



Metabolic Dysfunction in Parkinson’s Disease: Unraveling the Glucose–Lipid Connection


{kind=link}
{kind=link}
Abstract
1. Introduction
General Aspects of Parkinson’s Disease
2. Specific Aspects of PD and Metabolism: Mitochondria, Glucose, and Lipid Metabolism
2.1. Mitochondrial Dysfunction in PD
2.2. Implications of Mitochondrial Dysfunction and Disturbances in Cellular Bioenergetics of Glucose in PD
2.3. Mitochondrial Genes and PD
2.4. Is PD a “New Diabetes Type”—The Missing Link Between Insulin and Dopamine?
2.5. The Role of Hyperglycaemia in PD
2.6. The Effect of Cerebral Insulin Resistance in PD
2.7. Lipid Metabolism in PD
2.8. Bioenergetic Drug Treatment in Parkinson’s Disease
3. Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parkinson, J. An Essay on the Shaking Palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Birkmayer, W.; Riederer, P. The Clinical Pathology of Parkinson’s Disease. Biochemistry, Clinical Pathology, and Treatment; Springer: Berlin/Heidelberg, Germany, 1983; pp. 46–88. [Google Scholar]
- Rodriguez, M.; Morales, I.; Rodriguez-Sabate, C.; Sanchez, A.; Castro, R.; Brito, J.M.; Sabate, M. The degeneration and replacement of dopamine cells in Parkinson’s disease: The role of aging. Front. Neuroanat. 2014, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.V.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.; Klein, C. The Importance of Drosophila melanogaster Research to UnCover Cellular Pathways Underlying Parkinson’s Disease. Cells 2021, 10, 579. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.; Ballard, P.; Tetrud, J.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Mizuno, Y.; Ohta, S.; Tanaka, M.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1989, 163, 1450–1455. [Google Scholar] [CrossRef]
- Reichmann, H.; Riederer, P. Biochemische Analyse der Atmungskettenkomplexe verschiedener Hirnregionen von Patienten mit Morbus Parkinson. In Morbus Parkinson und Andere Basalganglienerkrankungen; BMFT Symposium Bad Kissingen: Bonn, Germany, 1989. [Google Scholar]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Gao, X.Y.; Yang, T.; Gu, Y.; Sun, X.-H. Mitochondrial Dysfunction in Parkinson’s Disease: From Mechanistic Insights to Therapy. Front. Aging Neurosci. 2022, 14, 885500. [Google Scholar] [CrossRef]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef]
- Borsche, M.; Märtens, A.; Hörmann, P.; Brückmann, T.; Lohmann, K.; Tunc, S.; Klein, C.; Hiller, K.; Balck, A. In Vivo Investigation of Glucose Metabolism in Idiopathic and PRKN -Related Parkinson’s disease. Mov. Disord. 2023, 38, 697. [Google Scholar] [CrossRef]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grünewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Park. Dis. 2020, 11, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Debashree, B.; Kumar, M.; Keshava-Prasad, T.S.; Natarajan, A.; Christopher, R.; Nalini, A.; Bindu, P.S.; Gayathri, N.; Srinivas-Bharath, M.M. Mitochondrial dysfunction in human skeletal muscle biopsies of lipid storage disorder. J. Neurochem. 2018, 145, 323–341. [Google Scholar] [CrossRef] [PubMed]
- Saxena, U. Bioenergetics failure in neurodegenerative diseases: Back to the future. Expert Opin. Ther. Targets 2012, 16, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.; Krieger, E.; Dekker, M.J.; Squitieri, F.; Ibanez, P.; Heutink, P. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2022, 299, 256–259. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Riederer, P.; Monoranu, C.; Strobel, S.; Iordache, T.; Sian-Huelsmann, J. Iron as the concert master in the pathogenic orchestra playing in sporadic Parkinson’s disease. J. Neural Transm. 2021, 128, 1577–1598. [Google Scholar] [CrossRef]
- Cooper, O.; Hallett, P.; Isacson, O. Upstream lipid and metabolic systems are potential causes of Alzheimer’s disease, Parkinson’s disease and dementias. FEBS J. 2024, 291, 632–645. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, H.; Zhang, J.; Shen, Y.; Ye, Y.; Jiang, Q.; Chen, L.; Sun, B.; Chen, Z.; Shen, L.; Fang, H.; et al. Targeting Mitochondrial Complex I Deficiency in MPP+/MPTP-induced Parkinson’s Disease Cell Culture and Mouse Models by Transducing Yeast NDI1 Gene. Biol. Proced. Online 2024, 26, 9. [Google Scholar] [CrossRef]
- Dexter, D.T.; Sian, J.; Rose, S.; Hindmarsh, J.G.; Mann, V.M.; Cooper, J.M.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Schapira, A.H.V.; et al. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann. Neurol. 1994, 35, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hulsmann, J.; Riederer, P. Virus-induced brain pathology and the neuroinflammation-inflammation continuum: The neurochemists view. J. Neural Transm. 2024, 131, 1429–1453. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, L.; Qin, Q.; Wang, D.; Zhao, J.; Gao, H.; Yuan, X.; Zhang, J.; Zou, Y.; Mao, Z.; et al. Upregulated hexokinase 2 expression induces the apoptosis of dopaminergic neurons by promoting lactate production in Parkinson’s disease. Neurobiol. Dis. 2022, 163, 105605. [Google Scholar] [CrossRef] [PubMed]
- Qi, A.; Liu, L.; Zhang, J.; Chen, S.; Xu, S.; Chen, Y.; Zhang, L.; Cai, C. Plasma Metabolic Analysis Reveals the Dysregulation of Short-Chain Fatty Acid Metabolism in Parkinson’s Disease. Mol. Neurobiol. 2023, 60, 2619–2631. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.L.; Sharif, H.; Griswold, A.; Fontana, P.; Mintseris, J.; Dagbay, K.B.; Paulo, J.A.; Gygi, S.P.; Bachovchin, D.A.; Wu, H. DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature 2021, 592, 778–783. [Google Scholar] [CrossRef]
- Kloss, O.; Eskin, N.A.M.; Suh, M. Thiamin deficiency on fetal brain development with and without prenatal alcohol exposure. Biochem. Cell Biol. 2018, 96, 169–177. [Google Scholar] [CrossRef]
- Mizuno, Y.; Matuda, S.; Yoshino, H.; Mori, H.; Hattori, N.; Ikebe, S. An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson’s disease. Ann. Neurol. 1994, 35, 204–210. [Google Scholar] [CrossRef]
- Constantini, A.; Immacolata-Pala, M.; Comagnoni, L.; Colangeli, M. High-dose thiamine as initial treatment of Parkinson’s disease. BMJ Case Rep. 2013. [Google Scholar] [CrossRef]
- Bartolák-Suki, E.; Imsirovic, J.; Nishibori, Y.; Krishnan, R.; Suki, B. Regulation of Mitochondrial Structure and Dynamics by the Cytoskeleton and Mechanical Factors. Int. J. Mol. Sci. 2017, 18, 1812. [Google Scholar] [CrossRef]
- Lass, A.; Agarwal, S.; Sohal, R.S. Mitochondrial Ubiquinone Homologues, Superoxide Radical Generation, and Longevity in Different Mammalian Species. J. Biol. Chem. 1997, 272, 19199–19204. [Google Scholar] [CrossRef]
- Keane, J.; Gray, B.; McNaughton, L. The Effect of rhGH and IGF-1 on Mitochondrial Superoxide Generation in Human Lymphocytes in Vitro. Med. Sci. Sports Exerc. 2011, 43, 434. [Google Scholar] [CrossRef]
- Pacelli, C.; Giguère, N.; Bourque, M.J.; Lévesque, M.; Slack, R.S.; Trudeau, L.-É. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Tan, C.; Zhao, L.; Liang, Y.; Liu, G.; Liu, H.; Zhong, Y.; Liu, Z.; Mo, L.; Liu, X.; et al. Glucose metabolism impairment in Parkinson’s disease. Brain Res. Bull. 2023, 199, 110672. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, S.; Kasai, T.; Mizuta, I.; Suematsu, M.; Osone, S.; Azuma, Y.; Imamura, T.; Tokuda, T.; Kanno, H.; El-Agnaf, O.M.A.; et al. Early-onset parkinsonism in a pedigree with phosphoglycerate kinase deficiency and a heterozygous carrier: Do PGK-1 mutations contribute to vulnerability to parkinsonism? npj Park. Dis. 2017, 3, 13. [Google Scholar] [CrossRef]
- Tang, B.L. Glucose, glycolysis, and neurodegenerative diseases. J. Cell. Physiol. 2020, 235, 8044–8805. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Yamashita, H.; Ohshita, T.; Sawamoto, N.; Nakamura, S. ATP increases extracellular dopamine level through stimulation of P2Y purinoceptors in the rat striatum. Brain Res. 1995, 691, 205–212. [Google Scholar] [CrossRef]
- Kokotos, A.C.; Antoniazzi, A.M.; Unda, S.R.; Ko, M.S.; Park, D.; Eliezer, D.; Kaplitt, M.G.; Camilli, P.D.; Ryan, T.A. Phosphoglycerate kinase is a central leverage point in Parkinson’s Disease driven neuronal metabolic deficits. bioRxiv 2023. [Google Scholar] [CrossRef]
- Cai, R.; Zhang, Y.; Simmering, J.E.; Schultz, J.L.; Li, Y.; Fernandez-Carasa, I.; Consiglio, A.; Raya, A.; Polgreen, P.M.; Narayanan, N.S.; et al. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J. Clin. Investig. 2019, 129, 4539–4549. [Google Scholar] [CrossRef]
- Akhmadi, A.; Yeskendir, A.; Dey, N.; Mussakhmetov, A.; Shatkenova, Z.; Kulyyassov, A.; Andreeva, A.; Utepbergenov, D. DJ-1 protects proteins from acylation by catalyzing the hydrolysis of highly reactive cyclic 3-phosphoglyceric anhydride. Nat. Commun. 2024, 15, 2004. [Google Scholar] [CrossRef]
- Wüllner, U.; Borghammer, P.; Choe, C.U.; Csoti, I.; Falkenburger, B.; Gasser, T.; Lingor, P.; Riederer, P. The heterogeneity of Parkinson’s disease. J. Neural Transm. 2023, 130, 827–838. [Google Scholar] [CrossRef]
- Shakya, S.; Prevett, J.; Hu, X.; Xiao, R. Characterization of Parkinson’s Disease Subtypes and Related Attributes. Front. Neurol. 2022, 13, 81003813. [Google Scholar] [CrossRef] [PubMed]
- Flønes, I.H.; Toker, L.; Sandnes, D.A.; Castelli, M.; Mostafavi, S.; Lura, N.; Shadad, O.; Fernandez-Vizarra, E.; Painous, C.; Pérez-Soriano, A.; et al. Mitochondrial complex I deficiency stratifies idiopathic Parkinson’s disease. Nat. Commun. 2024, 15, 3631. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Falck, B.; Hellman, B. Evidence for the presence of biogenic amines in pancreatic islets. Experientia 1963, 19, 139–140. [Google Scholar] [CrossRef]
- Lakomy, M.; Chodkowska, D. Cholinergic innervation of pig pancreas. Acta Histochem. 1984, 75, 63–68. [Google Scholar] [CrossRef]
- Cegrell, L. The occurrence of biogenic monoamines in the mammalian endocrine pancreas. Acta Physiol. Scand. Suppl. 1968, 314, 1–60. [Google Scholar]
- Malaisse, W.; Malaisse-Lagae, F.; Wright, P.H.; Ashmore, J. Effects of adrenergic and cholinergic agents upon insulin secretion in vitro. Endocrinology 1967, 80, 975–978. [Google Scholar] [CrossRef]
- De Iuliis, A.; Montinaro, E.; Fatati, G.; Plebani, M.; Colosimo, C. Diabetes mellitus and Parkinson’s disease: Dangerous liaisons between insulin and dopamine. Neural Regen. Res. 2022, 17, 523–533. [Google Scholar]
- Lu, Y.Q.; Yuan, L.; Sun, Y.; Dou, H.W.; Su, J.H.; Hou, Z.P.; Li, J.Y.; Li, W. Long-term hyperglycemia aggravates α-synuclein aggregation and dopaminergic neuronal loss in a Parkinson’s disease mouse model. Transl. Neurodegener. 2022, 11, 14. [Google Scholar]
- Anlauf, M.; Eissele, R.; Schäfer, M.K.-H.; Eiden, L.E.; Arnold, R.; Pauser, U.; Klöppel, G.; Weihe, E. Expression of the Two Isoforms of the Vesicular Monoamine Transporter (VMAT1 and VMAT2) in the Endocrine Pancreas and Pancreatic Endocrine Tumors. J. Histochem. Cytochem. 2003, 51, 1027–1040. [Google Scholar] [CrossRef]
- Mezey, E.; Eisenhofer, G.; Harta, G.; Hansson, S.; Gould, L.; Hunyady, B.; Hoffman, B.J. A Novel Nonneuronal Catecholaminergic System: Exocrine Pancreas Synthesizes and Releases Dopamine. Proc. Natl. Acad. Sci. USA 1996, 93, 10377–10382. [Google Scholar] [CrossRef] [PubMed]
- Bini, J.; Sanchez-Rangel, E.; Gallezot, J.D.; Naganawa, M.; Nabulsi, N.; Lim, K.; Najafzadeh, S.; Shirali, A.; Ropchan, J.; Matuskey, D.; et al. PET Imaging of Pancreatic Dopamine D2 and D3 Receptor Density with 11C-(+)-PHNO in Type 1 Diabetes. J. Nucl. Med. 2020, 61, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Rubí, B.; Ljubicic, S.; Pournourmohammadi, S.; Carobbio, S.; Armanet, M.; Bartley, C.; Maechler, P. Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion. J. Biol. Chem. 2005, 280, 36824–36832. [Google Scholar] [CrossRef] [PubMed]
- Figlewicz, D.P.; Evans, S.B.; Murphy, J.; Hoen, M.; Baskin, D.G. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res. 2003, 964, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef]
- Jurcovicova, J. Glucose transport in brain—Effect of inflammation. Endocr. Regul. 2014, 48, 35–48. [Google Scholar] [CrossRef]
- White, M.F.; Kahn, C.R. The insulin signaling system. J. Biol. Chem. 1994, 269, 1–4. [Google Scholar] [CrossRef]
- Duarte, A.I.; Santos, P.; Oliveira, C.R.; Santos, M.S.; Rego, A.C. Insulin neuroprotection against oxidative stress is mediated by Akt and GSK-3beta signaling pathways and changes in protein expression. Biochim. Biophys. Acta 2008, 1783, 994–1002. [Google Scholar] [CrossRef]
- Grünblatt, E.; Homolak, J.; Babic-Perhoc, A.; Davor, V.; Knezovic, A.; Osmanovic-Barilar, J.; Riederer, P.; Walitza, S.; Tackenberg, C.; Salkovic-Petrisic, M. From attention-deficit hyperactivity disorder to sporadic Alzheimer’s disease-Wnt/mTOR pathways hypothesis. Front. Neurosci. 2023, 17, 1104985. [Google Scholar] [CrossRef]
- Chen, L.; Saito, R.; Noda Narita, S.; Kassai, H.; Aiba, A. Hyperactive mTORC1 in striatum dysregulates dopamine receptor expression and odor preference behavior. Front. Neurosci. 2024, 30, 18. [Google Scholar] [CrossRef]
- Kosillo, P.; Ahmed, K.M.; Aisenberg, E.E.; Karalis, V.; Roberts, B.M.; Cragg, S.J.; Bateup, H.S. Dopamine neuron morphology and output are differentially controlled by mTORC1 and mTORC2. eLife 2022, 11, e75398. [Google Scholar] [CrossRef]
- Choi, K.C.; Kim, S.H.; Ha, J.Y.; Kim, S.T.; Son, J.H. A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J. Neurochem. 2010, 112, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ji, X.; Liu, J.; Li, Z.; Zhang, X. Dopamine Receptor Subtypes Differentially Regulate Autophagy. Int. J. Mol. Sci. 2018, 19, 1540. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Pan, R.-Y.; Guan, F.; Yuan, Z. Lactate metabolism in neurodegenerative diseases. Neural Regen. Res. 2024, 19, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Y.; Yu, L.; Vickstrom, C.R.; Liu, Q.S. VTA mTOR Signaling Regulates Dopamine Dynamics, Cocaine-Induced Synaptic Alterations, and Reward. Neuropsychopharmacology 2018, 43, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Mazei-Robison, M.S.; Koo, J.W.; Friedman, A.K.; Lansink, C.S.; Robison, A.J.; Vinish, M.; Krishnan, V.; Kim, S.; Siuta, M.A.; Galli, A.; et al. Role for mTOR signaling and neuronal activity in morphine-induced adaptations in ventral tegmental area dopamine neurons. Neuron 2011, 72, 977–990. [Google Scholar] [CrossRef]
- Barroso-Chinea, P.; Luis-Ravelo, D.; Fumagallo-Reading, F.; Castro-Hernandez, J.; Salas-Hernandez, J.; Rodriguez-Nuñez, J.; Febles-Casquero, A.; Cruz-Muros, I.; Afonso-Oramas, D.; Abreu-Gonzalez, P.; et al. DRD3 (dopamine receptor D3) but not DRD2 activates autophagy through MTORC1 inhibition preserving protein synthesis. Autophagy 2020, 16, 1279–1295. [Google Scholar] [CrossRef]
- Ramírez-Jarquín, U.N.; Shahani, N.; Pryor, W.; Usiello, A.; Subramaniam, S. The mammalian target of rapamycin (mTOR) kinase mediates haloperidol-induced cataleptic behavior. Transl. Psychiatry 2020, 10, 336. [Google Scholar] [CrossRef]
- Labandeira, C.M.; Fraga-Bau, A.; Arias-Ron, D.; Alvarez-Rodriguez, E.; Vicente-Alba, P.; Lago-Garma, J.; Rodriguez-Perez, A.I. Parkinson’s disease and diabetes mellitus: Common mechanisms and treatment repurposing. Neural Regen. Res. 2022, 17, 1652–1658. [Google Scholar] [CrossRef]
- Liu, M.; Jiao, Q.; Du, X.; Bi, M.; Chen, X.; Jiang, H. Potential Crosstalk Between Parkinson’s Disease and Energy Metabolism. Aging Dis. 2021, 12, 2003. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, S.; Chen, J.; Su, Z. Unraveling the Regulation of Hepatic Gluconeogenesis. Front. Endocrinol. 2019, 9, 802. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.; Bassareo, V.; Beaulieu, J.; Pinna, A.; Schlich, M.; Lavoie, C.; Murtas, D.; Simola, N.; Martinoli, M.G. Dopaminergic neurodegeneration in a rat model of long-term hyperglycemia: Preferential degeneration of the nigrostriatal motor pathway. Neurobiol. Dis. 2018, 69, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Münch, G.; Lüth, H.J.; Wong, A.; Arendt, T.; Hirsch, E.; Ravid, R.A.; Riederer, P. Crosslinking of α-synuclein by advanced glycation endproducts—An early pathophysiological step in Lewy body formation? J. Chem. Neuroanat. 2000, 20, 253–257. [Google Scholar] [CrossRef]
- Su, C.J.; Shen, Z.; Cui, R.X.; Huang, Y.; Xu, D.L.; Zhao, F.L.; Pan, J.; Shi, A.-M.; Liu, T.; Yu, Y.-L. Thioredoxin-Interacting Protein (TXNIP) Regulates Parkin/PINK1-mediated Mitophagy in Dopaminergic Neurons Under High-glucose Conditions: Implications for Molecular Links Between Parkinson’s Disease and Diabetes. Neurosci. Bull. 2020, 36, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.; Bournival, J.; Zottig, X.; Martinoli, M.G. Resveratrol Protects DAergic PC12 Cells from High Glucose-Induced Oxidative Stress and Apoptosis: Effect on p53 and GRP75 Localization. Neurotox. Res. 2013, 25, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Randell, E.; Han, Y.; Adeli, K.; Krahn, J.; Meng, Q.H. Increased plasma methylglyoxal level, inflammation, and vascular endothelial dysfunction in diabetic nephropathy. Clin. Biochem. 2011, 44, 307–311. [Google Scholar] [CrossRef]
- Xie, B.; Lin, F.; Ullah, K.; Peng, L.; Ding, W.; Dai, R.; Qing, H.; Deng, Y. A newly discovered neurotoxin ADTIQ associated with hyperglycemia and Parkinson’s disease. Biochem. Biophys. Res. Commun. 2015, 459, 361–366. [Google Scholar] [CrossRef]
- Lai, S.W.T.; Lopez Gonzalez, E.J.; Zoukari, T.; Ki, P.; Shuck, S.C. Methylglyoxal and its Adducts: Induction, Repair, and Association with Disease. Chem. Res. Toxicol. 2022, 35, 1720–1746. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Memon, M.A.; Khan, R.N.; Riaz, S.; Ain, Q.U.; Ahmed, M.; Kumar, N. Methylglyoxal and insulin resistance in berberine-treated type 2 diabetic patients. J. Res. Med. Sci. Off. J. Isfahan Univ. Med. Sci. 2018, 23, 110. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci. Lett. 1992, 142, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end products in age-related diseases. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, W.; Lu, H.; Cai, S. Methylglyoxal in the Brain: From Glycolytic Metabolite to Signalling Molecule. Molecules 2022, 27, 7905. [Google Scholar] [CrossRef]
- Chegão, A.; Guarda, M.; Alexandre, B.M.; Shvachiy, L.; Temido-Ferreira, M.; Marques-Morgado, I.; Fernandes Gomes, B.; Matthiesen, R.; Lopes, L.V.; Florindo, P.R.; et al. Glycation modulates glutamatergic signaling and exacerbates Parkinson’s disease-like phenotypes. npj Park. Dis. 2022, 8, 51. [Google Scholar] [CrossRef]
- Bantounou, M.; Shoaib, K.; Mazzoleni, A.; Modalavalasa, H.; Kumar, N.; Philip, S. The association between type 2 diabetes mellitus and Parkinson’s disease; a systematic review and meta-analysis. Brain Disord. 2014, 15, 100158. [Google Scholar] [CrossRef]
- Chohan, H.; Senkevich, K.; Patel, R.K.; Bestwick, J.P.; Jacobs, B.M.; Bandres Ciga, S.; Gan-Or, Z.; Noyce, A.J. Type 2 Diabetes as a Determinant of Parkinson’s Disease Risk and Progression. Mov. Disord. 2021, 36, 1420–1429. [Google Scholar] [CrossRef]
- Cullinane, P.W.; de Pablo Fernandez, E.; König, A.; Outeiro, T.F.; Jaunmuktane, Z.; Warner, T.T. Type 2 Diabetes and Parkinson’s Disease: A Focused Review of Current Concepts. Mov. Disord. Off. J. Mov. Disord. Soc. 2023, 38, 162–177. [Google Scholar] [CrossRef]
- Han, K.; Kim, B.; Lee, S.H.; Kim, M.K. A nationwide cohort study on diabetes severity and risk of Parkinson disease. npj Park. Dis. 2023, 9, 11. [Google Scholar] [CrossRef]
- Lipman, I.J.; Boykin, M.E.; Flora, R.E. Glucose intolerance in Parkinson’s disease. J. Chronic Dis. 1974, 27, 573–579. [Google Scholar] [CrossRef]
- Sabari, S.S.; Balasubramani, K.; Iyer, M.; Sureshbabu, H.W.; Venkatesan, D.; Gopalakrishnan, A.V.; Narayanaswamy, A.; Senthil Kumar, N.; Vellingiri, B. Type 2 Diabetes (T2DM) and Parkinson’s Disease (PD): A Mechanistic Approach. Mol. Neurobiol. 2023, 60, 4547–4573. [Google Scholar] [CrossRef] [PubMed]
- Sandyk, R. The relationship between diabetes mellitus and Parkinson’s disease. Int. J. Neurosci. 1993, 69, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Renaud, J.; Simola, N.; Martinoli, M.G. Diabetes, a Contemporary Risk for Parkinson’s Disease: Epidemiological and Cellular Evidences. Front. Aging Neurosci. 2019, 11, 302. [Google Scholar] [CrossRef] [PubMed]
- De Pablo-Fernández, E.; Courtney, R.; Rockliffe, A.; Gentleman, S.; Holton, J.L.; Warner, T.T. Faster disease progression in Parkinson’s disease with type 2 diabetes is not associated with increased α-synuclein, tau, amyloid-β or vascular pathology. Neuropathol. Appl. Neurobiol. 2021, 47, 1080–1091. [Google Scholar] [CrossRef]
- Meissner, W.G.; Remy, P.; Giordana, C.; Maltête, D.; Pascal-Derkinderen, J.L.; Houéto, M.A.; Benatru, I.; Boraud, T.; Brefel-Courbon, C.; Carrière, N.; et al. Trial of Lixisenatide in Early Parkinson’s Disease. N. Engl. J. Med. 2024, 390, 1176–1185. [Google Scholar] [CrossRef]
- Adam, D. Diabetes drug slows the development of Parkinson’s disease. Nature, 2024; epub ahead of print. [Google Scholar] [CrossRef]
- Hong, C.T.; Chen, K.Y.; Wang, W.; Chiu, J.Y.; Wu, D.; Chao, T.Y.; Hu, C.J.; Chau, K.D.; Bamodu, O.A. Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of α-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells 2020, 9, 740. [Google Scholar] [CrossRef]
- Ruiz-Pozo, V.A.; Tamayo-Trujillo, R.; Cadena-Ullauri, S.; Frias-Toral, E.; Guevara-Ramírez, P.; Paz-Cruz, E.; Chapela, S.; Montalván, M.; Morales-López, T.; Simancas-Racines, D.; et al. The Molecular Mechanisms of the Relationship between Insulin Resistance and Parkinson’s Disease Pathogenesis. Nutrients 2023, 5, 3585. [Google Scholar] [CrossRef]
- Wang, R.; Jin, Z.; Zhen, Q.; Qi, L.; Liu, C.; Wang, P.; Liu, Y.; Fang, J.; Liu, Y.; Su, Y.; et al. Hyperglycemia affects axial signs in patients with Parkinson’s disease through mechanisms of insulin resistance or non-insulin resistance. Neurol. Sci. 2024, 45, 2011–2019. [Google Scholar] [CrossRef]
- Soto, M.; Cai, W.; Konishi, M.; Kahn, C.R. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. USA 2019, 116, 6379–6384. [Google Scholar] [CrossRef]
- Reno, C.M.; Tanoli, T.; Bree, A.; Daphna-Iken, D.; Cui, C.; Maloney, S.E.; Wozniak, D.F.; Fisher, S.J. Antecedent glycemic control reduces severe hypoglycemia-induced neuronal damage in diabetic rats. Am. J. Physiol.-Endocrinol. Metab. 2013, 304, E1331–E1337. [Google Scholar] [CrossRef]
- Rensink, A.A.M.; Otte-Höller, I.; de Boer, R.; Bosch, R.R.; ten Donkelaar, H.J.; de Waal, R.M.W.; Verbeek, M.M.; Kremer, B. Insulin inhibits amyloid β-induced cell death in cultured human brain pericytes. Neurobiol. Aging 2024, 25, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Bassil, F.; Delamarre, A.; Canron, M.H.; Dutheil, N.; Vital, A.; Négrier-Leibreich, M.-L.; Bezard, E.; Fernagut, P.O.; Meissner, W.G. Impaired brain insulin signaling in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12760. [Google Scholar] [CrossRef]
- Kawahito, S.; Kitahata, H.; Oshita, S. Problems associated with glucose toxicity: Role of hyperglycemia-induced oxidative stress. World J. Gastroenterol. 2009, 15, 4137. [Google Scholar] [CrossRef] [PubMed]
- Al-Bachari, S.; Naish, J.H.; Parker, G.J.M.; Emsley, H.C.; Parkes, L.M. Blood–Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front. Physiol. 2020, 11, 593026. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chorell, E.; Steneberg, P.; Vernersson-Lindahl, E.; Edlund, H.; Wittung-Stafshede, P. Insulin-degrading enzyme prevents α-synuclein fibril formation in a nonproteolytical manner. Sci. Rep. 2015, 5, 12531. [Google Scholar] [CrossRef]
- Zhao, L. Insulin-Degrading Enzyme as a Downstream Target of Insulin Receptor Signaling Cascade: Implications for Alzheimer’s Disease Intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef]
- Anandhan, A.; Jacome, M.S.; Lei, S.; Hernandez-Franco, P.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Metabolic Dysfunction in Parkinson’s Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. Brain Res. Bull. 2017, 133, 12–30. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Athauda, D.; Foltynie, T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog. Neurobiol. 2016, 145–146, 98–120. [Google Scholar] [CrossRef]
- Baskin, D.G.; Porte, D.; Guest, K.; Dorsa, D.M. Regional Concentrations of Insulin in the Rat Brain. Endocrinology 1983, 112, 898–903. [Google Scholar] [CrossRef]
- Morris, J.K.; Vidoni, E.D.; Perea, R.D.; Rada, R.; Johnson, D.K.; Lyons, K.; Pahwa, R.; Burns, J.M.; Honea, R.A. Insulin resistance and gray matter volume in neurodegenerative disease. Neuroscience 2014, 13, 139–147. [Google Scholar] [CrossRef]
- Stouffer, M.A.; Woods, C.A.; Patel, J.C.; Lee, C.R.; Witkovsky, P.; Bao, L.; Machold, R.P.; Jones, K.T.; de Vaca, S.C.; Reith, M.E.A.; et al. Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat. Commun. 2015, 6, 8543. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, Y.; Lu, S.; Xu, J.; Liu, X.; Yang, D.; Yang, Y.; Hou, L.; Li, N. A crazy trio in Parkinson’s disease: Metabolism alteration, α-synuclein aggregation, and oxidative stress. Mol. Cell. Biochem. 2024; epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Sun, T.; He, X.; Wang, Z.; Zhao, K.; An, J.; Wen, L.; Li, J.; Li, W.; Feng, J. Association between Parkinson’s Disease and Diabetes Mellitus: From Epidemiology, Pathophysiology and Prevention to Treatment. Aging Dis. 2022, 13, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.P.; Thrush, A.B.; Heigenhauser, G.J.F.; Tandon, N.N.; Dyck, D.J.; Bonen, A.; Spriet, L.L. Skeletal muscle mitochondrial FAT/CD36 content and palmitate oxidation are not decreased in obese women. Am. J. Physiol.-Endocrinol. Metab. 2007, 292, E1782–E1789. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative Stress and Lipid Peroxidation Products in Cancer Progression and Therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Osmanovic, J.; Grünblatt, E.; Riederer, P.; Hoyer, S. Modeling sporadic Alzheimer’s disease: The insulin resistant brain state generates multiple long-term morphobiological abnormalities including hyperphosphorylated tau protein and amyloid-β. J. Alzheimer’s Dis. 2009, 18, 729–750. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Tribl, F.; Schmidt, M.; Hoyer, S.; Riederer, P. Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the insulin signalling pathway. J. Neurochem. 2006, 96, 1005–1015. [Google Scholar] [CrossRef]
- Aguer, C.; Harper, M.E. Skeletal muscle mitochondrial energetics in obesity and type 2 diabetes mellitus: Endocrine aspects. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 805–819. [Google Scholar] [CrossRef]
- Sukhorukov, V.N.; Orekhov, A.N. Molecular Aspects of Inflammation and Lipid Metabolism in Health and Disease: The Role of the Mitochondria. Int. J. Mol. Sci. 2024, 25, 6299. [Google Scholar] [CrossRef]
- Liu, D.; Ke, Z.; Luo, J. Thiamine Deficiency and Neurodegeneration: The Interplay Among Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy. Mol. Neurobiol. 2017, 54, 5440–5448. [Google Scholar] [CrossRef]
- Brekk, O.R.; Honey, J.R.; Lee, S.; Hallett, P.J.; Isacson, O. Cell type-specific lipid storage changes in Parkinson’s disease patient brains are recapitulated by experimental glycolipid disturbance. Proc. Natl. Acad. Sci. USA 2020, 117, 27646–27654. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Wei, L.; Su, Y.; Tang, Y.; Peng, G.; Wu, Y.; He, Y.; Liu, H.; Guo, W.; Wu, Z.; et al. Lipid Metabolism Disorder in Cerebrospinal Fluid Related to Parkinson’s Disease. Brain Sci. 2023, 13, 1166. [Google Scholar] [CrossRef] [PubMed]
- Mächler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martín, A.; Romero-Gómez, I.; et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef]
- Xu, B.; Liu, Y.; Li, N.; Geng, Q. Lactate and lactylation in macrophage metabolic reprogramming: Current progress and outstanding issues. Front. Immunol. 2024, 15, 1395786. [Google Scholar] [CrossRef] [PubMed]
- Liguori, C.; Stefani, A.; Fernandes, M.; Cerroni, R.; Mercuri, N.B.; Pierantozzi, M.X. Biomarkers of Cerebral Glucose Metabolism and Neurodegeneration in Parkinson’s Disease: A Cerebrospinal Fluid-Based Study. J. Park. Dis. 2022, 12, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y.; Liu, S.F.; Zhuang, J.L.; Li, M.M.; Huang, Z.P.; Chen, Y.H.; Chen, X.R.; Chen, C.N.; Lin, S.; Ye, L.C. Recent research progress on metabolic syndrome and risk of Parkinson’s disease. Rev. Neurosci. 2022, 34, 719–735. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Comito, G.; Parri, M.; Iozzo, M.; Duatti, A.; Di Virgilio, F.; Lorito, N.; Bacci, M.; Pardella, E.; Sandrini, G.; et al. Lactate Rewires Lipid Metabolism and Sustains a Metabolic–Epigenetic Axis in Prostate Cancer. Cells 2022, 82, 1267–1282. [Google Scholar] [CrossRef]
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2017, 1862, 1260–1272. [Google Scholar] [CrossRef]
- Chang, K.-H.; Chen, C.-M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletić-Savatić, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017, 26, 719–737. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Aviles-Olmos, I.; Limousin, P.; Lees, A.; Foltynie, T. Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain 2013, 136 Pt 2, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Mulvaney, C.A.; Duarte, G.S.; Handley, J.; Evans, D.J.; Menon, S.; Wyse, R.; Emsley, H.C. GLP-1 receptor agonists for Parkinson’s disease. Cochrane Database Syst. Rev. 2020, 23, CD012990. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Zhang, Q.; Guan, M.; Sheng, S.; Mo, W.; Zou, M.; Li, J.; Bi, J.; Tang, X.; et al. Exenatide and Renal Outcomes in Patients with Type 2 Diabetes and Diabetic Kidney Disease. Am. J. Nephrol. 2020, 51, 806–814. [Google Scholar] [CrossRef]
- Athauda, D.; Gulyani, S.; Karnati, H.K.; Li, Y.; Tweedie, D.; Mustapic, M.; Chawla, S.; Chowdhury, K.; Skene, S.S.; Greig, N.H.; et al. Utility of Neuronal-Derived Exosomes to Examine Molecular Mechanisms That Affect Motor Function in Patients with Parkinson Disease: A Secondary Analysis of the Exenatide-PD Trial. JAMA Neurol. 2019, 76, 420–429, 509. [Google Scholar] [CrossRef]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Brauer, R.; Wei, L.; Ma, T.; Athauda, D.; Girges, C.; Vijiaratnam, N.; Auld, G.; Whittlesea, C.; Wong, I.; Foltynie, T. Diabetes medications and risk of Parkinson’s disease: A cohort study of patients with diabetes. Brain 2020, 143, 3067–3076. [Google Scholar] [CrossRef]
- Svenningsson, P.; Wirdefeldt, K.; Yin, L.; Fang, F.; Markaki, I.; Efendic, S.; Ludvigsson, J. Reduced incidence of Parkinson’s disease after dipeptidyl peptidase-4 inhibitors-A nationwide case-control study. Mov. Disord. 2016, 31, 1904–1911. [Google Scholar] [CrossRef]
- Hussain, S.; Singh, A.; Baxi, H.; Taylor, B.; Burgess, J.; Antony, B. Thiazolidinedione use is associated with reduced risk of Parkinson’s disease in patients with diabetes: A meta-analysis of real-world evidence. Neurol. Sci. 2020, 41, 3697–3703. [Google Scholar] [CrossRef]
- Chang, Y.H.; Yen, S.J.; Chang, Y.H.; Wu, W.J.; Lin, K.D. Pioglitazone and statins lower incidence of Parkinson disease in patients with diabetes mellitus. Eur. J. Neurol. 2021, 28, 430–437. [Google Scholar] [CrossRef]
- Ping, F.; Jiang, N.; Li, Y. Association between metformin and neurodegenerative diseases of observational studies: Systematic review and meta-analysis. BMJ Open Diabetes Res. Care 2020, 8, e001370. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.; Pala, M.I.; Grossi, E.; Mondonico, S.; Cardelli, L.E.; Jenner, C.; Proietti, S.; Colangeli, M.; Fancellu, R. Long-Term Treatment with High-Dose Thiamine in Parkinson Disease: An Open-Label Pilot Study. J. Altern. Complement. Med. 2015, 21, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Luong, K.V.; Nguyên, L.T. The beneficial role of thiamine in Parkinson disease. CNS Neurosci. Ther. 2013, 19, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Mrowicka, M.; Mrowicki, J.; Dragan, G.; Majsterek, I. The importance of thiamine (vitamin B1) in humans. Biosci. Rep. 2023, 43, BSR20230374. [Google Scholar] [CrossRef]
- Bashir, B.; Mittal, S.; Muthukumar, A.; Vishwas, S.; Pandey, N.K.; Gulati, M.; Gupta, G.; Dhanasekaran, M.; Kumar, P.; Dureja, H.; et al. Harnessing the neuroprotective effect of oral administration of benfotiamine in MPTP induced Parkinson’s disease in rats. Eur. J. Pharmacol. 2024, 962, 176234. [Google Scholar] [CrossRef]
- Hernandez-Vazquez, A.J.; Garcia-Sanchez, J.A.; Moreno-Arriola, E.; Salvador-Adriano, A.; Ortega-Cuellar, D.; Velazquez-Arellano, A. Thiamine Deprivation Produces a Liver ATP Deficit and Metabolic and Genomic Effects in Mice: Findings Are Parallel to Those of Biotin Deficiency and Have Implications for Energy Disorders. J. Nutr. Nutr. 2016, 9, 287–299. [Google Scholar] [CrossRef]
- De Pablo-Fernández, E.; Lees, A.J.; Holton, J.L.; Warner, T.T. Prognosis and Neuropathologic Correlation of Clinical Subtypes of Parkinson Disease. JAMA Neurol. 2019, 76, 470–479. [Google Scholar] [CrossRef]
- Thenganatt, M.A.; Jankovic, J. Parkinson Disease Subtypes. JAMA Neurol. 2014, 71, 499–504. [Google Scholar] [CrossRef]
- Caggiu, E.; Arru, G.; Hosseini, S.; Niegowska, M.; Sechi, G.; Zarbo, I.R.; Sechi, L.A. Inflammation, Infectious Triggers, and Parkinson’s Disease. Front. Neurol. 2019, 10, 122. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Riederer, P.; Nagatsu, T.; Youdim, M.B.; Wulf, M.; Dijkstra, J.M.; Sian-Huelsmann, J. Lewy bodies, iron, inflammation and neuromelanin: Pathological aspects underlying Parkinson’s disease. J. Neural Transm. 2023, 130, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Knezovic, A.; Osmanovic-Barilar, J.; Curlin, M.; Hof, P.R.; Simic, G.; Riederer, P.; Salkovic-Petrisic, M. Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocin-induced rat model of Alzheimer’s disease. J. Neural Transm. 2015, 122, 577–592. [Google Scholar] [CrossRef] [PubMed]
- NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators. Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double blind, randomised trial. Lancet Neurol. 2015, 14, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Rom, S.; Heldt, N.A.; Gajghate, S.; Seliga, A.; Reichenbach, N.L.; Persidsky, Y. Hyperglycemia and advanced glycation end products disrupt BBB and promote occludin and claudin-5 protein secretion on extracellular microvesicles. Sci. Rep. 2020, 10, 7274. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Elabi, O.F.; Cunha, J.P.M.C.M.; Gaceb, A.; Fex, M.; Paul, G. High-fat diet-induced diabetes leads to vascular alterations, pericyte reduction, and perivascular depletion of microglia in a 6-OHDA toxin model of Parkinson disease. J. Neuroinflamm. 2021, 18, 175. [Google Scholar] [CrossRef]
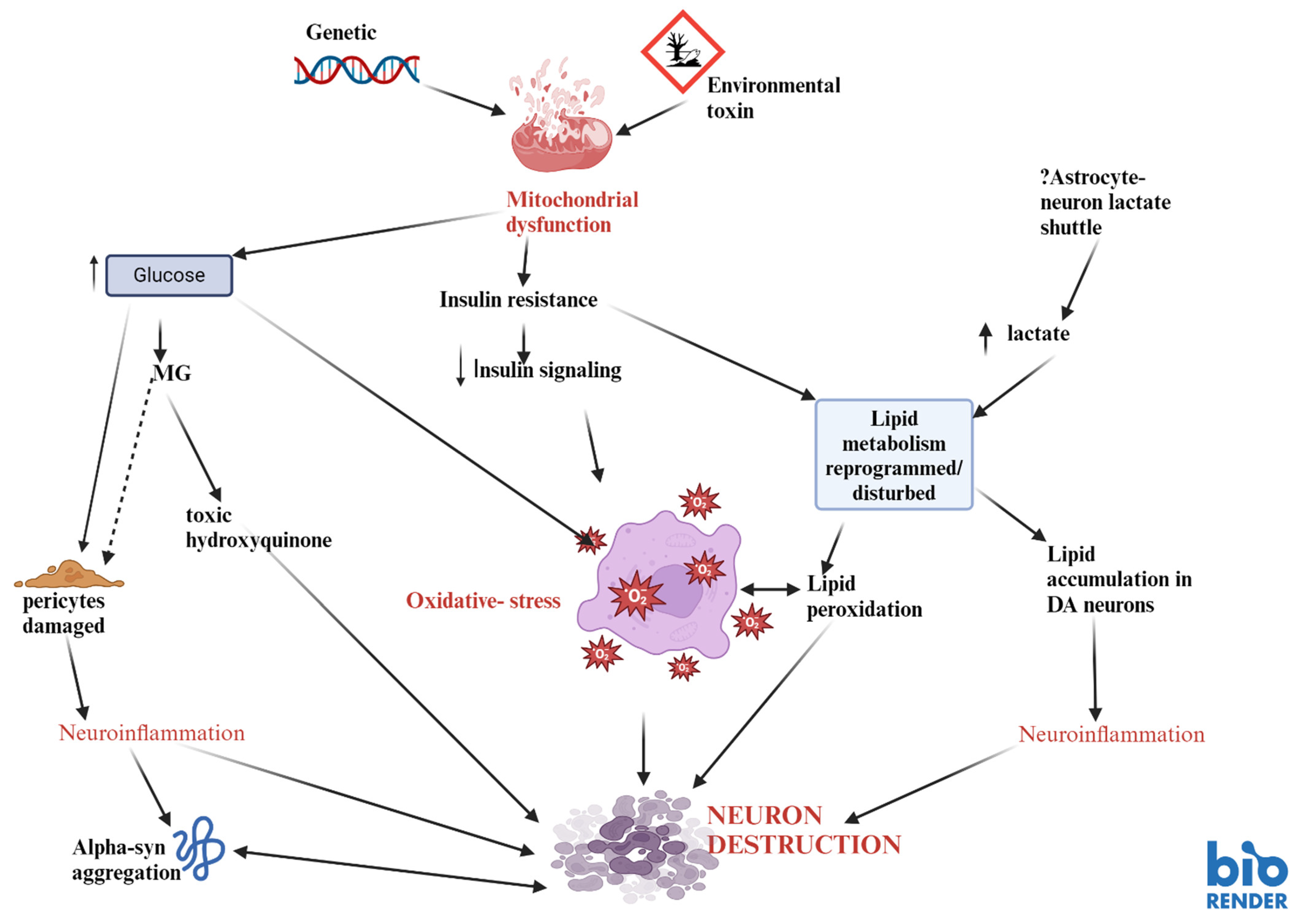
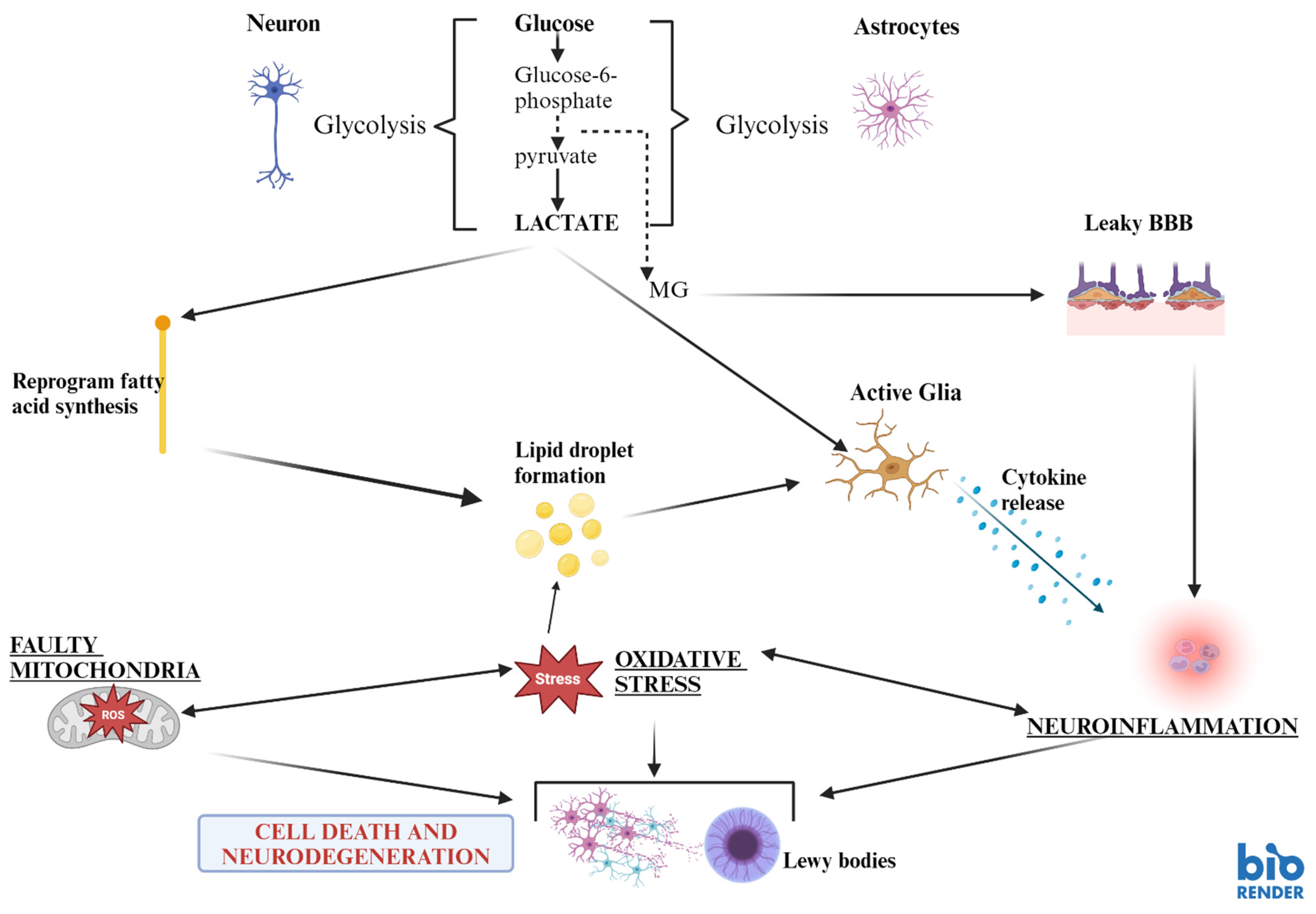
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sian-Hulsmann, J.; Riederer, P.; Michel, T.M. Metabolic Dysfunction in Parkinson’s Disease: Unraveling the Glucose–Lipid Connection. Biomedicines 2024, 12, 2841. https://doi.org/10.3390/biomedicines12122841
Sian-Hulsmann J, Riederer P, Michel TM. Metabolic Dysfunction in Parkinson’s Disease: Unraveling the Glucose–Lipid Connection. Biomedicines. 2024; 12(12):2841. https://doi.org/10.3390/biomedicines12122841
Chicago/Turabian StyleSian-Hulsmann, Jeswinder, Peter Riederer, and Tanja Maria Michel. 2024. "Metabolic Dysfunction in Parkinson’s Disease: Unraveling the Glucose–Lipid Connection" Biomedicines 12, no. 12: 2841. https://doi.org/10.3390/biomedicines12122841
APA StyleSian-Hulsmann, J., Riederer, P., & Michel, T. M. (2024). Metabolic Dysfunction in Parkinson’s Disease: Unraveling the Glucose–Lipid Connection. Biomedicines, 12(12), 2841. https://doi.org/10.3390/biomedicines12122841