
Sex and Gender Differences in Iron Chelation





Abstract
1. Introduction
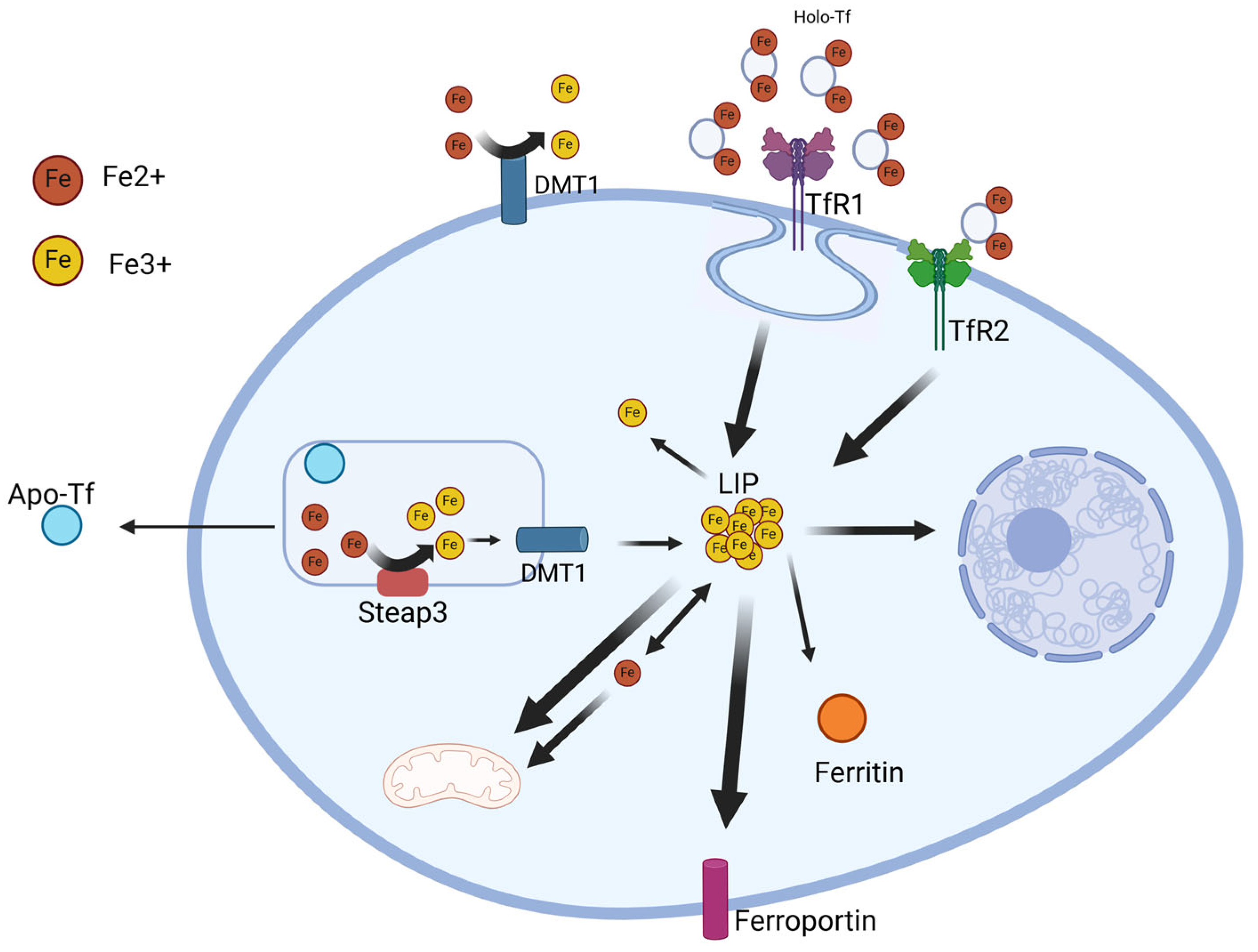
2. Iron Metabolism
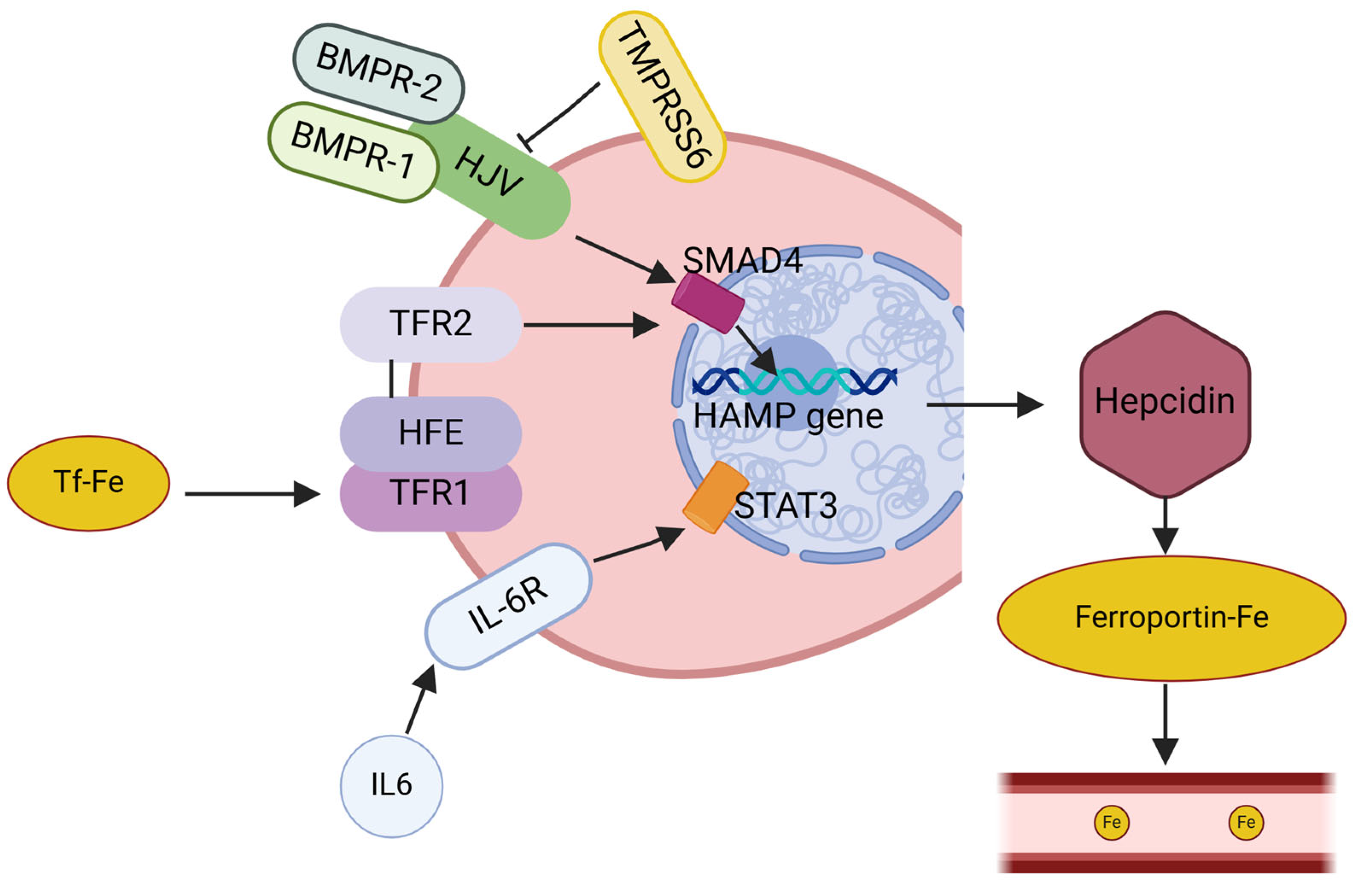
Hepcidin
3. Iron Level Measurement
3.1. Liver Biopsy
3.2. Ultrasonic Elastography
3.3. Superconducting Quantum Interference Devices
3.4. Magnetic Resonance Imaging
3.5. Serum Iron and Ferritin
3.6. Hepcidin Level Measurements
4. Animal Models
5. Chelation Therapy
5.1. Deferoxamine
5.2. Deferiprone
5.3. Deferasirox
5.4. Luspatercept
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Anderson, G.D. Gender differences in pharmacological response. Int. Rev. Neurobiol. 2008, 83, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Franconi, F.; Brunelleschi, S.; Steardo, L.; Cuomo, V. Gender differences in drug responses. Pharmacol. Res. 2007, 55, 81–95. [Google Scholar] [CrossRef]
- Franconi, F.; Carru, C.; Malorni, W.; Vella, S.; Mercuro, G. The effect of sex/gender on cardiovascular pharmacology. Curr. Pharm. Des. 2011, 17, 1095–1107. [Google Scholar] [CrossRef]
- Franconi, F.; Carru, C.; Spoletini, I.; Malorni, W.; Vella, S.; Mercuro, G.; Deidda, M.; Rosano, G. A GENS-based approach to cardiovascular pharmacology: Impact on metabolism, pharmacokinetics and pharmacodynamics. Ther. Deliv. 2011, 2, 1437–1453. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.; Aweeka, F.; Greenblatt, R.M.; Blaschke, T.F. Sex differences in pharmacokinetics and pharmacodynamics. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 499–523. [Google Scholar] [CrossRef]
- Barus, R.; Bergeron, S.; Chen, Y.; Gautier, S. Sex differences: From preclinical pharmacology to clinical pharmacology. Therapie 2023, 78, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Delahousse, J.; Wagner, A.D.; Borchmann, S.; Adjei, A.A.; Haanen, J.; Burgers, F.; Letsch, A.; Quaas, A.; Oertelt-Prigione, S.; Oezdemir, B.C.; et al. Sex differences in the pharmacokinetics of anticancer drugs: A systematic review. ESMO Open 2024, 9, 104002. [Google Scholar] [CrossRef] [PubMed]
- Glezerman, M. Gender and Sex Aspects in Pharmacology. Harefuah 2024, 163, 457–461. [Google Scholar]
- Brøsen, K. Sex differences in pharmacology. Ugeskr. Laeger 2007, 169, 2408–2411. [Google Scholar] [PubMed]
- Schwartz, J.B. The current state of knowledge on age, sex, and their interactions on clinical pharmacology. Clin. Pharmacol. Ther. 2007, 82, 87–96. [Google Scholar] [CrossRef]
- Schwartz, J.B. The influence of sex on pharmacokinetics. Clin. Pharmacokinet. 2003, 42, 107–121. [Google Scholar] [CrossRef]
- Harris, R.Z.; Benet, L.Z.; Schwartz, J.B. Gender effects in pharmacokinetics and pharmacodynamics. Drugs 1995, 50, 222–239. [Google Scholar] [CrossRef]
- Anthony, M.; Berg, M.J. Biologic and molecular mechanisms for sex differences in pharmacokinetics, pharmacodynamics, and pharmacogenetics: Part II. J. Womens Health Gend. Based Med. 2002, 11, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Soldin, O.P.; Mattison, D.R. Sex differences in pharmacokinetics and pharmacodynamics. Clin. Pharmacokinet. 2009, 48, 143–157. [Google Scholar] [CrossRef]
- Allegra, S.; Chiara, F.; Di Grazia, D.; Gaspari, M.; De Francia, S. Evaluation of Sex Differences in Preclinical Pharmacology Research: How Far Is Left to Go? Pharmaceuticals 2023, 16, 786. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Girelli, D. The changing landscape of iron deficiency. Mol. Asp. Med. 2020, 75, 100861. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Pantopoulos, K. Disorders associated with systemic or local iron overload: From pathophysiology to clinical practice. Metallomics 2011, 3, 971–986. [Google Scholar] [CrossRef]
- Olynyk, J.K.; Ramm, G.A. Hemochromatosis. N. Engl. J. Med. 2022, 387, 2159–2170. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2024, 25, 133–155. [Google Scholar] [CrossRef] [PubMed]
- Skolmowska, D.; Glabska, D. Analysis of Heme and Non-Heme Iron Intake and Iron Dietary Sources in Adolescent Menstruating Females in a National Polish Sample. Nutrients 2019, 11, 1049. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Bae, D.H.; Merlot, A.M.; Sahni, S.; Richardson, D.R. Duodenal cytochrome b (DCYTB) in iron metabolism: An update on function and regulation. Nutrients 2015, 7, 2274–2296. [Google Scholar] [CrossRef] [PubMed]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell 2014, 5, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.; Manceau, H.; Karim, Z. Iron metabolism and the role of the iron-regulating hormone hepcidin in health and disease. Presse Med. 2017, 46, e272–e278. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Hypoferremia of inflammation: Innate host defense against infections. Blood Cells Mol. Dis. 2024, 104, 102777. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Pelucchi, S.; Mariani, R. Inherited iron overload disorders. Transl. Gastroenterol. Hepatol. 2020, 5, 25. [Google Scholar] [CrossRef]
- Papanikolaou, G.; Pantopoulos, K. Systemic iron homeostasis and erythropoiesis. IUBMB Life 2017, 69, 399–413. [Google Scholar] [CrossRef]
- Musallam, K.M.; Motta, I.; Salvatori, M.; Fraquelli, M.; Marcon, A.; Taher, A.T.; Cappellini, M.D. Longitudinal changes in serum ferritin levels correlate with measures of hepatic stiffness in transfusion-independent patients with β-thalassemia intermedia. Blood Cells Mol. Dis. 2012, 49, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Richardson, K.J.; McNamee, A.P.; Simmonds, M.J. Haemochromatosis: Pathophysiology and the red blood cell. Clin. Hemorheol. Microcirc. 2018, 69, 295–304. [Google Scholar] [CrossRef]
- Cournane, S.; Browne, J.E.; Fagan, A.J. The effects of fatty deposits on the accuracy of the Fibroscan® liver transient elastography ultrasound system. Phys. Med. Biol. 2012, 57, 3901–3914. [Google Scholar] [CrossRef]
- Sandrin, L.; Fourquet, B.; Hasquenoph, J.M.; Yon, S.; Fournier, C.; Mal, F.; Christidis, C.; Ziol, M.; Poulet, B.; Kazemi, F.; et al. Transient elastography: A new noninvasive method for assessment of hepatic fibrosis. Ultrasound Med. Biol. 2003, 29, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Poynard, T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology 1996, 24, 289–293. [Google Scholar] [CrossRef]
- Castera, L.; Forns, X.; Alberti, A. Non-invasive evaluation of liver fibrosis using transient elastography. J. Hepatol. 2008, 48, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Tsochatzis, E.A.; Gurusamy, K.S.; Ntaoula, S.; Cholongitas, E.; Davidson, B.R.; Burroughs, A.K. Elastography for the diagnosis of severity of fibrosis in chronic liver disease: A meta-analysis of diagnostic accuracy. J. Hepatol. 2011, 54, 650–659. [Google Scholar] [CrossRef]
- Sharma, S.D.; Fischer, R.; Schoennagel, B.P.; Nielsen, P.; Kooijman, H.; Yamamura, J.; Adam, G.; Bannas, P.; Hernando, D.; Reeder, S.B. MRI-based quantitative susceptibility mapping (QSM) and R2* mapping of liver iron overload: Comparison with SQUID-based biomagnetic liver susceptometry. Magn. Reson. Med. 2017, 78, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.C. Guidelines for quantifying iron overload. Hematol. Am. Soc. Hematol. Educ. Program. 2014, 2014, 210–215. [Google Scholar] [CrossRef]
- Fovargue, D.; Nordsletten, D.; Sinkus, R. Stiffness reconstruction methods for MR elastography. NMR Biomed. 2018, 31, e3935. [Google Scholar] [CrossRef]
- Pennell, D.J.; Porter, J.B.; Piga, A.; Lai, Y.R.; El-Beshlawy, A.; Elalfy, M.; Yesilipek, A.; Kilinç, Y.; Habr, D.; Musallam, K.M.; et al. Sustained improvements in myocardial T2* over 2 years in severely iron-overloaded patients with beta thalassemia major treated with deferasirox or deferoxamine. Am. J. Hematol. 2015, 90, 91–96. [Google Scholar] [CrossRef]
- Anderson, L.J.; Holden, S.; Davis, B.; Prescott, E.; Charrier, C.C.; Bunce, N.H.; Firmin, D.N.; Wonke, B.; Porter, J.; Walker, J.M.; et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur. Heart J. 2001, 22, 2171–2179. [Google Scholar] [CrossRef]
- Wood, J.C.; Enriquez, C.; Ghugre, N.; Tyzka, J.M.; Carson, S.; Nelson, M.D.; Coates, T.D. MRI R2 and R2* mapping accurately estimates hepatic iron concentration in transfusion-dependent thalassemia and sickle cell disease patients. Blood 2005, 106, 1460–1465. [Google Scholar] [CrossRef]
- Merlo, F.; Groothof, D.; Khatami, F.; Ahanchi, N.S.; Wehrli, F.; Bakker, S.J.L.; Eisenga, M.F.; Muka, T. Changes in Iron Status Biomarkers with Advancing Age According to Sex and Menopause: A Population-Based Study. J. Clin. Med. 2023, 12, 5338. [Google Scholar] [CrossRef]
- Nicolas, G.; Viatte, L.; Bennoun, M.; Beaumont, C.; Kahn, A.; Vaulont, S. Hepcidin, a new iron regulatory peptide. Blood Cells Mol. Dis. 2002, 29, 327–335. [Google Scholar] [CrossRef]
- Roetto, A.; Papanikolaou, G.; Politou, M.; Alberti, F.; Girelli, D.; Christakis, J.; Loukopoulos, D.; Camaschella, C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 2003, 33, 21–22. [Google Scholar] [CrossRef]
- Ganz, T.; Olbina, G.; Girelli, D.; Nemeth, E.; Westerman, M. Immunoassay for human serum hepcidin. Blood 2008, 112, 4292–4297. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A.; Valentino, P.; Daraio, F.; Porporato, P.; Gramaglia, E.; Carturan, S.; Gottardi, E.; Camaschella, C.; Roetto, A. Hepatic expression of hemochromatosis genes in two mouse strains after phlebotomy and iron overload. Haematologica 2005, 90, 1161–1167. [Google Scholar]
- Galesloot, T.E.; Vermeulen, S.H.; Geurts-Moespot, A.J.; Klaver, S.M.; Kroot, J.J.; van Tienoven, D.; Wetzels, J.F.; Kiemeney, L.A.; Sweep, F.C.; den Heijer, M.; et al. Serum hepcidin: Reference ranges and biochemical correlates in the general population. Blood 2011, 117, e218–225. [Google Scholar] [CrossRef]
- Dos Santos, L.; Bertoli, S.R.; Ávila, R.A.; Marques, V.B. Iron overload, oxidative stress and vascular dysfunction: Evidences from clinical studies and animal models. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130172. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.E.; Feng, Q.; Britton, R.S. Knockout mouse models of iron homeostasis. Annu. Rev. Nutr. 2011, 31, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, L.C.; Nicolosi, R.J.; Renaud, M.M.; Finger, J.; Hegsted, M.; Peter, H.; Nathan, D.G. A non-human primate model for the study of oral iron chelators. Br. J. Haematol. 1989, 72, 456–461. [Google Scholar] [CrossRef]
- Nasrallah, G.K.; Younes, N.N.; Baji, M.H.; Shraim, A.M.; Mustafa, I. Zebrafish larvae as a model to demonstrate secondary iron overload. Eur. J. Haematol. 2018, 100, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Pullarkat, V. Deferasirox: Appraisal of safety and efficacy in long-term therapy. J. Blood Med. 2013, 4, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.R.; Liu, R.R.; Li, C.F.; Huang, S.L.; Li, Q.; Habr, D.; Martin, N.; Shen, Z.X. Efficacy of Deferasirox for the treatment of iron overload in Chinese thalassaemia major patients: Results from a prospective, open-label, multicentre clinical trial. Transfus. Med. 2013, 23, 389–396. [Google Scholar] [CrossRef]
- Naderi, M.; Sadeghi-Bojd, S.; Valeshabad, A.K.; Jahantigh, A.; Alizadeh, S.; Dorgalaleh, A.; Tabibian, S.; Bamedi, T. A prospective study of tubular dysfunction in pediatric patients with Beta thalassemia major receiving deferasirox. Pediatr. Hematol. Oncol. 2013, 30, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.C.; Jeffrey, G.; Ryan, J. Haemochromatosis. Lancet 2023, 401, 1811–1821. [Google Scholar] [CrossRef]
- Cappellini, M.D. Exjade(R) (deferasirox, ICL670) in the treatment of chronic iron overload associated with blood transfusion. Ther. Clin. Risk Manag. 2007, 3, 291–299. [Google Scholar] [CrossRef]
- Neufeld, E.J. Oral chelators deferasirox and deferiprone for transfusional iron overload in thalassemia major: New data, new questions. Blood 2006, 107, 3436–3441. [Google Scholar] [CrossRef]
- Galanello, R.; Origa, R. Beta-thalassemia. Orphanet J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.J.; Macklin, E.A.; Neufeld, E.J.; Cohen, A.R.; Network, T.C.R. Complications of beta-thalassemia major in North America. Blood 2004, 104, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Meng, Y.; Le, J.; Sun, Y.; Dian, Y.; Yao, L.; Xiong, Y.; Zeng, F.; Chen, X.; Deng, G. Ferroptosis: Mechanisms and therapeutic targets. MedComm (2020) 2024, 5, e70010. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.L.; Ding, S.Y.; Du, X.Z.; Wang, J.H.; Li, X.L. Ferroptosis-A Novel Mechanism With Multifaceted Actions on Stroke. Front. Neurol. 2022, 13, 881809. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Valenzuela, J.P.; Ward, R.; Abdelbary, M.; Dong, G.; Fagan, S.C.; Ergul, A. Post-stroke neovascularization and functional outcomes differ in diabetes depending on severity of injury and sex: Potential link to hemorrhagic transformation. Exp. Neurol. 2019, 311, 106–114. [Google Scholar] [CrossRef]
- Abdul, Y.; Li, W.; Ward, R.; Abdelsaid, M.; Hafez, S.; Dong, G.; Jamil, S.; Wolf, V.; Johnson, M.H.; Fagan, S.C.; et al. Deferoxamine Treatment Prevents Post-Stroke Vasoregression and Neurovascular Unit Remodeling Leading to Improved Functional Outcomes in Type 2 Male Diabetic Rats: Role of Endothelial Ferroptosis. Transl. Stroke Res. 2021, 12, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Abdul, Y.; Chandran, R.; Jamil, S.; Ward, R.A.; Abdelsaid, M.; Dong, G.; Fagan, S.C.; Ergul, A. Deferoxamine prevents poststroke memory impairment in female diabetic rats: Potential links to hemorrhagic transformation and ferroptosis. Am. J. Physiol. Heart Circ. Physiol. 2023, 324, H212–H225. [Google Scholar] [CrossRef]
- Sakamuri, S.S.V.P.; Sure, V.N.; Katakam, P.V.G. Iron chelation therapy to prevent poststroke cognitive impairments: Role of diabetes and sex. Am. J. Physiol. Heart Circ. Physiol. 2023, 324, H210–H211. [Google Scholar] [CrossRef] [PubMed]
- Pennell, D.J.; Berdoukas, V.; Karagiorga, M.; Ladis, V.; Piga, A.; Aessopos, A.; Gotsis, E.D.; Tanner, M.A.; Smith, G.C.; Westwood, M.A.; et al. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood 2006, 107, 3738–3744. [Google Scholar] [CrossRef]
- Taher, A.; Sheikh-Taha, M.; Sharara, A.; Inati, A.; Koussa, S.; Ellis, G.; Dhillon, A.P.; Hoffbrand, A.V. Safety and effectiveness of 100 mg/kg/day deferiprone in patients with thalassemia major: A two-year study. Acta Haematol. 2005, 114, 146–149. [Google Scholar] [CrossRef]
- Tricta, F.; Uetrecht, J.; Galanello, R.; Connelly, J.; Rozova, A.; Spino, M.; Palmblad, J. Deferiprone-induced agranulocytosis: 20 years of clinical observations. Am. J. Hematol. 2016, 91, 1026–1031. [Google Scholar] [CrossRef]
- Bellanti, F.; Danhof, M.; Della Pasqua, O. Population pharmacokinetics of deferiprone in healthy subjects. Br. J. Clin. Pharmacol. 2014, 78, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Farmaki, K.; Tzoumari, I.; Pappa, C.; Chouliaras, G.; Berdoukas, V. Normalisation of total body iron load with very intensive combined chelation reverses cardiac and endocrine complications of thalassaemia major. Br. J. Haematol. 2010, 148, 466–475. [Google Scholar] [CrossRef]
- Farmaki, K.; Tzoumari, I.; Pappa, C. Oral chelators in transfusion-dependent thalassemia major patients may prevent or reverse iron overload complications. Blood Cells Mol. Dis. 2011, 47, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R. Deferiprone in the treatment of transfusion-dependent thalassemia: A review and perspective. Ther. Clin. Risk Manag. 2007, 3, 795–805. [Google Scholar]
- Pharmaceuticals, N. Exjade (Deferasirox) Tablets for Oral Suspension [Prescribing Information]. Available online: https://www.novartis.com/us-en/sites/novartis_us/files/exjade.pdf (accessed on 1 October 2024).
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006, 107, 3455–3462. [Google Scholar] [CrossRef]
- Epar, E. Exjade (Deferasirox) Prescribing Information. Novartis Pharmaceuticals Corporation. Available online: https://www.ema.europa.eu/en/documents/product-information/exjade-epar-product-information_en.pdf (accessed on 22 October 2024).
- Vichinsky, E.; Onyekwere, O.; Porter, J.; Swerdlow, P.; Eckman, J.; Lane, P.; Files, B.; Hassell, K.; Kelly, P.; Wilson, F.; et al. A randomised comparison of deferasirox versus deferoxamine for the treatment of transfusional iron overload in sickle cell disease. Br. J. Haematol. 2007, 136, 501–508. [Google Scholar] [CrossRef]
- Bruin, G.J.; Faller, T.; Wiegand, H.; Schweitzer, A.; Nick, H.; Schneider, J.; Boernsen, K.O.; Waldmeier, F. Pharmacokinetics, distribution, metabolism, and excretion of deferasirox and its iron complex in rats. Drug Metab. Dispos. 2008, 36, 2523–2538. [Google Scholar] [CrossRef]
- Hershko, C.; Konijn, A.M.; Nick, H.P.; Breuer, W.; Cabantchik, Z.I.; Link, G. ICL670A: A new synthetic oral chelator: Evaluation in hypertransfused rats with selective radioiron probes of hepatocellular and reticuloendothelial iron stores and in iron-loaded rat heart cells in culture. Blood 2001, 97, 1115–1122. [Google Scholar] [CrossRef]
- Nick, H.; Acklin, P.; Lattmann, R.; Buehlmayer, P.; Hauffe, S.; Schupp, J.; Alberti, D. Development of tridentate iron chelators: From desferrithiocin to ICL670. Curr. Med. Chem. 2003, 10, 1065–1076. [Google Scholar] [CrossRef]
- Waldmeier, F.; Bruin, G.J.; Glaenzel, U.; Hazell, K.; Sechaud, R.; Warrington, S.; Porter, J.B. Pharmacokinetics, metabolism, and disposition of deferasirox in beta-thalassemic patients with transfusion-dependent iron overload who are at pharmacokinetic steady state. Drug Metab. Dispos. 2010, 38, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J. 2005, 7, E118–E133. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.; Das, S.; Janisch, L.; Wen, M.; Ramírez, J.; Karrison, T.; Fleming, G.F.; Vokes, E.E.; Schilsky, R.L.; Ratain, M.J. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenom. J. 2002, 2, 43–47. [Google Scholar] [CrossRef] [PubMed]
- De Francia, S.; Massano, D.; Piccione, F.M.; Pirro, E.; Racca, S.; Di Carlo, F.; Piga, A. A new HPLC UV validated method for therapeutic monitoring of deferasirox in thalassaemic patients. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2012, 893–894, 127–133. [Google Scholar] [CrossRef]
- Lee, J.W.; Kang, H.J.; Choi, J.Y.; Kim, N.H.; Jang, M.K.; Yeo, C.W.; Lee, S.S.; Kim, H.; Park, J.D.; Park, K.D.; et al. Pharmacogenetic study of deferasirox, an iron chelating agent. PLoS ONE 2013, 8, e64114. [Google Scholar] [CrossRef] [PubMed]
- Chirnomas, D.; Smith, A.L.; Braunstein, J.; Finkelstein, Y.; Pereira, L.; Bergmann, A.K.; Grant, F.D.; Paley, C.; Shannon, M.; Neufeld, E.J. Deferasirox pharmacokinetics in patients with adequate versus inadequate response. Blood 2009, 114, 4009–4013. [Google Scholar] [CrossRef] [PubMed]
- Cusato, J.; Allegra, S.; Massano, D.; De Francia, S.; Piga, A.; D’Avolio, A. Influence of single-nucleotide polymorphisms on deferasirox C trough levels and effectiveness. Pharmacogenom. J. 2014, 15, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Cusato, J.; Allegra, S.; De Francia, S.; Massano, D.; Piga, A.; D’Avolio, A. Role of pharmacogenetics on deferasirox AUC and efficacy. Pharmacogenomics 2016, 17, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E. Clinical application of deferasirox: Practical patient management. Am. J. Hematol. 2008, 83, 398–402. [Google Scholar] [CrossRef]
- Mattioli, F.; Puntoni, M.; Marini, V.; Fucile, C.; Milano, G.; Robbiano, L.; Perrotta, S.; Pinto, V.; Martelli, A.; Forni, G.L. Determination of deferasirox plasma concentrations: Do gender, physical and genetic differences affect chelation efficacy? Eur. J. Haematol. 2015, 94, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, L.; Ceccherini, F.; Fucile, C.; Marini, V.; Di Paolo, A.; Maximova, N.; Mattioli, F. Evaluation of Pharmacokinetics and Pharmacodynamics of Deferasirox in Pediatric Patients. Pharmaceutics 2021, 13, 1238. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Syed, Y.Y. Luspatercept: A Review in Transfusion-Dependent Anaemia due to Myelodysplastic Syndromes or β-Thalassaemia. Drugs 2021, 81, 945–952. [Google Scholar] [CrossRef]
- Wang, C.; Allegaert, K.; Peeters, M.Y.; Tibboel, D.; Danhof, M.; Knibbe, C.A. The allometric exponent for scaling clearance varies with age: A study on seven propofol datasets ranging from preterm neonates to adults. Br. J. Clin. Pharmacol. 2014, 77, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Suragani, R.N.; Cawley, S.M.; Li, R.; Wallner, S.; Alexander, M.J.; Mulivor, A.W.; Gardenghi, S.; Rivella, S.; Grinberg, A.V.; Pearsall, R.S.; et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine β-thalassemia. Blood 2014, 123, 3864–3872. [Google Scholar] [CrossRef]
- Suragani, R.N.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.; Loveday, K.S.; et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef]
- Chen, N.; Kassir, N.; Laadem, A.; Giuseppi, A.C.; Shetty, J.; Maxwell, S.E.; Sriraman, P.; Ritland, S.; Linde, P.G.; Budda, B.; et al. Population Pharmacokinetics and Exposure-Response Relationship of Luspatercept, an Erythroid Maturation Agent, in Anemic Patients With β-Thalassemia. J. Clin. Pharmacol. 2021, 61, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Motta, I.; Pinto, V.; Piolatto, A.; Ricchi, P.; Tartaglione, I.; Origa, R. Treating Thalassemia Patients with Luspatercept: An Expert Opinion Based on Current Evidence. J. Clin. Med. 2023, 12, 2584. [Google Scholar] [CrossRef] [PubMed]
- Attie, K.M.; Allison, M.J.; McClure, T.; Boyd, I.E.; Wilson, D.M.; Pearsall, A.E.; Sherman, M.L. A phase 1 study of ACE-536, a regulator of erythroid differentiation, in healthy volunteers. Am. J. Hematol. 2014, 89, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Lotinun, S.; Pearsall, R.S.; Davies, M.V.; Marvell, T.H.; Monnell, T.E.; Ucran, J.; Fajardo, R.J.; Kumar, R.; Underwood, K.W.; Seehra, J.; et al. A soluble activin receptor Type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone 2010, 46, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Moosavi, L. Luspatercept. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560635/ (accessed on 6 October 2024).
- Zaccheddu, E.; Zappu, A.; Barella, S.; Clemente, M.G.; Orecchia, V.; Pilia, M.P.; Piras, S.; Pitturru, C.; Scarano, M.; Origa, R. Unplanned pregnancy in women with beta-thalassaemia treated with luspatercept. Br. J. Haematol. 2024, 204, 2505–2507. [Google Scholar] [CrossRef] [PubMed]
- Origa, R.; Comitini, F. Pregnancy in Thalassemia. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019019. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Serum Iron (mcg/dL) | Serum Ferritin (ng/mL) | |
|---|---|---|
| Adul males | 65–176 | 12–300 |
| Adult females | 50–170 | 12–150 |
| Newborns | 100–250 | 25–200 |
| Children (6 m–15 y) | 50–250 | 7–140 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegra, S.; Comità, S.; Roetto, A.; De Francia, S. Sex and Gender Differences in Iron Chelation. Biomedicines 2024, 12, 2885. https://doi.org/10.3390/biomedicines12122885
Allegra S, Comità S, Roetto A, De Francia S. Sex and Gender Differences in Iron Chelation. Biomedicines. 2024; 12(12):2885. https://doi.org/10.3390/biomedicines12122885
Chicago/Turabian StyleAllegra, Sarah, Stefano Comità, Antonella Roetto, and Silvia De Francia. 2024. "Sex and Gender Differences in Iron Chelation" Biomedicines 12, no. 12: 2885. https://doi.org/10.3390/biomedicines12122885
APA StyleAllegra, S., Comità, S., Roetto, A., & De Francia, S. (2024). Sex and Gender Differences in Iron Chelation. Biomedicines, 12(12), 2885. https://doi.org/10.3390/biomedicines12122885