2. Materials and Methods
2.1. Materials
Fucoidan, a sulfated polysaccharide, was sourced from Hi-Q (OliFuco® Plus). Primary antibodies used in this study were directed against several key proteins, including Gal-3 (R&D, Cat number MAB1197); CD68 (Bioss, Cat number bs-0649R); connective tissue growth factor (CTGF) (Santa Cruz, Cat number sc-14939); discoidin domain receptor 2 (DDR2) (Santa Cruz, Cat number sc-8989); alpha-smooth muscle actin (alpha-SMA) (abcam®, Cat number ab5694); and CD44 (MCE, Cat number HY-P80062). Additionally, collagen I (Santa Cruz, Cat number sc-59772) was detected using primary antibodies. GAPDH (Cell Signaling, Cat number 2118) was utilized as a loading control. Secondary antibodies included horseradish peroxidase (HRP)-conjugated anti-mouse IgG (BioLegend, Cat number B426166), a common choice for enhanced signal detection via chemiluminescence. Protein quantification was conducted using the Bicinchoninic Acid (BCA) Protein Assay Kit (Thermo, Cat number 23228), a standard approach for determining protein concentration. For chemiluminescent visualization of Western blot results, Immobilon Western Chemiluminescent HRP Substrate from Millipore Corp (Billerica, MA, USA) was employed, allowing sensitive detection of HRP-conjugated secondary antibodies. Histological analysis employed Sirius red staining (SIGMA, Cat number 365548) and Masson Trichrome Staining Kits (SIGMA, Cat number 41116121), which, respectively, enabled the identification of cellular and fibrotic changes within the tissue samples.
2.2. Animals and Animal Models
All animal experiments adhered to the Guidelines for the Care and Use of Laboratory Animals by the U.S. National Institutes of Health (NIH Publication, revised 2011) and received approval from the Laboratory Animal Welfare and Ethics Committee at Taipei Medical University. Male C57BL/6J mice, aged 8–10 weeks and weighing between 23.5 and 27.5 g, were obtained from the Institute of Laboratory Animal Science, Academia Sinica, Taipei, Taiwan. To investigate the effects of fucoidan on transverse aortic constriction (TAC), mice were subjected to either a sham procedure, TAC surgery, or TAC surgery followed by treatment with 1.5 mg/kg/day fucoidan (FOL) or 7.5 mg/kg/day fucoidan (FOH). These procedures followed established protocols that reliably model pressure overload-induced cardiac hypertrophy and heart failure in vivo. The overall animal treatment protocol is illustrated in
Figure 1. For surgical preparation, anesthesia was induced via intraperitoneal injection of 80 mg/kg pentobarbital sodium (3% concentration; SIGMA, Cat number T48402), providing adequate depth for thoracic procedures. After induction, each mouse was intubated to secure the airway and mechanically ventilated with settings adjusted to 13–15 cmH
2O pressure and 105 breaths per minute to maintain stable respiration. The surgical site was sterilized to minimize infection risk, and a 5 mm incision was made at the left second or third intercostal space. Underlying muscle and soft tissue were carefully dissected to expose the thoracic cavity. After entering the thoracic cavity, the intercostal muscle was separated approximately 2 mm from the sternum, and the chest cavity was expanded by 5 mm with a small chest retractor, allowing adequate visualization of the descending aorta. To achieve aortic constriction, a 6-0 surgical suture was looped around the thoracic aorta to simulate pressure overload, and the ligature was tied securely before the pad needle was withdrawn. The thoracic cavity and skin were then closed layer by layer to maintain anatomical integrity. Postoperative care included recovery monitoring in a warmed environment. In sham-operated control mice, the procedure was identical except that the aorta was not ligated, providing baseline measurements for comparison.
2.3. Echocardiography and Hemodynamics
In this study, transthoracic echocardiography was performed to evaluate cardiac structure and function in mice subjected to either sham or TAC surgery. Using a Philip IE-33 high-resolution imaging system equipped with a 25 MHz RMV-710 transducer, key parameters—including left ventricular end-diastolic diameter (LVEDd), left ventricular end-systolic diameter (LVESd), and left ventricular ejection fraction (LVEF)—were measured at the mid-papillary level from M-mode tracings. These metrics provided detailed insights into ventricular remodeling and systolic function under pressure overload conditions. To ensure reproducibility and reliability of echocardiographic measurements, inter- and intra-observer variability coefficients were assessed. Inter-observer variability, determined by two independent investigators analyzing the same set of echocardiograms, yielded a coefficient of variation of 5.2% for LVEDd, 6.1% for LVESd, and 4.8% for LVEF. Intra-observer variability, assessed by reanalyzing the same images after a two-week interval, showed coefficients of variation of 4.6% for LVEDd, 5.3% for LVESd, and 4.2% for LVEF. These low variability values underscore the consistency and accuracy of the echocardiographic data. In addition to echocardiography, hemodynamic parameters were assessed via cardiac catheterization. Mice were anesthetized using 1.5% isoflurane to maintain stable physiological conditions. A microtip catheter transducer was introduced through the right carotid artery into the left ventricle to continuously monitor pressure and heart rate. Hemodynamic data, including left ventricular systolic pressure (LVSP) and the maximum and minimum rates of pressure change (dP/dt_max and dP/dt_min), were analyzed using LabChart 7 software. This approach provided a high-resolution evaluation of cardiac performance, capturing both structural and functional responses to pressure overload. The combined use of echocardiography and invasive hemodynamic measurements offers a comprehensive assessment of cardiac remodeling and function. The integration of inter- and intra-observer variability coefficients ensures transparency and underscores the robustness of the echocardiographic methodology employed in this study.
2.4. Histological Analysis
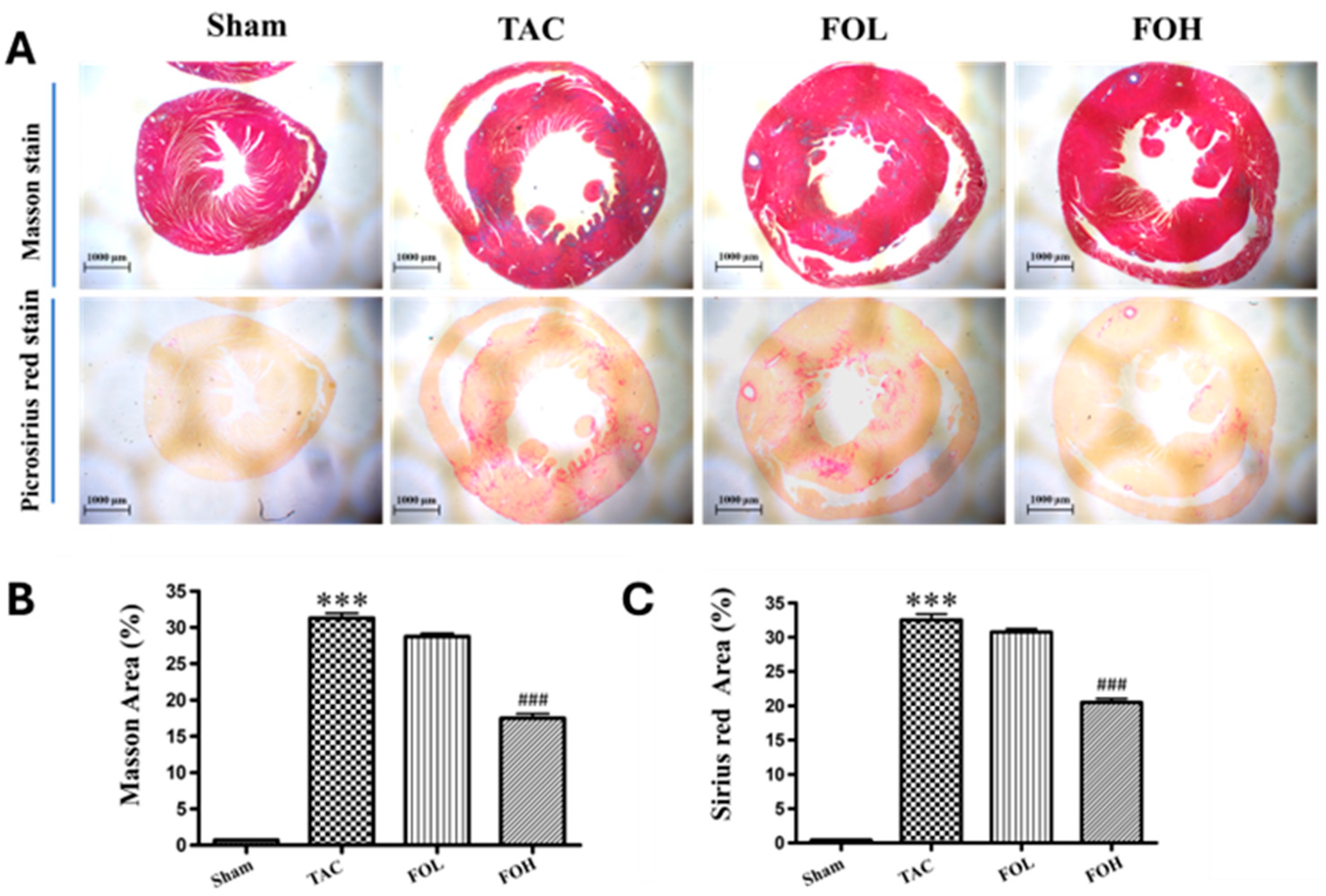
Cardiac tissue samples were processed through a comprehensive series of steps to assess histopathological changes and collagen deposition accurately. Initially, the tissues underwent dehydration and embedding in paraffin to maintain structural integrity. Sections were cut into thin slices of approximately 5 μm and stained using Sirius red staining to evaluate general histological features, and Masson’s trichrome staining to highlight collagen distribution, which is critical in assessing myocardial fibrosis [
12]. The presence of collagen within myocardial tissues, indicating fibrotic remodeling, was quantified by measuring the area of collagen deposition in relation to the total myocardial area, following established procedures in myocardial tissue analysis [
8]. Images of the stained sections were obtained using a Leica DM4000B light microscope (Leica Microsystems, Wetzlar, Germany), allowing for a clear visualization of histopathological features. These images provided a basis for quantitative analysis, wherein the myocardial damage was assessed by calculating the ratio of the area infiltrated by inflammatory cells to the total myocardial area. Similarly, collagen deposition, a marker of fibrosis, was quantified to evaluate the progression of fibrotic remodeling. This assessment was executed with Image Pro Plus software (version 6.0; Media Cybernetics, Bethesda, MD, USA), which facilitated precise measurement of both inflammatory and fibrotic changes within the cardiac tissue. The quantitative evaluation of myocardial tissue using Sirius red staining and Masson’s trichrome staining offers insights into the extent of histological alterations and provides a framework to interpret anti-inflammatory and anti-fibrotic interventions in cardiovascular pathology.
2.5. Western Blot Analysis
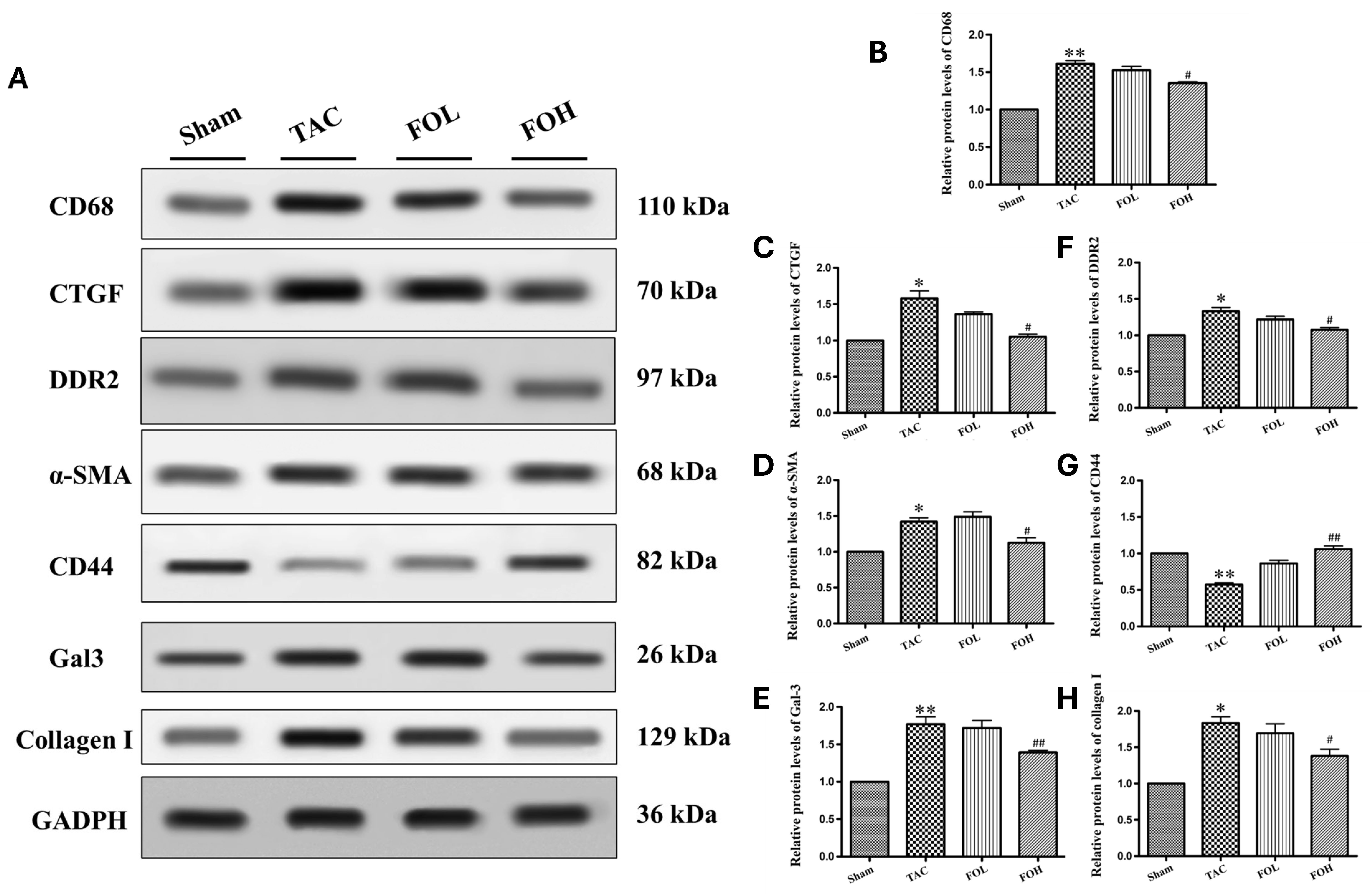
Heart tissue was collected from animal models, immediately rinsed with ice-cold PBS to maintain integrity, and lysed using radioimmunoprecipitation assay (RIPA) buffer. The buffer was enriched with protease inhibitors, including phenylmethylsulfonyl fluoride and phosphatase inhibitors, to protect against protein degradation, as seen in methodologies for examining protein activity under various inflammatory conditions [
8]. Protein concentrations in the lysates were quantified with a Bicinchoninic Acid Protein Assay Kit, ensuring accurate measurements for consistent loading [
11]. Following quantification, equal protein amounts were loaded onto a 10% SDS-PAGE gel for separation by electrophoresis, a standard technique used to resolve proteins by molecular weight. After electrophoresis, proteins were transferred onto polyvinylidene fluoride (PVDF) membranes, optimizing binding efficiency for subsequent detection [
10]. To minimize nonspecific antibody interactions, membranes were blocked in a 5% non-fat milk solution for 90 min, which serves as a general protein blocker. Primary antibodies targeting specific proteins associated with cardiac function and inflammation, such as Gal-3 (1:2000), CD68, CTGF, DDR2, alpha-SAM, CD44, and collagen I (all at 1:1000), were diluted in Tris-buffered saline with Tween-20 (TBST), and applied overnight at 4 °C. Each primary antibody was selected based on its relevance to the pathways of interest in inflammation and fibrosis, as shown in related studies of cardiovascular markers [
13]. GAPDH (1:10,000) was used as a housekeeping protein for normalization, providing a control for equal protein loading across samples. Following overnight incubation, membranes were washed with TBST to eliminate excess unbound primary antibodies, then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at a dilution of 1:20,000 for one hour. The HRP enzyme on the secondary antibody allows for chemiluminescent detection of target proteins when combined with an enhanced chemiluminescence (ECL) substrate [
9]. Protein bands were visualized using the Fusion FX5 Spectra imaging system (Vilber Lourmat, Collégien, France), a highly sensitive method for capturing low-abundance proteins.
2.6. Enzyme-Linked Immunosorbent Assay
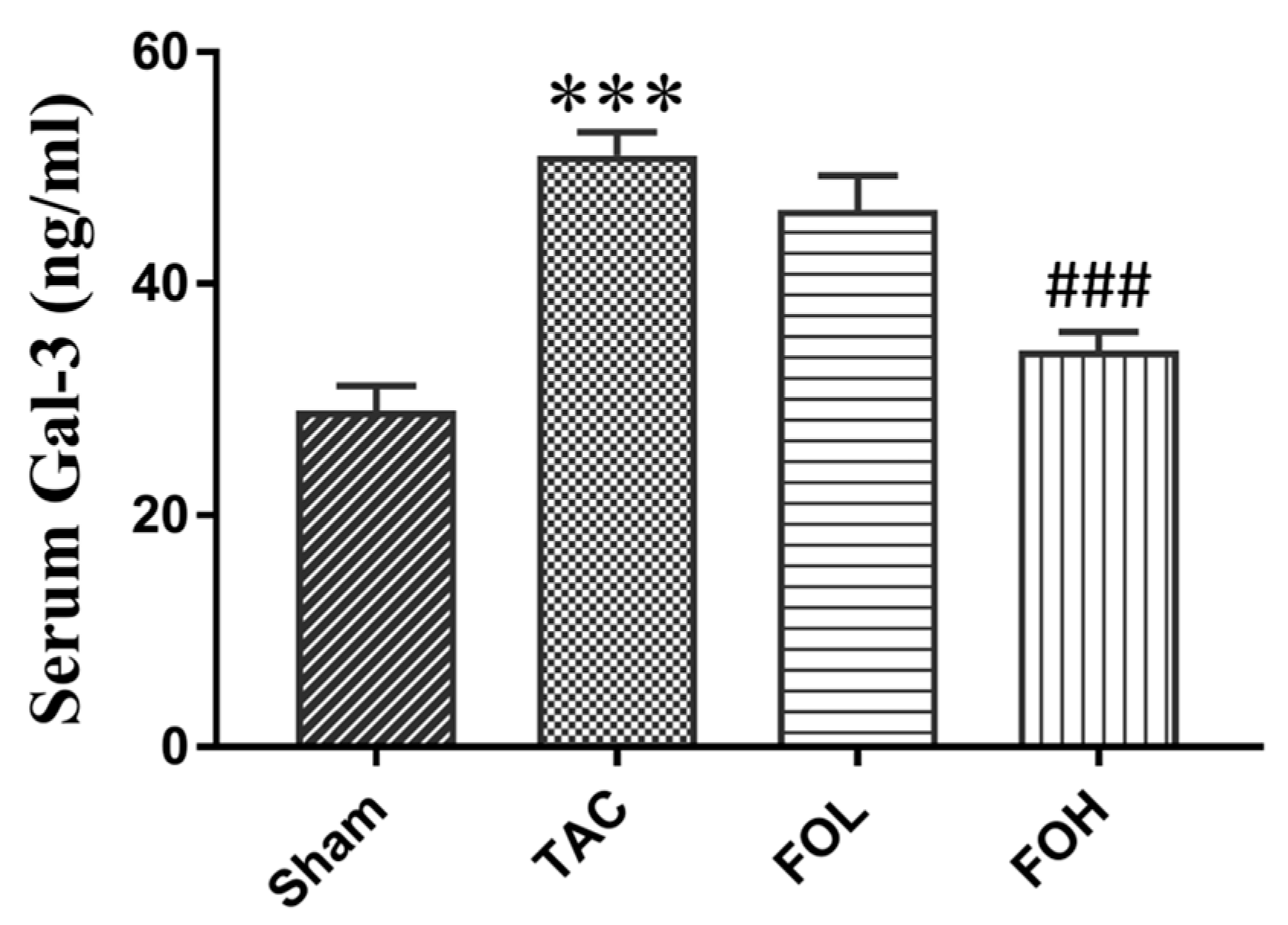
On day 22 of the experiment, blood samples were collected and centrifuged to separate supernatants. The serum was then added to the wells and incubated with enzyme-conjugated anti-Gal-3 antibodies (R&D, Cat number MAB1197). Unbound conjugate was removed by washing, and an enzyme substrate was added to each well. The color generated was proportional to the amount of Gal-3 in the serum. Absorbance was measured immediately using an ELISA reader (TECAN, Infinite 200 Pro) at 450 nm.
2.7. Statistical Analysis
Data analysis was conducted using SPSS 22.0 software (SPSS Inc., Chicago, IL, USA) and GraphPad Prism 8 software (GraphPad, San Diego, CA, USA). The results are presented as mean ± standard deviation (SD). Normality of the data was verified, after which one-way analysis of variance (ANOVA) was used to determine statistical significance among groups. For pairwise group comparisons, post hoc multiple comparison testing was employed, with a p-value < 0.05 considered statistically significant.
4. Discussion
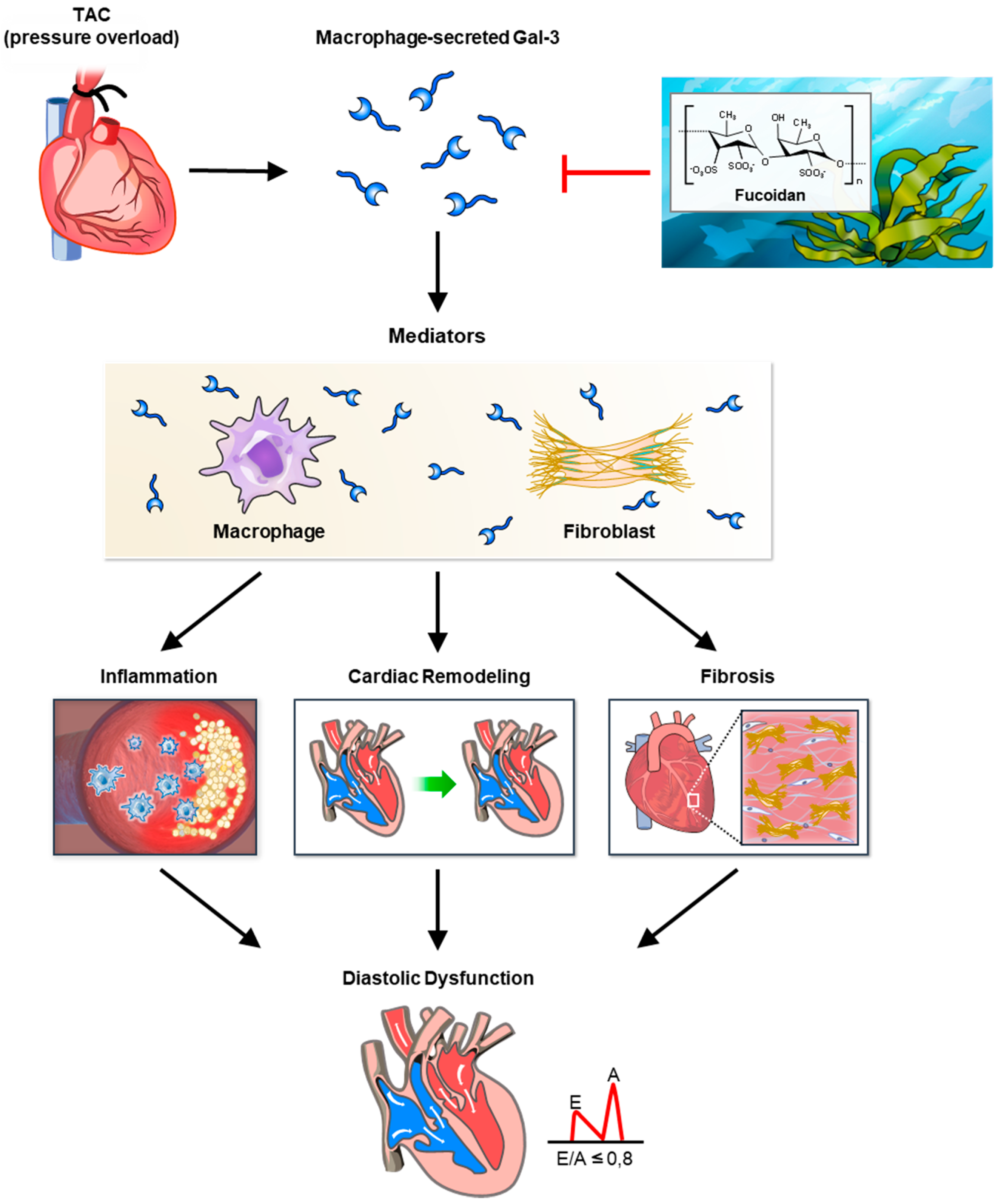
In this study, fucoidan was shown to significantly reduce Gal-3 expression, fibrosis, and inflammation in a mouse model of pressure overload, illustrating its potential therapeutic effects against cardiac remodeling (
Figure 7). The findings indicate that fucoidan’s suppression of Gal-3, a protein strongly associated with fibrosis and inflammation, likely mitigates fibrotic processes in the myocardium, reducing the extent of structural remodeling under pressure overload conditions. Notably, previous research supports these anti-inflammatory and antifibrotic effects, as demonstrated by fucoidan’s modulation of immune responses and its influence on inflammation-related signaling pathways, including CD44 and Nrf2/GPX4, which are crucial for cellular protection under oxidative stress conditions. Fucoidan’s effect on inflammation was also apparent through its reduction in markers associated with immune cell infiltration and fibrotic signaling. Studies on high-molecular-weight fucoidan, such as those by Park et al. (2024) [
13], highlight its immune-enhancing properties, which might contribute to the attenuation of cardiac inflammation observed in this model. Additionally, work by Zhang et al. (2024) underscores fucoidan’s capacity to downregulate lipopolysaccharide-induced inflammatory markers [
11], suggesting a broad anti-inflammatory role in cardiovascular settings. These results position fucoidan as a promising agent for controlling cardiac hypertrophy and fibrosis by targeting multifaceted mechanisms involved in inflammatory and fibrotic signaling pathways.
Fucoidan’s cardioprotective effects in pressure-overload conditions appear to involve key antioxidative and anti-inflammatory pathways that modulate Gal-3 and fibrosis-related markers. One critical mechanism includes the activation of the Nrf2 pathway, which enhances cellular defenses against oxidative stress. By upregulating Nrf2 signaling, fucoidan helps increase the expression of antioxidant enzymes, thereby reducing oxidative damage—a factor that can drive fibrosis and inflammation in cardiac tissue. Studies demonstrate that Nrf2 activation also plays a role in inhibiting ferroptosis, a form of cell death linked to cardiac remodeling, which may further contribute to Gal-3 modulation in fucoidan-treated hearts [
9,
14]. Furthermore, fucoidan’s anti-inflammatory properties may counteract fibrosis progression by modulating the CD44 pathway, a receptor associated with inflammation-induced tissue changes. Research by Chen et al. (2024) suggests that pressure-induced fibrosis and inflammation involve CD44 signaling, which is downregulated by fucoidan [
10]. This modulation likely inhibits pathways leading to Gal-3 upregulation, reducing the pro-fibrotic responses in cardiac tissue. Studies on fucoidan’s action in immune cells have also highlighted its ability to decrease inflammatory mediators [
11,
13], suggesting a broader role in attenuating inflammatory pathways central to cardiac fibrosis. The reduction in Gal-3 secretion and fibrosis markers indicates that fucoidan may limit oxidative stress and inflammatory signals that exacerbate remodeling. Its influence on these molecular targets presents a multifaceted approach to mitigating cardiac remodeling, supporting the compound’s potential as a therapeutic intervention against fibrosis in pressure-overload contexts.
This study aligns with prior research on fucoidan’s cardioprotective effects but extends the understanding of its potential by demonstrating a more direct impact on Gal-3 expression and fibrosis in pressure overload-induced cardiac remodeling. Previous studies have shown that fucoidan from various brown algae, such as those explored by Ersoydan and Rustemeyer (2024) [
8], can inhibit inflammation and reduce oxidative stress in cardiovascular models, suggesting a broad anti-inflammatory and antioxidant role. However, while earlier research focused on general anti-inflammatory actions, the current findings specifically indicate that fucoidan suppresses Gal-3 expression, a key fibrosis-associated protein. This adds to the literature by highlighting Gal-3 as a targeted mechanism in fucoidan’s antifibrotic actions. In addition, studies by Zhang et al. (2024) demonstrated that fucoidan from Costaria costata mitigates inflammation in lipopolysaccharide-induced mouse models, which supports the notion that fucoidan’s immunomodulatory effects contribute to cardiac resilience [
11]. Unlike Zhang’s work, which primarily addresses systemic inflammation, this study focuses on cardiac-specific fibrotic and inflammatory pathways, suggesting that fucoidan’s effects may be particularly beneficial for preventing fibrosis in hypertrophic cardiac conditions. Furthermore, other studies on fucoidan-loaded hydrogel coatings, such as those by Wang et al. (2024), highlight the compound’s capacity to promote endothelial growth and improve cardiovascular outcomes through antioxidant mechanisms, particularly through the Nrf2/GPX4 pathway [
9]. The present study not only confirms these antioxidative pathways but also specifies that fucoidan’s Nrf2-related antioxidative action likely inhibits Gal-3, thereby reducing fibrotic remodeling in pressure-overloaded myocardium. This insight advances our understanding of how fucoidan can specifically prevent cardiac hypertrophy by integrating antioxidative and antifibrotic effects focused on Gal-3 modulation. Collectively, this study contributes to the existing literature by reinforcing fucoidan’s utility in fibrosis inhibition while providing novel mechanistic insights into its ability to directly target Gal-3. This potentially positions fucoidan as an essential therapeutic option for cardiac hypertrophy, particularly in conditions marked by inflammation and oxidative stress.
Fucoidan shows promising therapeutic potential for conditions associated with cardiac hypertrophy and fibrosis. Its ability to modulate multiple cellular pathways, such as reducing oxidative stress via the Nrf2 pathway and dampening inflammation through CD44-related mechanisms, indicates a comprehensive impact on processes underlying cardiac remodeling. By inhibiting Ga-3 and thereby reducing fibrotic activity, fucoidan could directly address fibrosis, a key factor in cardiac dysfunction in hypertrophic hearts [
10,
14]. Therapeutically, fucoidan’s multi-targeted effects make it especially relevant for cardiovascular diseases where inflammation and oxidative stress are prominent. Research by Wang et al. (2024) illustrates that fucoidan’s antioxidative effects extend beyond simple antioxidant activity [
9], engaging cellular defenses to improve mitochondrial resilience and reduce apoptosis, which is critical for preserving cardiac tissue under stress. Moreover, studies on high-molecular-weight fucoidan suggest immune-enhancing effects, indicating that it may strengthen the body’s overall defense mechanisms, which are often compromised by chronic inflammatory states associated with heart disease [
13]. Fucoidan’s broad therapeutic potential makes it a promising candidate for managing cardiac hypertrophy and fibrosis, especially in clinical settings where oxidative stress and inflammation exacerbate disease progression. Its ability to modulate key pathways such as Keap1/Nrf2 and CD44 contributes not only to reduction in fibrosis markers like Gal-3 but also to systemic antioxidative and anti-inflammatory defenses, which are critical for maintaining cardiac health [
10,
14]. In clinical practice, these properties could translate into meaningful therapeutic outcomes. For instance, by enhancing the antioxidative response via the Nrf2 pathway, fucoidan may help protect cardiomyocytes and other cardiac cells from oxidative damage, which is a key driver of maladaptive remodeling and heart failure. Studies have demonstrated that this pathway plays a role in mitigating ferroptosis—a type of programmed cell death linked to cardiac injury—highlighting its potential to preserve cardiac function [
9,
11]. Also, fucoidan’s influence on CD44 signaling, which mediates inflammation-induced fibrosis, suggests its utility in reducing structural and functional deterioration in conditions of chronic pressure overload [
10]. By targeting CD44 and Gal-3 simultaneously, fucoidan may disrupt the feedback loops between inflammation and fibrosis, preventing progressive damage to cardiac tissue. Fucoidan’s benefits extend beyond direct cardiac effects. Its broad-spectrum immune-enhancing properties, as shown in both preclinical and clinical studies, indicate its potential to improve systemic health, particularly in patients with comorbid conditions such as diabetes or metabolic syndrome, which often exacerbate cardiac pathology [
13,
15]. For example, its ability to modulate gut microbiota and enhance systemic antioxidative capacity could complement its cardioprotective effects, creating a multifaceted therapeutic approach [
16]. From a translational perspective, fucoidan could serve as an adjunctive therapy to standard treatments for heart failure and other cardiovascular diseases, particularly those characterized by inflammatory and oxidative stress components. Its incorporation into advanced delivery systems, such as hydrogel coatings for cardiovascular stents, has already shown promise in improving biocompatibility and promoting vascular healing, underscoring its versatility in various clinical applications [
17]. Further clinical studies are needed to establish optimal dosing regimens, delivery methods, and long-term safety profiles for fucoidan. Nonetheless, its ability to address multiple pathways involved in cardiac remodeling positions it as a valuable addition to the arsenal of therapies for hypertrophic and fibrotic cardiovascular diseases.
This study has several limitations that should be acknowledged. Firstly, the absence of a sham group with fucoidan supplementation limits our ability to evaluate its basal effects on cardiac health. While this was not included in the current experimental design, future studies should incorporate such a group to provide insights into the baseline impact of fucoidan, independent of stress conditions. Evidence from prior research suggests that fucoidan exerts anti-inflammatory and antioxidative effects, even under non-stressed conditions, potentially enhancing cardiac and systemic health by modulating gut microbiota and reducing systemic inflammation [
11,
13]. Secondly, while this study employed the TAC model to induce pressure overload, we did not measure the pressure gradient across the aortic constriction at key time points. This represents a limitation in ensuring consistency in the severity of pressure overload among groups. Future experiments should include pressure gradient measurements both one week post-surgery and before the final sacrifice, as this would verify the uniformity of the model and improve its robustness. Prior studies have emphasized the importance of such assessments in correlating hemodynamic parameters with structural and molecular changes in cardiac remodeling [
18]. Thirdly, this study did not employ wheat germ agglutinin (WGA) staining to assess cardiomyocyte hypertrophy or terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining to quantify apoptosis. Both methods could have provided additional cellular context to the observed effects of fucoidan. Future studies should include these techniques to validate and extend our findings, particularly in light of evidence showing fucoidan’s protective effects against hypertrophy and apoptosis via pathways like Nrf2/GPX4 and Notch signaling [
9,
16]. Fourthly, the causal relationship between fucoidan’s effects and Gal-3 suppression was not established in this study. While our results demonstrate a correlation, further mechanistic studies using Gal-3 supplementation or silencing in the presence and absence of fucoidan are necessary to clarify whether Gal-3 suppression is a direct or indirect effect mediated by other signaling proteins. Such experiments in isolated cardiomyocytes or fibroblasts would provide definitive insights into fucoidan’s molecular mechanisms [
10]. Fifthly, the use of an animal model, while valuable for preclinical investigation, poses challenges in translating findings to human applications. Variability in metabolism, physiological response, and immune modulation between species could influence how fucoidan behaves in human systems compared to mice [
8]. Additionally, dosage levels used in animal studies may not correspond linearly to safe and effective dosages in humans, necessitating careful recalibration through clinical trials [
9,
11]. Another limitation relates to the bioavailability of fucoidan when administered in different forms. While fucoidan-loaded hydrogel coatings and oral supplementation have shown promise in experimental settings, differences in absorption, distribution, and degradation in human systems may affect efficacy and dosing requirements [
10]. Lastly, while this study focused on specific markers such as Gal-3, a more comprehensive approach examining additional inflammatory and oxidative markers would further clarify fucoidan’s therapeutic potential and inform its broader applicability in human cardiology.
Future research should prioritize elucidating the molecular mechanisms by which fucoidan influences cardiac remodeling, with particular focus on its effects on cardiomyocytes and fibroblasts. While in vitro experiments on isolated cells were beyond the scope of the current study, incorporating such models in future studies could enhance understanding of fucoidan’s cellular targets. Specifically, cultured neonatal rat cardiomyocytes or fibroblasts, or cells isolated from adult hearts, could be valuable tools for this purpose. One promising avenue is the interaction between Gal-3 and interleukin-1 receptor like-1 (IL-1RL1), also known as suppression of tumorigenicity 2 (ST2), a receptor for IL-33. The soluble form, sST2, is a critical mediator of cardiac fibrosis and inflammation, serving as a biomarker for heart failure severity [
18]. Elevated sST2 inhibits the protective IL-33/transmembrane form ST2 (ST2L) signaling axis, exacerbating maladaptive remodeling. Fucoidan’s effects on Gal-3 expression suggest it may attenuate this pathological cascade. Future studies should explore whether fucoidan modulates sST2 levels or restores IL-33/ST2L signaling, potentially amplifying its cardioprotective effects. Mechanistically, fucoidan’s antioxidative properties, mediated through the Nrf2/GPX4 pathway, and its anti-inflammatory effects, potentially through CD44 signaling, could indirectly downregulate sST2 by mitigating upstream oxidative and inflammatory triggers [
9,
10]. These pathways are central to cardiac fibrosis and remodeling, and their modulation by fucoidan aligns with findings from studies highlighting its ability to suppress lipid accumulation, enhance mitochondrial function, and regulate gut microbiota-related inflammation [
11,
14]. The antifibrotic effects of fucoidan may extend to its potential to influence fibroblast proliferation and extracellular matrix production. Research indicates that fucoidan can suppress fibrosis-related pathways via Notch signaling and reduce oxidative damage in various models [
15,
16]. Furthermore, its bioactivity in altering macrophage polarization and reducing inflammatory cytokines highlights its broader immunomodulatory role [
11,
13]. For clinical translation, studies should assess changes in sST2 and Gal-3 levels following fucoidan treatment in animal models of heart failure. Preclinical experiments could include hydrogel-based controlled-release systems to improve the bioavailability and sustained therapeutic effects of fucoidan [
17]. Such systems could be particularly effective in modulating both Gal-3 and ST2 pathways, as demonstrated by advances in cardiovascular biomaterials. Finally, clinical trials are necessary to evaluate fucoidan’s long-term effects on cardiac remodeling and function, particularly in patients with elevated sST2 and Gal-3 levels. Investigating the synergy of targeting both pathways could reveal new dual-targeted therapeutic strategies. By integrating these molecular and cellular insights, future research will better define fucoidan’s potential as a multifaceted treatment for heart failure, addressing the interplay between fibrosis, oxidative stress, and inflammation.



 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}