions. Mol. Cell. Probes 2016, 30, 218–224. [Google Scholar]
- Daoud, H.; Belzil, V.; Martins, S.; Sabbagh, M.; Provencher, P.; Lacomblez, L.; Meininger, V.; Camu, W.; Dupré, N.; Dion, P.A.; et al. Association of Long ATXN2 CAG Repeat Sizes With Increased Risk of Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 739–742. [Google Scholar]
- Gispert, S.; Kurz, A.; Waibel, S.; Bauer, P.; Liepelt, I.; Geisen, C.; Gitler, A.D.; Becker, T.; Weber, M.; Berg, D.; et al. The modulation of Amyotrophic Lateral Sclerosis risk by Ataxin-2 intermediate polyglutamine expansions is a specific effect. Neurobiol. Dis. 2012, 45, 356–361. [Google Scholar]
- Conforti, F.L.; Spataro, R.; Sproviero, W.; Mazzei, R.; Cavalcanti, F.; Condino, F.; Simone, I.L.; Logroscino, G.; Patitucci, A.; Magariello, A.; et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology 2012, 79, 2315–2320. [Google Scholar]
- De Andrade, H.M.T.; Cintra, V.P.; de Albuquerque, M.; Piccinin, C.C.; Bonadia, L.C.; Couteiro, R.E.D.; de Oliveira, D.S.; Claudino, R.; Gonçalves, M.V.M.; Dourado, M.E.T.J.; et al. Intermediate-length CAG repeat in ATXN2 is associated with increased risk for amyotrophic lateral sclerosis in Brazilian patients. Neurobiol. Aging 2018, 69, 292.e15–292.e18. [Google Scholar]
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113. [Google Scholar]
- Lattante, S.; Millecamps, S.; Stevanin, G.; Rivaud-Péchoux, S.; Moigneu, C.; Camuzat, A.; Da Barroca, S.; Mundwiller, E.; Couarch, P.; Salachas, F.; et al. Contribution of ATXN2 intermediary polyQ expansions in a spectrum of neurodegenerative disorders. Neurology 2014, 83, 990–995. [Google Scholar]
- Tan, R.H.; Kril, J.J.; McGinley, C.; Hassani, M.; Masuda-Suzukake, M.; Hasegawa, M.; Mito, R.; Kiernan, M.C.; Halliday, G.M. Cerebellar neuronal loss in amyotrophic lateral sclerosis cases with ATXN2 intermediate repeat expansions. Ann. Neurol. 2016, 79, 295–305. [Google Scholar]
- Yang, Y.; Halliday, G.M.; Kiernan, M.C.; Tan, R.H. TDP-43 levels in the brain tissue of ALS cases with and without C9ORF72 or ATXN2 gene expansions. Neurology 2019, 93, e1748–e1755. [Google Scholar]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar]
- Chiò, A.; Mora, G.; Sabatelli, M.; Caponnetto, C.; Lunetta, C.; Traynor, B.J.; Johnson, J.O.; Nalls, M.A.; Calvo, A.; Moglia, C.; et al. ATNX2 is not a regulatory gene in Italian amyotrophic lateral sclerosis patients with C9ORF72 GGGGCC expansion. Neurobiol. Aging 2016, 39, 218.e5–218.e8. [Google Scholar]
- Van Blitterswijk, M.; Mullen, B.; Heckman, M.G.; Baker, M.C.; DeJesus-Hernandez, M.; Brown, P.H.; Murray, M.E.; Hsiung, G.Y.R.; Stewart, H.; Karydas, A.M.; et al. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol. Aging 2014, 35, 2421.e13–2421.e17. [Google Scholar]
- Van Wijk, I.F.; Van Eijk, R.P.; Van Boxmeer, L.; Westeneng, H.-J.; Van Es, M.A.; Van Rheenen, W.; Berg, L.H.V.D.; Eijkemans, M.J.; Veldink, J.H. Assessment of risk of ALS conferred by the GGGGCC hexanucleotide repeat expansion in C9orf72 among first-degree relatives of patients with ALS carrying the repeat expansion. Amyotroph. Lateral Scler. Front. Degener. 2023, 25, 188–196. [Google Scholar]
- Sellier, C.; Campanari, M.L.; Julie Corbier, C.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | ALS | FTD | Controls |
|---|---|---|---|
| Comorbidity, n (%) | 87 (14.03%) | 18 (13.14%) | - |
| Male sex, n (%) | 337 (54.35%) | 81 (59.12%) | 175 (48.34%) |
| Limb onset, n (%) | 415 (66.94%) | - | - |
| Familial, n (%) | 142 (22.90%) | 38 (27.74%) | - |
| ATXN2 ≥ 27 CAG repeats, n (%) | 56 (9.03%) | 7 (5.11%) | 13 (3.59%) |
| C9orfF72 expansion, n (%) | 60 (9.68%) | 7 (5.11%) | 0 (0.00%) |
| ATXN2 ≥ 27 CAG repeats and C9orfF72 expansion, n (%) | 6 (10.0%) | 1 (14.3%) | 0 (0.00%) |
| Age of onset (years, mean ± SD (range) | 59.87 ± 13.45 (16.4–95.6) | 63.16 ± 9.85 (39.3–81.8) | 56.32 ± 15.64 (18.5–85.8) * |
| Survival (months, mean ± SD) (range) | 39.67 ± 27.30 (5.3–156.1) | 99.32 ± 51.90 (21.8–216.0) | - |
| Total ALS samples | 620 | 137 | 362 |
| CAG Repeats | ALS | Control | p-Value a | Odds Ratio b |
|---|---|---|---|---|
| ≥27 CAG repeats | 56 (9.03%) | 13 (3.59%) | 0.0006 | 2.666 [1.471–4.882] |
| ≥28 CAG repeats | 49 (7.90%) | 10 (2.76%) | 0.0005 | 3.021 [1.547–6.309] |
| ≥29 CAG repeats | 25 (4.03%) | 2 (0.55%) | 0.0005 | 7.563 [2.098–32.550] |
| ALS | ||||||
|---|---|---|---|---|---|---|
| <27 | ≥27 | <28 | ≥28 | <29 | ≥29 | |
| Mean age of onset (years) | 59.95 | 59.16 | 59.96 | 58.66 | 59.81 | 61.35 |
| Mean survival (months) | 40.69 | 29.23 | 40.47 | 29.68 | 39.90 | 33.66 |
| C9orf72 + (n) | 54 (9.57%) | 6 (10.71%) | 54 (9.46%) | 6 (12.24%) | 58 (9.75%) | 2 (8%) |
| Other genes + (n) | 94 (16.66%) | 10 (18.52%) | 94 (16.46%) | 10 (20.41%) | 100 (16.81%) | 4 (16%) |
| Male/female (n/n; ratio) | 309/254 (1.217) | 28/28 (1) | 310/260 (1.192) | 27/22 (1.227) | 324/270 (1.2) | 13/12 (1.083) |
| Limb/bulbar (n/n; ratio) | 374/176 (2.125) | 41/11 (3.727) | 378/179 (2.111) | 37/8 (4.625) | 396/184 (1.467) | 29/3 (6.333) |
| Family history (+/−; %) | 122/442 (21.63%) | 20/36 (35.71%) | 122/449 (21.37%) | 20/29 (40.28%) | 131/464 (22.02%) | 11/14 (44.00%) |
| FTD (yes/no; %) | 79/485 (14.01%) | 7/49 (12.5%) | 79/492 (13.84%) | 7/42 (14.29%) | 83/512 (13.95%) | 3/22 (12.00%) |
| Univariable | Multivariable | |||||
|---|---|---|---|---|---|---|
| Variable | HR | CI (95%) | p-Value | HR | CI (95%) | p-Value |
| ≥27 repeats | 1.74 | 1.181–2.563 | 0.005 | 1.782 | 1.209–2.628 | 0.004 |
| Bulbar onset | 1.415 | 0.538–0.856 | 0.001 | 1.400 | 1.105–1.775 | 0.004 |
| Age of onset | 1.016 | 1.007–1.026 | 0.001 | 1.015 | 1.005–1.824 | 0.003 |
| FTD | 0.992 | 0.7748–1.270 | 0.949 | - | - | - |
| Female sex | 1.253 | 1.003–1.565 | 0.046 | - | - | - |
| CAG Repeats | FTD | Control | p-Value a | Odds Ratio b |
|---|---|---|---|---|
| ≥27 CAG repeats | 7 (5.11%) | 13 (3.59%) | 0.220 | 1.446 [0.558–3.574] |
| ≥28 CAG repeats | 7 (5.11%) | 10 (2.76%) | 0.099 | 1.895 [0.7295–5.241] |
| ≥29 CAG repeats | 2 (1.46%) | 2 (0.55%) | 0.155 | 2.667 [0.4134–17.12] |
| Study | CAG Repeat Number | p-Value a | Odds Ratio b |
|---|---|---|---|
| North America [15] | 27–33 | 0.000036 | 2.8 [1.54–5.12] |
| Europe [22] | >30 | 0.0062 | - |
| France and Canada [39] | ≥29 | 0.00024 | 5.5 [1.9–15.9] |
| Europe [40] | >30 | 0.004 | 5.74 [4.26–7.22] |
| Flanders [23] | 27–33 | 0.012 | - |
| South Italy [41] | ≥28 | 0.001 | 5.832 [1.71–9.78] |
| Turkey [25] | >30 | 0.01721 | - |
| Meta-analysis [24] | 29–33 | - | >1 |
| Europe and North America [26] | 30–33 | 0.0001 | 4.44 [2.91–6.76] |
| Sardinia [27] | ≥31 | 0.0001 | - |
| Brazil [42] | ≥26 | 0.005 | 2.56 [1.29–5.08] |
| Europe [32] | ≥31 | 9.50 × 10−7 | 6.93 [3.19–15.02] |
| Spain (present study) | ≥27 | 0.0013 | 2.666 [1.471–4.882] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrego-Hernández, D.; Vázquez-Costa, J.F.; Domínguez-Rubio, R.; Expósito-Blázquez, L.; Aller, E.; Padró-Miquel, A.; García-Casanova, P.; Colomina, M.J.; Martín-Arriscado, C.; Osta, R.; et al. Intermediate Repeat Expansion in the ATXN2 Gene as a Risk Factor in the ALS and FTD Spanish Population. Biomedicines 2024, 12, 356. https://doi.org/10.3390/biomedicines12020356
Borrego-Hernández D, Vázquez-Costa JF, Domínguez-Rubio R, Expósito-Blázquez L, Aller E, Padró-Miquel A, García-Casanova P, Colomina MJ, Martín-Arriscado C, Osta R, et al. Intermediate Repeat Expansion in the ATXN2 Gene as a Risk Factor in the ALS and FTD Spanish Population. Biomedicines. 2024; 12(2):356. https://doi.org/10.3390/biomedicines12020356
Chicago/Turabian StyleBorrego-Hernández, Daniel, Juan Francisco Vázquez-Costa, Raúl Domínguez-Rubio, Laura Expósito-Blázquez, Elena Aller, Ariadna Padró-Miquel, Pilar García-Casanova, María J. Colomina, Cristina Martín-Arriscado, Rosario Osta, and et al. 2024. "Intermediate Repeat Expansion in the ATXN2 Gene as a Risk Factor in the ALS and FTD Spanish Population" Biomedicines 12, no. 2: 356. https://doi.org/10.3390/biomedicines12020356
APA StyleBorrego-Hernández, D., Vázquez-Costa, J. F., Domínguez-Rubio, R., Expósito-Blázquez, L., Aller, E., Padró-Miquel, A., García-Casanova, P., Colomina, M. J., Martín-Arriscado, C., Osta, R., Cordero-Vázquez, P., Esteban-Pérez, J., Povedano-Panadés, M., & García-Redondo, A. (2024). Intermediate Repeat Expansion in the ATXN2 Gene as a Risk Factor in the ALS and FTD Spanish Population. Biomedicines, 12(2), 356. https://doi.org/10.3390/biomedicines12020356