
Proteins Associated with Neurodegenerative Diseases: Link to DNA Repair



Abstract
1. Introduction
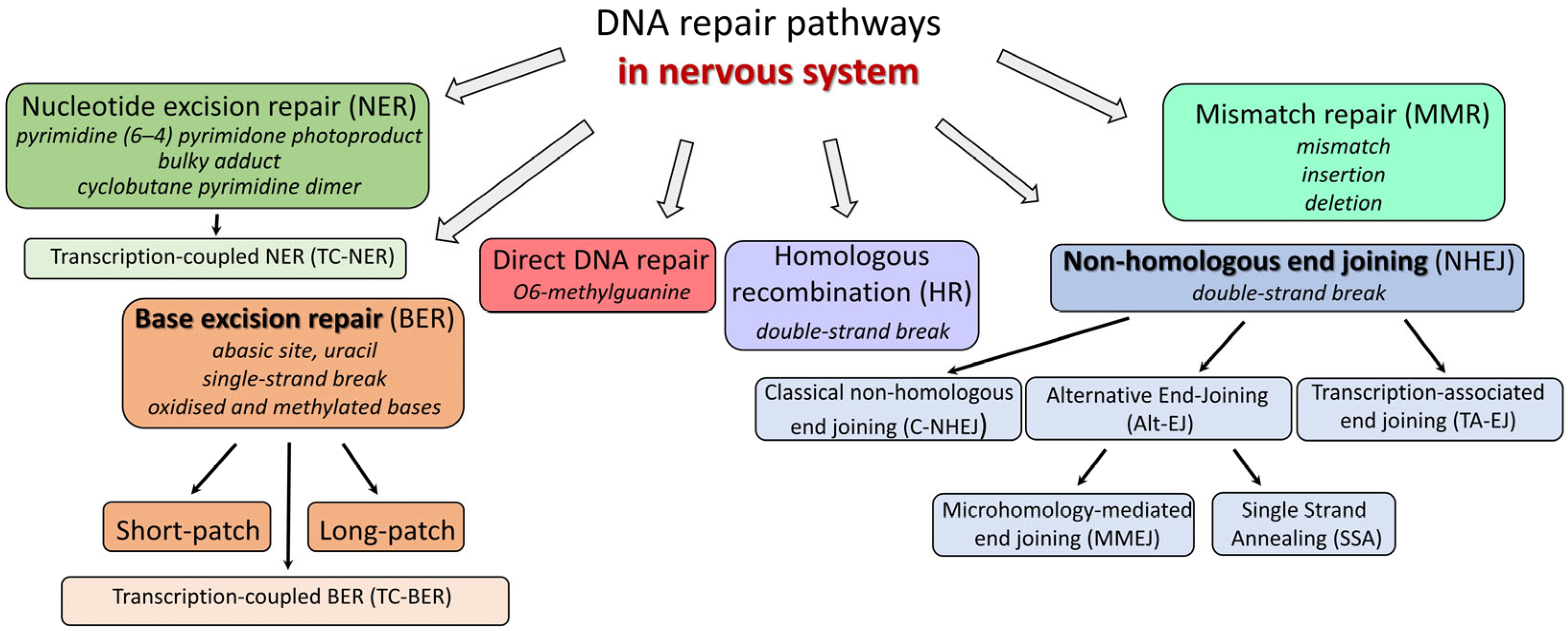
2. DNA Repair Pathways
2.1. Base Excision Repair (BER)
2.1.1. Minor Pathways of BER
2.1.2. Transcription-Coupled Base Excision Repair (TC-BER)
2.2. Nucleotide Excision Repair (NER)
2.3. Double Strand Break Repair (DSBR)
2.3.1. Homologous Recombination (HR)
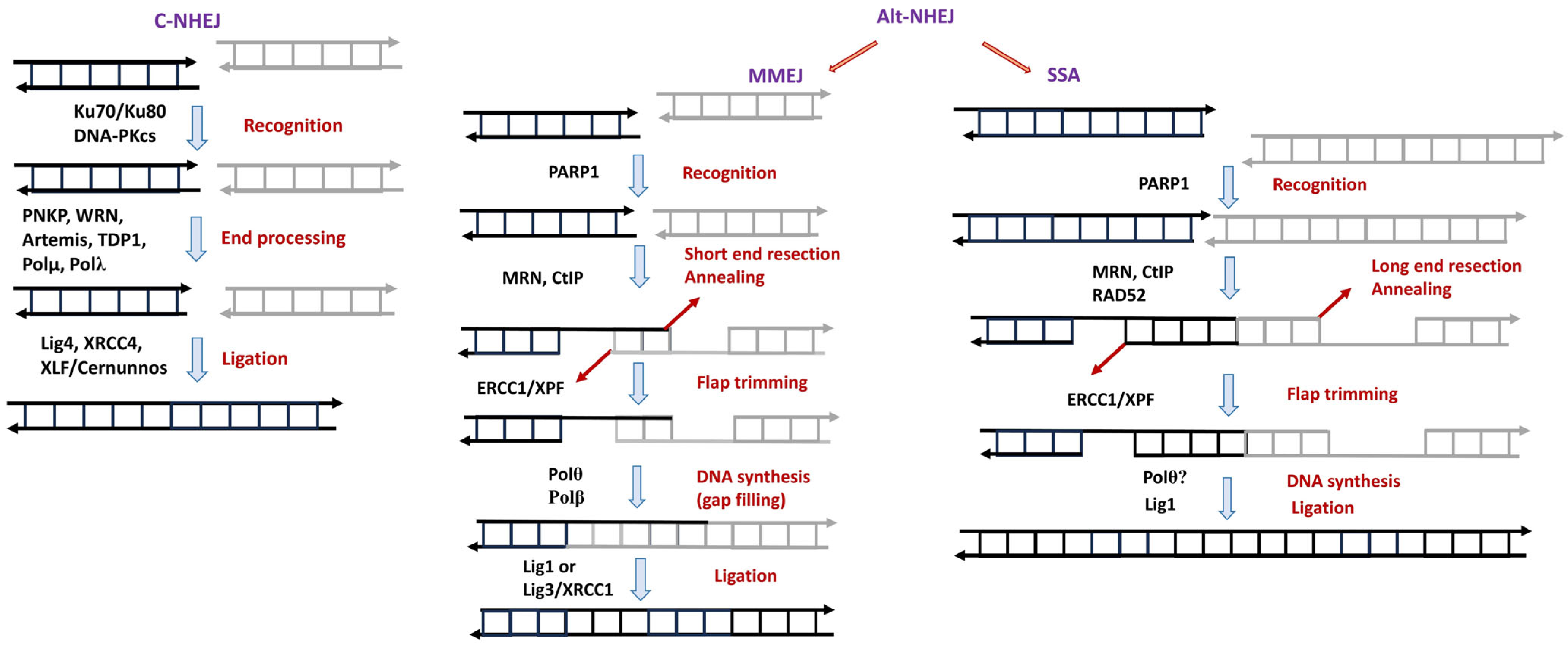
2.3.2. Nonhomologous End Joining (NHEJ)
2.4. Direct DNA Repair
2.5. Mismatch Repair (MMR)
3. Liquid–Liquid Phase Separation (LLPS) as Factor Influencing DNA Repair
4. Proteins Associated with Neurodegenerative Disorders
4.1. Poly(ADP-Ribose) Polymerase 1 (PARP1)
4.2. Role of Proteins Associated with Neurodegenerative Disorders in the DNA Repair Pathways in Neurons
4.2.1. FUS
4.2.2. TDP-43
4.2.3. C9orf72
4.2.4. α-Synuclein
4.2.5. Tau Protein
4.2.6. Amyloid β
4.2.7. NONO and SFPQ
4.2.8. Huntingtin
5. Conclusions
Funding
Conflicts of Interest
References
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA Double-Strand Breaks: Production, Fidelity of Repair, and Induction of Cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, Antioxidants, and the Degenerative Diseases of Aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.E. DNA Repair. Gatekeepers of Recombination. Nature 1999, 398, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and Decay of the Primary Structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Dizdaroglu, M. Oxidatively Induced DNA Damage: Mechanisms, Repair and Disease. Cancer Lett. 2012, 327, 26–47. [Google Scholar] [CrossRef]
- Lee, H.-C.; Wei, Y.-H. Oxidative Stress, Mitochondrial DNA Mutation, and Apoptosis in Aging. Exp. Biol. Med. 2007, 232, 592–606. [Google Scholar]
- Martínez, M.C.; Andriantsitohaina, R. Reactive Nitrogen Species: Molecular Mechanisms and Potential Significance in Health and Disease. Antioxid. Redox Signal. 2009, 11, 669–702. [Google Scholar] [CrossRef]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial Defects and Oxidative Stress in Alzheimer Disease and Parkinson Disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-Ros Crosstalk in the Control of Cell Death and Aging. J. Signal Transduct. 2012, 2012, 329635. [Google Scholar] [CrossRef]
- Slupphaug, G.; Kavli, B.; Krokan, H.E. The Interacting Pathways for Prevention and Repair of Oxidative DNA Damage. Mutat. Res. 2003, 531, 231–251. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Gesuete, R.; Orsini, F.; Zanier, E.R.; Albani, D.; Deli, M.A.; Bazzoni, G.; De Simoni, M.-G. Glial Cells Drive Preconditioning-Induced Blood-Brain Barrier Protection. Stroke 2011, 42, 1445–1453. [Google Scholar] [CrossRef]
- Shanbhag, N.M.; Evans, M.D.; Mao, W.; Nana, A.L.; Seeley, W.W.; Adame, A.; Rissman, R.A.; Masliah, E.; Mucke, L. Early Neuronal Accumulation of DNA Double Strand Breaks in Alzheimer’s Disease. Acta Neuropathol. Commun. 2019, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic Brain Activity Causes DNA Double-Strand Breaks in Neurons, with Exacerbation by Amyloid-β. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA Damage and Its Links to Neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J.A. Oxidative DNA Damage & Repair: An Introduction. Free Radic. Biol. Med. 2017, 107, 2–12. [Google Scholar] [CrossRef]
- Marlatt, M.; Lee, H.-G.; Perry, G.; Smith, M.A.; Zhu, X. Sources and Mechanisms of Cytoplasmic Oxidative Damage in Alzheimer’s Disease. Acta Neurobiol. Exp. 2004, 64, 81–87. [Google Scholar] [CrossRef]
- Bermúdez-Guzmán, L.; Leal, A. DNA Repair Deficiency in Neuropathogenesis: When All Roads Lead to Mitochondria. Transl. Neurodegener. 2019, 8, 14. [Google Scholar] [CrossRef]
- Norat, P.; Soldozy, S.; Sokolowski, J.D.; Gorick, C.M.; Kumar, J.S.; Chae, Y.; Yağmurlu, K.; Prada, F.; Walker, M.; Levitt, M.R.; et al. Mitochondrial Dysfunction in Neurological Disorders: Exploring Mitochondrial Transplantation. npj Regen. Med. 2020, 5, 22. [Google Scholar] [CrossRef]
- Gohil, D.; Sarker, A.H.; Roy, R. Base Excision Repair: Mechanisms and Impact in Biology, Disease, and Medicine. Int. J. Mol. Sci. 2023, 24, 14186. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Mantha, A.K.; Hazra, T.K.; Bhakat, K.K.; Mitra, S.; Szczesny, B. Oxidative Genome Damage and Its Repair: Implications in Aging and Neurodegenerative Diseases. Mech. Ageing Dev. 2012, 133, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Wilson, D.M.; Bohr, V.A. Base Excision Repair in the Mammalian Brain: Implication for Age Related Neurodegeneration. Mech. Ageing Dev. 2013, 134, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Talhaoui, I.; Matkarimov, B.T.; Tchenio, T.; Zharkov, D.O.; Saparbaev, M.K. Aberrant Base Excision Repair Pathway of Oxidatively Damaged DNA: Implications for Degenerative Diseases. Free Radic. Biol. Med. 2017, 107, 266–277. [Google Scholar] [CrossRef]
- Lautrup, S.; Myrup Holst, C.; Yde, A.; Asmussen, S.; Thinggaard, V.; Larsen, K.; Laursen, L.S.; Richner, M.; Vaegter, C.B.; Prieto, G.A.; et al. The Role of Aging and Brain-Derived Neurotrophic Factor Signaling in Expression of Base Excision Repair Genes in the Human Brain. Aging Cell 2023, 22, e13905. [Google Scholar] [CrossRef]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef]
- Dantzer, F.; De La Rubia, G.; Ménissier-de Murcia, J.; Hostomsky, Z.; De Murcia, G.; Schreiber, V. Base Excision Repair Is Impaired in Mammalian Cells Lacking Poly(ADP-Ribose) Polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early Steps in the DNA Base Excision/Single-Strand Interruption Repair Pathway in Mammalian Cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hegde, P.M.; Holthauzen, L.M.F.; Hazra, T.K.; Rao, K.S.J.; Mitra, S. Specific Inhibition of NEIL-Initiated Repair of Oxidized Base Damage in Human Genome by Copper and Iron: Potential Etiological Linkage to Neurodegenerative Diseases. J. Biol. Chem. 2010, 285, 28812–28825. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian DNA Base Excision Repair: Dancing in the Moonlight. DNA Repair 2020, 93, 102921. [Google Scholar] [CrossRef]
- Lavrik, O.I. PARPs’ Impact on Base Excision DNA Repair. DNA Repair 2020, 93, 102911. [Google Scholar] [CrossRef] [PubMed]
- Moor, N.; Lavrik, O. Coordination of DNA Base Excision Repair by Protein-Protein Interactions. In DNA Repair–An Update; IntechOpen: London, UK, 2019; ISBN 978-1-83880-783-2. [Google Scholar]
- Lindahl, T. An N-Glycosidase from Escherichia Coli That Releases Free Uracil from DNA Containing Deaminated Cytosine Residues. Proc. Natl. Acad. Sci. USA 1974, 71, 3649–3653. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, K.; Bjørås, K.Ø.; Bjørås, M. Diverse Functions of DNA Glycosylases Processing Oxidative Base Lesions in Brain. DNA Repair 2019, 81, 102665. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; McNeill, D.R.; Gleichmann, M.; Mattson, M.P.; Wilson, D.M. XRCC1 Protects against the Lethality of Induced Oxidative DNA Damage in Nondividing Neural Cells. Nucleic Acids Res. 2008, 36, 5111–5121. [Google Scholar] [CrossRef]
- Mitra, S.; Boldogh, I.; Izumi, T.; Hazra, T.K. Complexities of the DNA Base Excision Repair Pathway for Repair of Oxidative DNA Damage. Environ. Mol. Mutagen. 2001, 38, 180–190. [Google Scholar] [CrossRef]
- Peña-Diaz, J.; Jiricny, J. Mammalian Mismatch Repair: Error-Free or Error-Prone? Trends Biochem. Sci. 2012, 37, 206–214. [Google Scholar] [CrossRef]
- Wilson, D.M.; Barsky, D. The Major Human Abasic Endonuclease: Formation, Consequences and Repair of Abasic Lesions in DNA. Mutat. Res. 2001, 485, 283–307. [Google Scholar] [CrossRef]
- Krokeide, S.Z.; Bolstad, N.; Laerdahl, J.K.; Bjørås, M.; Luna, L. Expression and Purification of NEIL3, a Human DNA Glycosylase Homolog. Protein Expr. Purif. 2009, 65, 160–164. [Google Scholar] [CrossRef]
- Liu, M.; Doublié, S.; Wallace, S.S. Neil3, the Final Frontier for the DNA Glycosylases That Recognize Oxidative Damage. Mutat. Res. 2013, 743–744, 4–11. [Google Scholar] [CrossRef]
- Gajewski, S.; Comeaux, E.Q.; Jafari, N.; Bharatham, N.; Bashford, D.; White, S.W.; van Waardenburg, R.C.A.M. Analysis of the Active-Site Mechanism of Tyrosyl-DNA Phosphodiesterase I: A Member of the Phospholipase D Superfamily. J. Mol. Biol. 2012, 415, 741–758. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.N.; Das, B.B.; Dexheimer, T.S.; Takeda, S.; Pommier, Y. Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Repairs DNA Damage Induced by Topoisomerases I and II and Base Alkylation in Vertebrate Cells. J. Biol. Chem. 2012, 287, 12848–12857. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Lee, J.W.; Tatavarthi, H.; Lupski, J.R.; Valerie, K.; Povirk, L.F. Deficiency in 3’-Phosphoglycolate Processing in Human Cells with a Hereditary Mutation in Tyrosyl-DNA Phosphodiesterase (TDP1). Nucleic Acids Res. 2005, 33, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Interthal, H.; Chen, H.J.; Champoux, J.J. Human Tdp1 Cleaves a Broad Spectrum of Substrates, Including Phosphoamide Linkages. J. Biol. Chem. 2005, 280, 36518–36528. [Google Scholar] [CrossRef] [PubMed]
- Allinson, S.L.; Dianova, I.I.; Dianov, G.L. DNA Polymerase Beta Is the Major dRP Lyase Involved in Repair of Oxidative Base Lesions in DNA by Mammalian Cell Extracts. EMBO J. 2001, 20, 6919–6926. [Google Scholar] [CrossRef] [PubMed]
- Sobol, R.W.; Prasad, R.; Evenski, A.; Baker, A.; Yang, X.P.; Horton, J.K.; Wilson, S.H. The Lyase Activity of the DNA Repair Protein Beta-Polymerase Protects from DNA-Damage-Induced Cytotoxicity. Nature 2000, 405, 807–810. [Google Scholar] [CrossRef]
- Piersen, C.E.; Prasad, R.; Wilson, S.H.; Lloyd, R.S. Evidence for an Imino Intermediate in the DNA Polymerase Beta Deoxyribose Phosphate Excision Reaction. J. Biol. Chem. 1996, 271, 17811–17815. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Kim, K. Excision of Deoxyribose Phosphate Residues by DNA Polymerase Beta during DNA Repair. Science 1995, 269, 699–702. [Google Scholar] [CrossRef]
- Grin, I.R.; Khodyreva, S.N.; Nevinsky, G.A.; Zharkov, D.O. Deoxyribophosphate Lyase Activity of Mammalian Endonuclease VIII-like Proteins. FEBS Lett. 2006, 580, 4916–4922. [Google Scholar] [CrossRef]
- Tomkinson, A.E.; Chen, L.; Dong, Z.; Leppard, J.B.; Levin, D.S.; Mackey, Z.B.; Motycka, T.A. Completion of Base Excision Repair by Mammalian DNA Ligases. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 151–164. [Google Scholar] [CrossRef]
- Fortini, P.; Dogliotti, E. Base Damage and Single-Strand Break Repair: Mechanisms and Functional Significance of Short- and Long-Patch Repair Subpathways. DNA Repair 2007, 6, 398–409. [Google Scholar] [CrossRef]
- Liu, Y.; Beard, W.A.; Shock, D.D.; Prasad, R.; Hou, E.W.; Wilson, S.H. DNA Polymerase Beta and Flap Endonuclease 1 Enzymatic Specificities Sustain DNA Synthesis for Long Patch Base Excision Repair. J. Biol. Chem. 2005, 280, 3665–3674. [Google Scholar] [CrossRef] [PubMed]
- Chagovetz, A.M.; Sweasy, J.B.; Preston, B.D. Increased Activity and Fidelity of DNA Polymerase β on Single-Nucleotide Gapped DNA. J. Biol. Chem. 1997, 272, 27501–27504. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Bohr, V.A. The Mechanics of Base Excision Repair, and Its Relationship to Aging and Disease. DNA Repair 2007, 6, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Sukhanova, M.; Khodyreva, S.; Lavrik, O. Poly(ADP-Ribose) Polymerase 1 Regulates Activity of DNA Polymerase Beta in Long Patch Base Excision Repair. Mutat. Res. 2010, 685, 80–89. [Google Scholar] [CrossRef]
- Dalhus, B.; Laerdahl, J.K.; Backe, P.H.; Bjørås, M. DNA Base Repair—Recognition and Initiation of Catalysis. FEMS Microbiol. Rev. 2009, 33, 1044–1078. [Google Scholar] [CrossRef]
- Srivastava, D.K.; Berg, B.J.; Prasad, R.; Molina, J.T.; Beard, W.A.; Tomkinson, A.E.; Wilson, S.H. Mammalian Abasic Site Base Excision Repair. Identification of the Reaction Sequence and Rate-Determining Steps. J. Biol. Chem. 1998, 273, 21203–21209. [Google Scholar] [CrossRef]
- Horton, J.K.; Prasad, R.; Hou, E.; Wilson, S.H. Protection against Methylation-Induced Cytotoxicity by DNA Polymerase Beta-Dependent Long Patch Base Excision Repair. J. Biol. Chem. 2000, 275, 2211–2218. [Google Scholar] [CrossRef]
- Akbari, M.; Peña-Diaz, J.; Andersen, S.; Liabakk, N.-B.; Otterlei, M.; Krokan, H.E. Extracts of Proliferating and Non-Proliferating Human Cells Display Different Base Excision Pathways and Repair Fidelity. DNA Repair 2009, 8, 834–843. [Google Scholar] [CrossRef]
- Wei, W.; Englander, E.W. DNA Polymerase Beta-Catalyzed-PCNA Independent Long Patch Base Excision Repair Synthesis: A Mechanism for Repair of Oxidatively Damaged DNA Ends in Post-Mitotic Brain. J. Neurochem. 2008, 107, 734–744. [Google Scholar] [CrossRef]
- Wilson, S.H. Mammalian Base Excision Repair and DNA Polymerase Beta. Mutat. Res. 1998, 407, 203–215. [Google Scholar] [CrossRef]
- Woodrick, J.; Gupta, S.; Camacho, S.; Parvathaneni, S.; Choudhury, S.; Cheema, A.; Bai, Y.; Khatkar, P.; Erkizan, H.V.; Sami, F.; et al. A New Sub-Pathway of Long-Patch Base Excision Repair Involving 5’ Gap Formation. EMBO J. 2017, 36, 1605–1622. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.A.; Rechkunova, N.I.; Lavrik, O.I. AP-Site Cleavage Activity of Tyrosyl-DNA Phosphodiesterase 1. FEBS Lett. 2011, 585, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Vasil’eva, I.A.; Anarbaev, R.O.; Moor, N.A.; Lavrik, O.I. Dynamic Light Scattering Study of Base Excision DNA Repair Proteins and Their Complexes. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Wiederhold, L.; Leppard, J.B.; Kedar, P.; Prasad, R.; Wang, H.; Boldogh, I.; Karimi-Busheri, F.; Weinfeld, M.; Tomkinson, A.E.; et al. NEIL2-Initiated, APE-Independent Repair of Oxidized Bases in DNA: Evidence for a Repair Complex in Human Cells. DNA Repair 2006, 5, 1439–1448. [Google Scholar] [CrossRef]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP Endonuclease-Independent DNA Base Excision Repair in Human Cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef]
- Gros, L.; Ishchenko, A.A.; Ide, H.; Elder, R.H.; Saparbaev, M.K. The Major Human AP Endonuclease (Ape1) Is Involved in the Nucleotide Incision Repair Pathway. Nucleic Acids Res. 2004, 32, 73–81. [Google Scholar] [CrossRef]
- Ischenko, A.A.; Saparbaev, M.K. Alternative Nucleotide Incision Repair Pathway for Oxidative DNA Damage. Nature 2002, 415, 183–187. [Google Scholar] [CrossRef]
- Hanawalt, P.C.; Spivak, G. Transcription-Coupled DNA Repair: Two Decades of Progress and Surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef]
- Portman, J.R.; Strick, T.R. Transcription-Coupled Repair and Complex Biology. J. Mol. Biol. 2018, 430, 4496–4512. [Google Scholar] [CrossRef]
- Chakraborty, A.; Tapryal, N.; Islam, A.; Mitra, S.; Hazra, T. Transcription Coupled Base Excision Repair in Mammalian Cells: So Little Is Known and so Much to Uncover. DNA Repair 2021, 107, 103204. [Google Scholar] [CrossRef]
- Guo, J.; Hanawalt, P.C.; Spivak, G. Comet-FISH with Strand-Specific Probes Reveals Transcription-Coupled Repair of 8-oxoGuanine in Human Cells. Nucleic Acids Res. 2013, 41, 7700–7712. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Wakamiya, M.; Venkova-Canova, T.; Pandita, R.K.; Aguilera-Aguirre, L.; Sarker, A.H.; Singh, D.K.; Hosoki, K.; Wood, T.G.; Sharma, G.; et al. Neil2-Null Mice Accumulate Oxidized DNA Bases in the Transcriptionally Active Sequences of the Genome and Are Susceptible to Innate Inflammation. J. Biol. Chem. 2015, 290, 24636–24648. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Mandal, S.M.; Das, A.; Hegde, M.L.; Das, S.; Bhakat, K.K.; Boldogh, I.; Sarkar, P.S.; Mitra, S.; Hazra, T.K. Preferential Repair of Oxidized Base Damage in the Transcribed Genes of Mammalian Cells. J. Biol. Chem. 2011, 286, 6006–6016. [Google Scholar] [CrossRef] [PubMed]
- Englander, E.W.; Ma, H. Differential Modulation of Base Excision Repair Activities during Brain Ontogeny: Implications for Repair of Transcribed DNA. Mech. Ageing Dev. 2006, 127, 64–69. [Google Scholar] [CrossRef]
- Krasikova, Y.; Rechkunova, N.; Lavrik, O. Nucleotide Excision Repair: From Molecular Defects to Neurological Abnormalities. Int. J. Mol. Sci. 2021, 22, 6220. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, M.; Hu, J. Nucleotide Excision Repair: A Versatile and Smart Toolkit. Acta Biochim. Biophys. Sin. 2022, 54, 807–819. [Google Scholar] [CrossRef]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Spivak, G. Nucleotide Excision Repair in Humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding Nucleotide Excision Repair and Its Roles in Cancer and Ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Schärer, O.D. Achieving Broad Substrate Specificity in Damage Recognition by Binding Accessible Nondamaged DNA. Mol. Cell 2007, 28, 184–186. [Google Scholar] [CrossRef]
- van den Heuvel, D.; van der Weegen, Y.; Boer, D.E.C.; Ogi, T.; Luijsterburg, M.S. Transcription-Coupled DNA Repair: From Mechanism to Human Disorder. Trends Cell Biol. 2021, 31, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, F.; Paccosi, E.; Proietti-De-Santis, L.; Egly, J.M. CS Proteins and Ubiquitination: Orchestrating DNA Repair with Transcription and Cell Division. Trends Cell Biol. 2024, 34, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Nieto Moreno, N.; Olthof, A.M.; Svejstrup, J.Q. Transcription-Coupled Nucleotide Excision Repair and the Transcriptional Response to UV-Induced DNA Damage. Annu. Rev. Biochem. 2023, 92, 81–113. [Google Scholar] [CrossRef] [PubMed]
- Selby, C.P.; Lindsey-Boltz, L.A.; Li, W.; Sancar, A. Molecular Mechanisms of Transcription-Coupled Repair. Annu. Rev. Biochem. 2023, 92, 115–144. [Google Scholar] [CrossRef]
- Kajitani, G.S.; de Souza Nascimento, L.L.; de Camargo Neves, M.R.; da Silva Leandro, G.; Garcia, C.C.M.; Menck, C.F.M. Transcription Blockage by DNA Damage in Nucleotide Excision Repair-Related Neurological Dysfunctions. Semin. Cell Dev. Biol. 2021, 114, 20–35. [Google Scholar] [CrossRef]
- Nouspikel, T.; Hanawalt, P.C. Terminally Differentiated Human Neurons Repair Transcribed Genes but Display Attenuated Global DNA Repair and Modulation of Repair Gene Expression. Mol. Cell. Biol. 2000, 20, 1562–1570. [Google Scholar] [CrossRef]
- Li, W.; Liu, W.; Kakoki, A.; Wang, R.; Adebali, O.; Jiang, Y.; Sancar, A. Nucleotide Excision Repair Capacity Increases during Differentiation of Human Embryonic Carcinoma Cells into Neurons and Muscle Cells. J. Biol. Chem. 2019, 294, 5914–5922. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Schaich, M.A.; Smith, M.R.; Flynn, T.S.; Freudenthal, B.D. Base Excision Repair of Oxidative DNA Damage: From Mechanism to Disease. Front. Biosci. 2017, 22, 1493–1522. [Google Scholar] [CrossRef]
- Shafirovich, V.; Geacintov, N.E. Removal of Oxidatively Generated DNA Damage by Overlapping Repair Pathways. Free Radic. Biol. Med. 2017, 107, 53–61. [Google Scholar] [CrossRef]
- Kumar, N.; Raja, S.; Van Houten, B. The Involvement of Nucleotide Excision Repair Proteins in the Removal of Oxidative DNA Damage. Nucleic Acids Res. 2020, 48, 11227–11243. [Google Scholar] [CrossRef]
- Melis, J.P.M.; van Steeg, H.; Luijten, M. Oxidative DNA Damage and Nucleotide Excision Repair. Antioxid. Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Soulas-Sprauel, P.; Rivera-Munoz, P.; Malivert, L.; Le Guyader, G.; Abramowski, V.; Revy, P.; de Villartay, J.-P. V(D)J and Immunoglobulin Class Switch Recombinations: A Paradigm to Study the Regulation of DNA End-Joining. Oncogene 2007, 26, 7780–7791. [Google Scholar] [CrossRef] [PubMed]
- Malu, S.; Malshetty, V.; Francis, D.; Cortes, P. Role of Non-Homologous End Joining in V(D)J Recombination. Immunol. Res. 2012, 54, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Schatz, D.G.; Swanson, P.C. V(D)J Recombination: Mechanisms of Initiation. Annu. Rev. Genet. 2011, 45, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Qing, X.; Zhang, G.; Wang, Z.-Q. DNA Damage Response in Neurodevelopment and Neuromaintenance. FEBS J. 2023, 290, 3300–3310. [Google Scholar] [CrossRef]
- Weber Boutros, S.; Unni, V.K.; Raber, J. An Adaptive Role for DNA Double-Strand Breaks in Hippocampus-Dependent Learning and Memory. Int. J. Mol. Sci. 2022, 23, 8352. [Google Scholar] [CrossRef]
- Alt, F.W.; Schwer, B. DNA Double-Strand Breaks as Drivers of Neural Genomic Change, Function, and Disease. DNA Repair 2018, 71, 158–163. [Google Scholar] [CrossRef]
- Gospodinov, A.; Ugrinova, I. Chromatin Control in Double Strand Break Repair. Adv. Protein Chem. Struct. Biol. 2019, 115, 69–94. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Boboila, C.; Alt, F.W.; Schwer, B. Classical and Alternative End-Joining Pathways for Repair of Lymphocyte-Specific and General DNA Double-Strand Breaks. Adv. Immunol. 2012, 116, 1–49. [Google Scholar]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of Eukaryotic Homologous Recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef]
- Wei, L.; Nakajima, S.; Böhm, S.; Bernstein, K.A.; Shen, Z.; Tsang, M.; Levine, A.S.; Lan, L. DNA Damage during the G0/G1 Phase Triggers RNA-Templated, Cockayne Syndrome B-Dependent Homologous Recombination. Proc. Natl. Acad. Sci. USA 2015, 112, E3495–E3504. [Google Scholar] [CrossRef]
- Welty, S.; Teng, Y.; Liang, Z.; Zhao, W.; Sanders, L.H.; Greenamyre, J.T.; Rubio, M.E.; Thathiah, A.; Kodali, R.; Wetzel, R.; et al. RAD52 Is Required for RNA-Templated Recombination Repair in Post-Mitotic Neurons. J. Biol. Chem. 2018, 293, 1353–1362. [Google Scholar] [CrossRef]
- Stinson, B.M.; Loparo, J.J. Repair of DNA Double-Strand Breaks by the Nonhomologous End Joining Pathway. Annu. Rev. Biochem. 2021, 90, 137–164. [Google Scholar] [CrossRef]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The Molecular Basis and Disease Relevance of Non-Homologous DNA End Joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef]
- Ghosh, D.; Raghavan, S.C. Nonhomologous End Joining: New Accessory Factors Fine Tune the Machinery. Trends Genet. 2021, 37, 582–599. [Google Scholar] [CrossRef]
- Dylgjeri, E.; Knudsen, K.E. DNA-PKcs: A Targetable Protumorigenic Protein Kinase. Cancer Res. 2022, 82, 523–533. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Watanabe, G.; Gerodimos, C.A.; Ochi, T.; Blundell, T.L.; Jackson, S.P.; Lieber, M.R. Different DNA End Configurations Dictate Which NHEJ Components Are Most Important for Joining Efficiency. J. Biol. Chem. 2016, 291, 24377–24389. [Google Scholar] [CrossRef]
- Watanabe, G.; Lieber, M.R. Dynamics of the Artemis and DNA-PKcs Complex in the Repair of Double-Strand Breaks. J. Mol. Biol. 2022, 434, 167858. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Perrault, A.R.; Takeda, Y.; Qin, W.; Wang, H.; Iliakis, G. Biochemical Evidence for Ku-Independent Backup Pathways of NHEJ. Nucleic Acids Res. 2020, 48, 5200. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku Compete for Repair of DNA Double Strand Breaks by Distinct NHEJ Pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Liu, C.; Chen, S.-H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-Resolution Imaging Identifies PARP1 and the Ku Complex Acting as DNA Double-Strand Break Sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of Microhomology-Mediated End-Joining Promoted by Human DNA Polymerase θ. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef]
- Paul, K.; Wang, M.; Mladenov, E.; Bencsik-Theilen, A.; Bednar, T.; Wu, W.; Arakawa, H.; Iliakis, G. DNA Ligases I and III Cooperate in Alternative Non-Homologous End-Joining in Vertebrates. PLoS ONE 2013, 8, e59505. [Google Scholar] [CrossRef]
- Della-Maria, J.; Zhou, Y.; Tsai, M.-S.; Kuhnlein, J.; Carney, J.P.; Paull, T.T.; Tomkinson, A.E. Human Mre11/Human Rad50/Nbs1 and DNA Ligase IIIα/XRCC1 Protein Complexes Act Together in an Alternative Nonhomologous End Joining Pathway. J. Biol. Chem. 2011, 286, 33845–33853. [Google Scholar] [CrossRef]
- Singh, S.K.; Bednar, T.; Zhang, L.; Wu, W.; Mladenov, E.; Iliakis, G. Inhibition of B-NHEJ in Plateau-Phase Cells Is Not a Direct Consequence of Suppressed Growth Factor Signaling. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, e237–e243. [Google Scholar] [CrossRef]
- Hendrickson, E.A. RAD52: Viral Friend or Foe? Cancers 2020, 12, 399. [Google Scholar] [CrossRef]
- Mladenov, E.; Mladenova, V.; Stuschke, M.; Iliakis, G. New Facets of DNA Double Strand Break Repair: Radiation Dose as Key Determinant of HR versus c-NHEJ Engagement. Int. J. Mol. Sci. 2023, 24, 14956. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and Its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed]
- van de Kooij, B.; Kruswick, A.; van Attikum, H.; Yaffe, M.B. Multi-Pathway DNA-Repair Reporters Reveal Competition between End-Joining, Single-Strand Annealing and Homologous Recombination at Cas9-Induced DNA Double-Strand Breaks. Nat. Commun. 2022, 13, 5295. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Tapryal, N.; Venkova, T.; Horikoshi, N.; Pandita, R.K.; Sarker, A.H.; Sarkar, P.S.; Pandita, T.K.; Hazra, T.K. Classical Non-Homologous End-Joining Pathway Utilizes Nascent RNA for Error-Free Double-Strand Break Repair of Transcribed Genes. Nat. Commun. 2016, 7, 13049. [Google Scholar] [CrossRef] [PubMed]
- Giannini, M.; Bayona-Feliu, A.; Sproviero, D.; Barroso, S.I.; Cereda, C.; Aguilera, A. TDP-43 Mutations Link Amyotrophic Lateral Sclerosis with R-Loop Homeostasis and R Loop-Mediated DNA Damage. PLoS Genet. 2020, 16, e1009260. [Google Scholar] [CrossRef]
- Cristini, A.; Gromak, N.; Sordet, O. Transcription-Dependent DNA Double-Strand Breaks and Human Disease. Mol. Cell. Oncol. 2020, 7, 1691905. [Google Scholar] [CrossRef]
- Yasuhara, T.; Kato, R.; Yamauchi, M.; Uchihara, Y.; Zou, L.; Miyagawa, K.; Shibata, A. RAP80 Suppresses the Vulnerability of R-Loops during DNA Double-Strand Break Repair. Cell. Rep. 2022, 38, 110335. [Google Scholar] [CrossRef]
- Chakraborty, A.; Tapryal, N.; Islam, A.; Sarker, A.H.; Manohar, K.; Mitra, J.; Hegde, M.L.; Hazra, T. Human DNA Polymerase η Promotes RNA-Templated Error-Free Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2023, 299, 102991. [Google Scholar] [CrossRef]
- Pradhan, S.; Bush, K.; Zhang, N.; Pandita, R.K.; Tsai, C.-L.; Smith, C.; Pandlebury, D.F.; Gaikwad, S.; Leonard, F.; Nie, L.; et al. Chromatin Remodeler BRG1 Recruits Huntingtin to Repair DNA Double-Strand Breaks in Neurons. bioRxiv 2024. [Google Scholar] [CrossRef]
- Aquilina, G.; Biondo, R.; Dogliotti, E.; Meuth, M.; Bignami, M. Expression of the Endogenous O6-Methylguanine-DNA-Methyltransferase Protects Chinese Hamster Ovary Cells from Spontaneous G:C to A:T Transitions. Cancer Res. 1992, 52, 6471–6475. [Google Scholar]
- Kang, H.; Konishi, C.; Kuroki, T.; Huh, N. A Highly Sensitive and Specific Method for Quantitation of O-Alkylated DNA Adducts and Its Application to the Analysis of Human Tissue DNA. Environ. Health Perspect. 1993, 99, 269–271. [Google Scholar] [CrossRef]
- Kang, H.; Konishi, C.; Kuroki, T.; Huh, N. Detection of O6-Methylguanine, O4-Methylthymine and O4-Ethylthymine in Human Liver and Peripheral Blood Leukocyte DNA. Carcinogenesis 1995, 16, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Nakamura, T.; Maki, H.; Sekiguchi, M. Roles of Transcription and Repair in Alkylation Mutagenesis. Mutat. Res. 1994, 314, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.A.; Dreij, K.; Cartularo, L.; Scicchitano, D.A. O6-Methylguanine Induces Altered Proteins at the Level of Transcription in Human Cells. Nucleic Acids Res. 2010, 38, 8178–8187. [Google Scholar] [CrossRef] [PubMed]
- Dimitri, A.; Burns, J.A.; Broyde, S.; Scicchitano, D.A. Transcription Elongation Past O6-Methylguanine by Human RNA Polymerase II and Bacteriophage T7 RNA Polymerase. Nucleic Acids Res. 2008, 36, 6459–6471. [Google Scholar] [CrossRef] [PubMed]
- Xu-Welliver, M.; Pegg, A.E. Degradation of the Alkylated Form of the DNA Repair Protein, O(6)-Alkylguanine-DNA Alkyltransferase. Carcinogenesis 2002, 23, 823–830. [Google Scholar] [CrossRef]
- Iyer, R.R.; Pluciennik, A. DNA Mismatch Repair and Its Role in Huntington’s Disease. J. Huntingt. Dis. 2021, 10, 75–94. [Google Scholar] [CrossRef]
- Provasek, V.E.; Kodavati, M.; Kim, B.; Mitra, J.; Hegde, M.L. TDP43 Interacts with MLH1 and MSH6 Proteins in a DNA Damage-Inducible Manner. Mol. Brain 2024, 17, 32. [Google Scholar] [CrossRef]
- Provasek, V.E.; Bacolla, A.; Rangaswamy, S.; Mitra, J.; Kodavati, M.; Yusuf, I.O.; Malojirao, V.H.; Vasquez, V.; Britz, G.W.; Li, G.-M.; et al. RNA/DNA Binding Protein TDP43 Regulates DNA Mismatch Repair Genes with Implications for Genome Stability. bioRxiv 2024. [Google Scholar] [CrossRef]
- Bhattacharya, M.R.C. A Nerve-Wracking Buzz: Lessons from Drosophila Models of Peripheral Neuropathy and Axon Degeneration. Front. Aging Neurosci. 2023, 15, 1166146. [Google Scholar] [CrossRef]
- Naranjo-Galindo, F.J.; Ai, R.; Fang, E.F.; Nilsen, H.L.; SenGupta, T.C. Elegans as an Animal Model to Study the Intersection of DNA Repair, Aging and Neurodegeneration. Front. Aging 2022, 3, 916118. [Google Scholar] [CrossRef]
- Abugable, A.A.; Morris, J.L.M.; Palminha, N.M.; Zaksauskaite, R.; Ray, S.; El-Khamisy, S.F. DNA Repair and Neurological Disease: From Molecular Understanding to the Development of Diagnostics and Model Organisms. DNA Repair 2019, 81, 102669. [Google Scholar] [CrossRef] [PubMed]
- Andressoo, J.-O.; Weeda, G.; De Wit, J.; Mitchell, J.R.; Beems, R.B.; Van Steeg, H.; Van Der Horst, G.T.J.; Hoeijmakers, J.H. An Xpb Mouse Model for Combined Xeroderma Pigmentosum and Cockayne Syndrome Reveals Progeroid Features upon Further Attenuation of DNA Repair. Mol. Cell. Biol. 2009, 29, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, G.T.; van Steeg, H.; Berg, R.J.; van Gool, A.J.; de Wit, J.; Weeda, G.; Morreau, H.; Beems, R.B.; van Kreijl, C.F.; de Gruijl, F.R.; et al. Defective Transcription-Coupled Repair in Cockayne Syndrome B Mice Is Associated with Skin Cancer Predisposition. Cell 1997, 89, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Laposa, R.R.; Huang, E.J.; Cleaver, J.E. Increased Apoptosis, P53 up-Regulation, and Cerebellar Neuronal Degeneration in Repair-Deficient Cockayne Syndrome Mice. Proc. Natl. Acad. Sci. USA 2007, 104, 1389–1394. [Google Scholar] [CrossRef]
- Murai, M.; Enokido, Y.; Inamura, N.; Yoshino, M.; Nakatsu, Y.; van der Horst, G.T.; Hoeijmakers, J.H.; Tanaka, K.; Hatanaka, H. Early Postnatal Ataxia and Abnormal Cerebellar Development in Mice Lacking Xeroderma Pigmentosum Group A and Cockayne Syndrome Group B DNA Repair Genes. Proc. Natl. Acad. Sci. USA 2001, 98, 13379–13384. [Google Scholar] [CrossRef]
- Katyal, S.; El-Khamisy, S.F.; Russell, H.R.; Li, Y.; Ju, L.; Caldecott, K.W.; McKinnon, P.J. TDP1 Facilitates Chromosomal Single-Strand Break Repair in Neurons and Is Neuroprotective in Vivo. EMBO J. 2007, 26, 4720–4731. [Google Scholar] [CrossRef]
- Walker, C.; El-Khamisy, S.F. Perturbed Autophagy and DNA Repair Converge to Promote Neurodegeneration in Amyotrophic Lateral Sclerosis and Dementia. Brain 2018, 141, 1247–1262. [Google Scholar] [CrossRef]
- Hoch, N.C.; Hanzlikova, H.; Rulten, S.L.; Tétreault, M.; Komulainen, E.; Ju, L.; Hornyak, P.; Zeng, Z.; Gittens, W.; Rey, S.A.; et al. XRCC1 Mutation Is Associated with PARP1 Hyperactivation and Cerebellar Ataxia. Nature 2017, 541, 87–91. [Google Scholar] [CrossRef]
- Dumitrache, L.C.; McKinnon, P.J. Polynucleotide Kinase-Phosphatase (PNKP) Mutations and Neurologic Disease. Mech. Ageing Dev. 2017, 161, 121–129. [Google Scholar] [CrossRef]
- Sykora, P.; Misiak, M.; Wang, Y.; Ghosh, S.; Leandro, G.S.; Liu, D.; Tian, J.; Baptiste, B.A.; Cong, W.-N.; Brenerman, B.M.; et al. DNA Polymerase β Deficiency Leads to Neurodegeneration and Exacerbates Alzheimer Disease Phenotypes. Nucleic Acids Res. 2015, 43, 943–959. [Google Scholar] [CrossRef]
- Gao, Y.; Katyal, S.; Lee, Y.; Zhao, J.; Rehg, J.E.; Russell, H.R.; McKinnon, P.J. DNA Ligase III Is Critical for mtDNA Integrity but Not Xrcc1-Mediated Nuclear DNA Repair. Nature 2011, 471, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Bunting, S.; Montaña, M.F.; Soria, R.; Mulero, F.; Cañamero, M.; Lee, Y.; McKinnon, P.J.; Nussenzweig, A.; Fernandez-Capetillo, O. A Mouse Model of ATR-Seckel Shows Embryonic Replicative Stress and Accelerated Aging. Nat. Genet. 2009, 41, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.M.; Sharpless, N.E.; Gao, Y.; Sekiguchi, J.M.; Ferguson, D.O.; Zhu, C.; Manis, J.P.; Horner, J.; DePinho, R.A.; Alt, F.W. DNA Ligase IV Deficiency in Mice Leads to Defective Neurogenesis and Embryonic Lethality via the P53 Pathway. Mol. Cell. 2000, 5, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Quek, H.; Luff, J.; Cheung, K.; Kozlov, S.; Gatei, M.; Lee, C.S.; Bellingham, M.C.; Noakes, P.G.; Lim, Y.C.; Barnett, N.L.; et al. Rats with a Missense Mutation in Atm Display Neuroinflammation and Neurodegeneration Subsequent to Accumulation of Cytosolic DNA Following Unrepaired DNA Damage. J. Leukoc. Biol. 2017, 101, 927–947. [Google Scholar] [CrossRef]
- Xu, Y.; Ashley, T.; Brainerd, E.E.; Bronson, R.T.; Meyn, M.S.; Baltimore, D. Targeted Disruption of ATM Leads to Growth Retardation, Chromosomal Fragmentation during Meiosis, Immune Defects, and Thymic Lymphoma. Genes Dev. 1996, 10, 2411–2422. [Google Scholar] [CrossRef]
- Kuljis, R.O.; Xu, Y.; Aguila, M.C.; Baltimore, D. Degeneration of Neurons, Synapses, and Neuropil and Glial Activation in a Murine Atm Knockout Model of Ataxia-Telangiectasia. Proc. Natl. Acad. Sci. USA 1997, 94, 12688–12693. [Google Scholar] [CrossRef]
- Shacham, T.; Galron, R.; Bihari, O.; Kanner, S.; Barzilai, A. The Role of Malfunctioning DNA Damage Response (DDR) in Brain Degeneration. Opera Med. Physiol. 2016, 2, 141–152. [Google Scholar] [CrossRef]
- McConnell, M.J.; Kaushal, D.; Yang, A.H.; Kingsbury, M.A.; Rehen, S.K.; Treuner, K.; Helton, R.; Annas, E.G.; Chun, J.; Barlow, C. Failed Clearance of Aneuploid Embryonic Neural Progenitor Cells Leads to Excess Aneuploidy in the Atm-Deficient but Not the Trp53-Deficient Adult Cerebral Cortex. J. Neurosci. 2004, 24, 8090–8096. [Google Scholar] [CrossRef]
- Herzog, K.H.; Chong, M.J.; Kapsetaki, M.; Morgan, J.I.; McKinnon, P.J. Requirement for Atm in Ionizing Radiation-Induced Cell Death in the Developing Central Nervous System. Science 1998, 280, 1089–1091. [Google Scholar] [CrossRef]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef]
- Ryan, V.H.; Fawzi, N.L. Physiological, Pathological, and Targetable Membraneless Organelles in Neurons. Trends Neurosci. 2019, 42, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active Liquid-like Behavior of Nucleoli Determines Their Size and Shape in Xenopus Laevis Oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Biological Liquid-Liquid Phase Separation, Biomolecular Condensates, and Membraneless Organelles: Now You See Me, Now You Don’t. Int. J. Mol. Sci. 2023, 24, 13150. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhou, F.; Zhang, L. PARP1-DNA Co-Condensation: The Driver of Broken DNA Repair. Signal Transduct. Target. Ther. 2024, 9, 135. [Google Scholar] [CrossRef]
- Spannl, S.; Tereshchenko, M.; Mastromarco, G.J.; Ihn, S.J.; Lee, H.O. Biomolecular Condensates in Neurodegeneration and Cancer. Traffic 2019, 20, 890–911. [Google Scholar] [CrossRef]
- Khorsand, F.R.; Uversky, V.N. Liquid-Liquid Phase Separation as Triggering Factor of Fibril Formation. Prog. Mol. Biol. Transl. Sci. 2024, 206, 143–182. [Google Scholar] [CrossRef]
- Mehta, S.; Zhang, J. Liquid-Liquid Phase Separation Drives Cellular Function and Dysfunction in Cancer. Nat. Rev. Cancer 2022, 22, 239–252. [Google Scholar] [CrossRef]
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase Separation of 53BP1 Determines Liquid-like Behavior of DNA Repair Compartments. EMBO J. 2019, 38, e101379. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.; Chen, H.; Yang, Y.; Mu, C.; Ren, H.; Liu, Y.; Yu, L.; Fang, Q.; Wang, G.; et al. Disrupted Phase Behavior of FUS Underlies Poly-PR-Induced DNA Damage in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2023, 33, 64–77. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Kriwacki, R.W. Phase Separation in Biology; Functional Organization of a Higher Order. Cell Commun. Signal 2016, 14, 1. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically Unstructured Proteins and Their Functions. Nat. Rev. Mol. Cell. Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Forman-Kay, J.D.; Mittag, T. From Sequence and Forces to Structure, Function, and Evolution of Intrinsically Disordered Proteins. Structure 2013, 21, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Kodavati, M.; Maloji Rao, V.H.; Provasek, V.E.; Hegde, M.L. Regulation of DNA Damage Response by RNA/DNA-Binding Proteins: Implications for Neurological Disorders and Aging. Ageing Res. Rev. 2024, 100, 102413. [Google Scholar] [CrossRef]
- Tsoi, P.S.; Quan, M.D.; Ferreon, J.C.; Ferreon, A.C.M. Aggregation of Disordered Proteins Associated with Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 3380. [Google Scholar] [CrossRef] [PubMed]
- Vassallu, F.; Igaz, L.M. TDP-43 Nuclear Condensation and Neurodegenerative Proteinopathies. Trends Neurosci. 2024, 47, 849–850. [Google Scholar] [CrossRef]
- Milicevic, K.; Rankovic, B.; Andjus, P.R.; Bataveljic, D.; Milovanovic, D. Emerging Roles for Phase Separation of RNA-Binding Proteins in Cellular Pathology of ALS. Front. Cell Dev. Biol. 2022, 10, 840256. [Google Scholar] [CrossRef]
- Levone, B.R.; Lenzken, S.C.; Antonaci, M.; Maiser, A.; Rapp, A.; Conte, F.; Reber, S.; Mechtersheimer, J.; Ronchi, A.E.; Mühlemann, O.; et al. FUS-Dependent Liquid-Liquid Phase Separation Is Important for DNA Repair Initiation. J. Cell Biol. 2021, 220, e202008030. [Google Scholar] [CrossRef]
- Carey, J.L.; Guo, L. Liquid-Liquid Phase Separation of TDP-43 and FUS in Physiology and Pathology of Neurodegenerative Diseases. Front. Mol. Biosci. 2022, 9, 826719. [Google Scholar] [CrossRef]
- Naskar, A.; Nayak, A.; Salaikumaran, M.R.; Vishal, S.S.; Gopal, P.P. Phase Separation and Pathologic Transitions of RNP Condensates in Neurons: Implications for Amyotrophic Lateral Sclerosis, Frontotemporal Dementia and Other Neurodegenerative Disorders. Front. Mol. Neurosci. 2023, 16, 1242925. [Google Scholar] [CrossRef]
- Duan, Y.; Du, A.; Gu, J.; Duan, G.; Wang, C.; Gui, X.; Ma, Z.; Qian, B.; Deng, X.; Zhang, K.; et al. PARylation Regulates Stress Granule Dynamics, Phase Separation, and Neurotoxicity of Disease-Related RNA-Binding Proteins. Cell Res. 2019, 29, 233–247. [Google Scholar] [CrossRef]
- Boyko, S.; Qi, X.; Chen, T.-H.; Surewicz, K.; Surewicz, W.K. Liquid–Liquid Phase Separation of Tau Protein: The Crucial Role of Electrostatic Interactions. J. Biol. Chem. 2019, 294, 11054–11059. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Wei, M.; Xu, F.; Niu, Z. Aggregation and Phase Separation of α-Synuclein in Parkinson’s Disease. Chem. Commun. 2024, 60, 6581–6590. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, R.; Nabika, H. Liquid-Liquid Phase Separation Induced by Crowding Condition Affects Amyloid-β Aggregation Mechanism. Soft. Matter. 2024, 20, 5331–5342. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, S.; Manohar, A.; Mani, E. Liquid-Liquid Phase Separation (LLPS)-Driven Fibrilization of Amyloid-β Protein. ACS Chem. Neurosci. 2023, 14, 3655–3664. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Lavrik, O.I. A sePARate Phase? Poly(ADP-Ribose) versus RNA in the Organization of Biomolecular Condensates. Nucleic Acids Res. 2022, 50, 10817–10838. [Google Scholar] [CrossRef]
- Leung, A.K.L. Poly(ADP-Ribose): A Dynamic Trigger for Biomolecular Condensate Formation. Trends Cell Biol. 2020, 30, 370–383. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-Ribose) in Condensates: The PARtnership of Phase Separation and Site-Specific Interactions. Int. J. Mol. Sci. 2022, 23, 14075. [Google Scholar] [CrossRef]
- Rhine, K.; Odeh, H.M.; Shorter, J.; Myong, S. Regulation of Biomolecular Condensates by Poly(ADP-Ribose). Chem. Rev. 2023, 123, 9065–9093. [Google Scholar] [CrossRef]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grøfte, M.; Rask, M.-B.D.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid Demixing of Intrinsically Disordered Proteins Is Seeded by Poly(ADP-Ribose). Nat. Commun. 2015, 6, 8088. [Google Scholar] [CrossRef]
- Sukhanova, M.V.; Anarbaev, R.O.; Maltseva, E.A.; Kutuzov, M.M.; Lavrik, O.I. Divalent and Multivalent Cations Control Liquid-like Assembly of Poly(ADP-Ribosyl)Ated PARP1 into Multimolecular Associates in Vitro. Commun. Biol. 2024, 7, 1148. [Google Scholar] [CrossRef] [PubMed]
- Singatulina, A.S.; Hamon, L.; Sukhanova, M.V.; Desforges, B.; Joshi, V.; Bouhss, A.; Lavrik, O.I.; Pastré, D. PARP-1 Activation Directs FUS to DNA Damage Sites to Form PARG-Reversible Compartments Enriched in Damaged DNA. Cell Rep. 2019, 27, 1809–1821.e5. [Google Scholar] [CrossRef] [PubMed]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)—Function in Genome Maintenance and Relevance of Inhibitors for Anti-Cancer Therapy. Front. Mol. Biosci. 2020, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-L.; Pan, Y.-R.; Yong, Y.-Y.; Liu, Y.; Yu, L.; Qin, D.-L.; Qiao, G.; Law, B.Y.-K.; Wu, J.-M.; Zhou, X.-G.; et al. Poly (ADP-Ribose) Polymerase 1 and Neurodegenerative Diseases: Past, Present, and Future. Ageing Res. Rev. 2023, 91, 102078. [Google Scholar] [CrossRef]
- Feltes, B.C.; de Oliveira Alvares, L. PARP1 in the Intersection of Different DNA Repair Pathways, Memory Formation, and Sleep Pressure in Neurons. J. Neurochem. 2024, 168, 2351–2362. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-Ribosyl)Ation by PARP1: Reaction Mechanism and Regulatory Proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef]
- Khodyreva, S.N.; Lavrik, O.I. Poly(ADP-Ribose) polymerase 1 as a key regulator of DNA repair. Mol. Biol. 2016, 50, 655–673. [Google Scholar] [CrossRef]
- Herrmann, G.K.; Russell, W.K.; Garg, N.J.; Yin, Y.W. Poly(ADP-Ribose) Polymerase 1 Regulates Mitochondrial DNA Repair in an NAD-Dependent Manner. J. Biol. Chem. 2021, 296, 100309. [Google Scholar] [CrossRef]
- Arruri, V.K.; Gundu, C.; Khan, I.; Khatri, D.K.; Singh, S.B. PARP Overactivation in Neurological Disorders. Mol. Biol. Rep. 2021, 48, 2833–2841. [Google Scholar] [CrossRef]
- McGurk, L.; Mojsilovic-Petrovic, J.; Van Deerlin, V.M.; Shorter, J.; Kalb, R.G.; Lee, V.M.; Trojanowski, J.Q.; Lee, E.B.; Bonini, N.M. Nuclear Poly(ADP-Ribose) Activity Is a Therapeutic Target in Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 2018, 6, 84. [Google Scholar] [CrossRef]
- Hossain, M.I.; Lee, J.H.; Gagné, J.-P.; Khan, J.; Poirier, G.G.; King, P.H.; Dawson, V.L.; Dawson, T.M.; Andrabi, S.A. Poly(ADP-Ribose) Mediates Bioenergetic Defects and Redox Imbalance in Neurons Following Oxygen and Glucose Deprivation. FASEB J. 2024, 38, e23556. [Google Scholar] [CrossRef] [PubMed]
- Nosella, M.L.; Kim, T.H.; Huang, S.K.; Harkness, R.W.; Goncalves, M.; Pan, A.; Tereshchenko, M.; Vahidi, S.; Rubinstein, J.L.; Lee, H.O.; et al. Poly(ADP-Ribosyl)Ation Enhances Nucleosome Dynamics and Organizes DNA Damage Repair Components within Biomolecular Condensates. Mol. Cell 2024, 84, 429–446.e17. [Google Scholar] [CrossRef]
- Liu, C.; Fang, Y. New Insights of Poly(ADP-Ribosylation) in Neurodegenerative Diseases: A Focus on Protein Phase Separation and Pathologic Aggregation. Biochem. Pharmacol. 2019, 167, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ge, P. Parthanatos in the Pathogenesis of Nervous System Diseases. Neuroscience 2020, 449, 241–250. [Google Scholar] [CrossRef]
- Puentes, L.N.; Lengyel-Zhand, Z.; Lee, J.Y.; Hsieh, C.-J.; Schneider, M.E.; Edwards, K.J.; Luk, K.C.; Lee, V.M.-Y.; Trojanowski, J.Q.; Mach, R.H. Poly (ADP-Ribose) Interacts With Phosphorylated α-Synuclein in Post Mortem PD Samples. Front. Aging Neurosci. 2021, 13, 704041. [Google Scholar] [CrossRef]
- McGurk, L.; Gomes, E.; Guo, L.; Mojsilovic-Petrovic, J.; Tran, V.; Kalb, R.G.; Shorter, J.; Bonini, N.M. Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization. Mol. Cell. 2018, 71, 703–717. [Google Scholar] [CrossRef]
- Martire, S.; Mosca, L.; d’Erme, M. PARP-1 Involvement in Neurodegeneration: A Focus on Alzheimer’s and Parkinson’s Diseases. Mech. Ageing Dev. 2015, 146–148, 53–64. [Google Scholar] [CrossRef]
- Kam, T.-I.; Mao, X.; Park, H.; Chou, S.-C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-Ribose) Drives Pathologic α-Synuclein Neurodegeneration in Parkinson’s Disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef]
- Mamontova, E.M.; Clément, M.-J.; Sukhanova, M.V.; Joshi, V.; Bouhss, A.; Rengifo-Gonzalez, J.C.; Desforges, B.; Hamon, L.; Lavrik, O.I.; Pastré, D. FUS RRM Regulates Poly(ADP-Ribose) Levels after Transcriptional Arrest and PARP-1 Activation on DNA Damage. Cell. Rep. 2023, 42, 113199. [Google Scholar] [CrossRef]
- Vasil’eva, I.; Moor, N.; Anarbaev, R.; Kutuzov, M.; Lavrik, O. Functional Roles of PARP2 in Assembling Protein-Protein Complexes Involved in Base Excision DNA Repair. Int. J. Mol. Sci. 2021, 22, 4679. [Google Scholar] [CrossRef]
- Chappidi, N.; Quail, T.; Doll, S.; Vogel, L.T.; Aleksandrov, R.; Felekyan, S.; Kühnemuth, R.; Stoynov, S.; Seidel, C.A.M.; Brugués, J.; et al. PARP1-DNA Co-Condensation Drives DNA Repair Site Assembly to Prevent Disjunction of Broken DNA Ends. Cell 2024, 187, 945–961.e18. [Google Scholar] [CrossRef] [PubMed]
- Thadathil, N.; Delotterie, D.F.; Xiao, J.; Hori, R.; McDonald, M.P.; Khan, M.M. DNA Double-Strand Break Accumulation in Alzheimer’s Disease: Evidence from Experimental Models and Postmortem Human Brains. Mol. Neurobiol. 2021, 58, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hegde, M.L. New Mechanisms of DNA Repair Defects in Fused in Sarcoma-Associated Neurodegeneration: Stage Set for DNA Repair-Based Therapeutics? J. Exp. Neurosci. 2019, 13, 1179069519856358. [Google Scholar] [CrossRef] [PubMed]
- Kodavati, M.; Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Provasek, V.; Rao, V.H.M.; Vedula, I.; Zhang, A.; Mitra, S.; et al. FUS Unveiled in Mitochondrial DNA Repair and Targeted Ligase-1 Expression Rescues Repair-Defects in FUS-Linked Motor Neuron Disease. Nat. Commun. 2024, 15, 2156. [Google Scholar] [CrossRef]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-Binding Protein Fused in Sarcoma (FUS) Functions Downstream of Poly(ADP-Ribose) Polymerase (PARP) in Response to DNA Damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef]
- Reber, S.; Jutzi, D.; Lindsay, H.; Devoy, A.; Mechtersheimer, J.; Levone, B.R.; Domanski, M.; Bentmann, E.; Dormann, D.; Mühlemann, O.; et al. The Phase Separation-Dependent FUS Interactome Reveals Nuclear and Cytoplasmic Function of Liquid-Liquid Phase Separation. Nucleic Acids Res. 2021, 49, 7713–7731. [Google Scholar] [CrossRef]
- Sukhanova, M.V.; Singatulina, A.S.; Pastré, D.; Lavrik, O.I. Fused in Sarcoma (FUS) in DNA Repair: Tango with Poly(ADP-Ribose) Polymerase 1 and Compartmentalisation of Damaged DNA. Int. J. Mol. Sci. 2020, 21, 7020. [Google Scholar] [CrossRef]
- Provasek, V.E.; Mitra, J.; Malojirao, V.H.; Hegde, M.L. DNA Double-Strand Breaks as Pathogenic Lesions in Neurological Disorders. Int. J. Mol. Sci. 2022, 23, 4653. [Google Scholar] [CrossRef]
- Bai, D.; Zhu, L.; Jia, Q.; Duan, X.; Chen, L.; Wang, X.; Hou, J.; Jiang, G.; Yang, S.; Li, S.; et al. Loss of TDP-43 Promotes Somatic CAG Repeat Expansion in Huntington’s Disease Knock-in Mice. Prog. Neurobiol. 2023, 227, 102484. [Google Scholar] [CrossRef]
- Fang, M.; Deibler, S.K.; Nana, A.L.; Vatsavayai, S.C.; Banday, S.; Zhou, Y.; Almeida, S.; Weiss, A.; Brown, R.H.; Seeley, W.W.; et al. Loss of TDP-43 Function Contributes to Genomic Instability in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2023, 17, 1251228. [Google Scholar] [CrossRef]
- Konopka, A.; Whelan, D.R.; Jamali, M.S.; Perri, E.; Shahheydari, H.; Toth, R.P.; Parakh, S.; Robinson, T.; Cheong, A.; Mehta, P.; et al. Impaired NHEJ Repair in Amyotrophic Lateral Sclerosis Is Associated with TDP-43 Mutations. Mol. Neurodegener. 2020, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor Neuron Disease-Associated Loss of Nuclear TDP-43 Is Linked to DNA Double-Strand Break Repair Defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef] [PubMed]
- Mitra, J.; Dharmalingam, P.; Kodavati, M.; Guerrero, E.N.; Rao, K.S.; Garruto, R.M.; Hegde, M.L. Endogenous TDP-43 Mislocalization in a Novel Knock-in Mouse Model Reveals DNA Repair Impairment, Inflammation, and Neuronal Senescence. Res. Sq. 2024, rs.3.rs-3879966. [Google Scholar] [CrossRef]
- Lopez-Herdoiza, M.-B.; Bauché, S.; Wilmet, B.; Le Duigou, C.; Roussel, D.; Frah, M.; Béal, J.; Devely, G.; Boluda, S.; Frick, P.; et al. C9ORF72 Knockdown Triggers FTD-like Symptoms and Cell Pathology in Mice. Front. Cell Neurosci. 2023, 17, 1155929. [Google Scholar] [CrossRef]
- Andrade, N.S.; Ramic, M.; Esanov, R.; Liu, W.; Rybin, M.J.; Gaidosh, G.; Abdallah, A.; Del’Olio, S.; Huff, T.C.; Chee, N.T.; et al. Dipeptide Repeat Proteins Inhibit Homology-Directed DNA Double Strand Break Repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 2020, 15, 13. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Yang, D.; Pribadi, M.; Kim, T.S.; Krishnan, G.; Choi, S.Y.; Lee, S.; Coppola, G.; Gao, F.-B. Partial Inhibition of the Overactivated Ku80-Dependent DNA Repair Pathway Rescues Neurodegeneration in C9ORF72-ALS/FTD. Proc. Natl. Acad. Sci. USA 2019, 116, 9628–9633. [Google Scholar] [CrossRef]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 Poly(GR) Aggregation Induces TDP-43 Proteinopathy. Sci. Transl. Med. 2020, 12, eabb3774. [Google Scholar] [CrossRef]
- Farg, M.A.; Konopka, A.; Soo, K.Y.; Ito, D.; Atkin, J.D. The DNA Damage Response (DDR) Is Induced by the C9orf72 Repeat Expansion in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2017, 26, 2882–2896. [Google Scholar] [CrossRef]
- He, L.; Liang, J.; Chen, C.; Chen, J.; Shen, Y.; Sun, S.; Li, L. C9orf72 Functions in the Nucleus to Regulate DNA Damage Repair. Cell Death Differ. 2023, 30, 716–730. [Google Scholar] [CrossRef]
- Khosravi, B.; Hartmann, H.; May, S.; Möhl, C.; Ederle, H.; Michaelsen, M.; Schludi, M.H.; Dormann, D.; Edbauer, D. Cytoplasmic Poly-GA Aggregates Impair Nuclear Import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet. 2017, 26, 790–800. [Google Scholar] [CrossRef]
- Konopka, A.; Atkin, J.D. The Emerging Role of DNA Damage in the Pathogenesis of the C9orf72 Repeat Expansion in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2018, 19, 3137. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Verma, H.; Dhiman, M.; Mantha, A.K. Activation of Multifunctional DNA Repair APE1/Ref-1 Enzyme by the Dietary Phytochemical Ferulic Acid Protects Human Neuroblastoma SH-SY5Y Cells against Aβ(25-35)-Induced Oxidative Stress and Inflammatory Responses. Mitochondrion 2024, 79, 101947. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Duchen, M.R. Activation of PARP by Oxidative Stress Induced by β-Amyloid: Implications for Alzheimer’s Disease. Neurochem. Res. 2012, 37, 2589–2596. [Google Scholar] [CrossRef] [PubMed]
- Forestier, A.; Douki, T.; Sauvaigo, S.; De Rosa, V.; Demeilliers, C.; Rachidi, W. Alzheimer’s Disease-Associated Neurotoxic Peptide Amyloid-β Impairs Base Excision Repair in Human Neuroblastoma Cells. Int. J. Mol. Sci. 2012, 13, 14766–14787. [Google Scholar] [CrossRef]
- Lu, J.; Li, Y.; Mollinari, C.; Garaci, E.; Merlo, D.; Pei, G. Amyloid-β Oligomers-Induced Mitochondrial DNA Repair Impairment Contributes to Altered Human Neural Stem Cell Differentiation. Curr. Alzheimer Res. 2019, 16, 934–949. [Google Scholar] [CrossRef]
- Cardinale, A.; Racaniello, M.; Saladini, S.; De Chiara, G.; Mollinari, C.; De Stefano, M.C.; Pocchiari, M.; Garaci, E.; Merlo, D. Sublethal Doses of β-Amyloid Peptide Abrogate DNA-Dependent Protein Kinase Activity. J. Biol. Chem. 2012, 287, 2618–2631. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The Physiological Roles of Tau and Aβ: Implications for Alzheimer’s Disease Pathology and Therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- D’Andrea, L.; Stringhi, R.; Di Luca, M.; Marcello, E. Looking at Alzheimer’s Disease Pathogenesis from the Nuclear Side. Biomolecules 2021, 11, 1261. [Google Scholar] [CrossRef]
- Kanungo, J. DNA-PK Deficiency in Alzheimer’s Disease. J. Neurol. Neuromed. 2016, 1, 17–22. [Google Scholar] [CrossRef]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-Synuclein Is a DNA Binding Protein That Modulates DNA Repair with Implications for Lewy Body Disorders. Sci. Rep. 2019, 9, 10919. [Google Scholar] [CrossRef] [PubMed]
- Dent, S.E.; King, D.P.; Osterberg, V.R.; Adams, E.K.; Mackiewicz, M.R.; Weissman, T.A.; Unni, V.K. Phosphorylation of the Aggregate-Forming Protein Alpha-Synuclein on Serine-129 Inhibits Its DNA-Bending Properties. J. Biol. Chem. 2022, 298, 101552. [Google Scholar] [CrossRef] [PubMed]
- Rose, E.P.; Osterberg, V.R.; Gorbunova, V.; Unni, V.K. Alpha-Synuclein Modulates the Repair of Genomic DNA Double-Strand Breaks in a DNA-PKcs-Regulated Manner. Neurobiol. Dis. 2024, 201, 106675. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent Developments in Clinical Biomarkers and Therapies Targeting Tau Phosphorylation in Alzheimer’s Disease and Other Tauopathies. Mol. Neurodegener. 2021, 16, 37. [Google Scholar] [CrossRef]
- Abasi, L.S.; Elathram, N.; Movva, M.; Deep, A.; Corbett, K.D.; Debelouchina, G.T. Phosphorylation Regulates Tau’s Phase Separation Behavior and Interactions with Chromatin. Commun. Biol. 2024, 7, 251. [Google Scholar] [CrossRef]
- Asada-Utsugi, M.; Urushitani, M. Tau beyond Tangles: DNA Damage Response and Cytoskeletal Protein Crosstalk on Neurodegeneration. Int. J. Mol. Sci. 2024, 25, 7906. [Google Scholar] [CrossRef]
- Asada-Utsugi, M.; Uemura, K.; Ayaki, T.; Uemura, M.T.; Minamiyama, S.; Hikiami, R.; Morimura, T.; Shodai, A.; Ueki, T.; Takahashi, R.; et al. Failure of DNA Double-Strand Break Repair by Tau Mediates Alzheimer’s Disease Pathology in Vitro. Commun. Biol. 2022, 5, 358. [Google Scholar] [CrossRef]
- Fu, Q.; Zhang, B.; Chen, X.; Chu, L. Liquid-Liquid Phase Separation in Alzheimer’s Disease. J. Mol. Med. 2024, 102, 167–181. [Google Scholar] [CrossRef]
- Lin, Y.; Fichou, Y.; Longhini, A.P.; Llanes, L.C.; Yin, P.; Bazan, G.C.; Kosik, K.S.; Han, S. Liquid-Liquid Phase Separation of Tau Driven by Hydrophobic Interaction Facilitates Fibrillization of Tau. J. Mol. Biol. 2021, 433, 166731. [Google Scholar] [CrossRef]
- Sharma, H.; Koirala, S.; Chew, Y.L.; Konopka, A. DNA Damage and Chromatin Rearrangement Work Together to Promote Neurodegeneration. Mol. Neurobiol. 2024, 1–9. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and Tau Oligomers in Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef] [PubMed]
- Tomé, S.O.; Gomes, L.A.; Li, X.; Vandenberghe, R.; Tousseyn, T.; Thal, D.R. TDP-43 Interacts with Pathological τ Protein in Alzheimer’s Disease. Acta Neuropathol. 2021, 141, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Vitet, H.; Brandt, V.; Saudou, F. Traffic Signaling: New Functions of Huntingtin and Axonal Transport in Neurological Disease. Curr. Opin. Neurobiol. 2020, 63, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Coudert, L.; Nonaka, T.; Bernard, E.; Hasegawa, M.; Schaeffer, L.; Leblanc, P. Phosphorylated and Aggregated TDP-43 with Seeding Properties Are Induced upon Mutant Huntingtin (mHtt) Polyglutamine Expression in Human Cellular Models. Cell. Mol. Life Sci. 2019, 76, 2615–2632. [Google Scholar] [CrossRef]
- Enokido, Y.; Tamura, T.; Ito, H.; Arumughan, A.; Komuro, A.; Shiwaku, H.; Sone, M.; Foulle, R.; Sawada, H.; Ishiguro, H.; et al. Mutant Huntingtin Impairs Ku70-Mediated DNA Repair. J. Cell Biol. 2010, 189, 425–443. [Google Scholar] [CrossRef]
- Gao, R.; Chakraborty, A.; Geater, C.; Pradhan, S.; Gordon, K.L.; Snowden, J.; Yuan, S.; Dickey, A.S.; Choudhary, S.; Ashizawa, T.; et al. Mutant Huntingtin Impairs PNKP and ATXN3, Disrupting DNA Repair and Transcription. elife 2019, 8, e42988. [Google Scholar] [CrossRef]
- Maiuri, T.; Mocle, A.J.; Hung, C.L.; Xia, J.; van Roon-Mom, W.M.C.; Truant, R. Huntingtin Is a Scaffolding Protein in the ATM Oxidative DNA Damage Response Complex. Hum. Mol. Genet. 2017, 26, 395–406. [Google Scholar] [CrossRef]
- Maiuri, T.; Suart, C.E.; Hung, C.L.K.; Graham, K.J.; Barba Bazan, C.A.; Truant, R. DNA Damage Repair in Huntington’s Disease and Other Neurodegenerative Diseases. Neurotherapeutics 2019, 16, 948–956. [Google Scholar] [CrossRef]
- Makeeva, V.S.; Dyrkheeva, N.S.; Lavrik, O.I.; Zakian, S.M.; Malakhova, A.A. Mutant-Huntingtin Molecular Pathways Elucidate New Targets for Drug Repurposing. Int. J. Mol. Sci. 2023, 24, 16798. [Google Scholar] [CrossRef]
- Tamura, T.; Sone, M.; Iwatsubo, T.; Tagawa, K.; Wanker, E.E.; Okazawa, H. Ku70 Alleviates Neurodegeneration in Drosophila Models of Huntington’s Disease. PLoS ONE 2011, 6, e27408. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Zhao, W.-W.; Bai, S.-M.; Ma, Y.; Yin, X.-K.; Feng, L.-L.; Zeng, G.-D.; Wang, F.; Feng, W.-X.; Zheng, J.; et al. DNA Damage-Induced Paraspeckle Formation Enhances DNA Repair and Tumor Radioresistance by Recruiting Ribosomal Protein P0. Cell Death Dis. 2022, 13, 709. [Google Scholar] [CrossRef] [PubMed]
- Belur, N.R.; Bustos, B.I.; Lubbe, S.J.; Mazzulli, J.R. Nuclear Aggregates of NONO/SFPQ and A-to-I-Edited RNA in Parkinson’s Disease and Dementia with Lewy Bodies. Neuron 2024, 112, 2558–2580.e13. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.-J.; Wang, Y.-L.; Zhao, W.-W.; Bai, S.-M.; Ma, Y.; Yin, X.-K.; Feng, L.-L.; Feng, W.-X.; Wang, Y.-N.; Liu, Q.; et al. NONO Phase Separation Enhances DNA Damage Repair by Accelerating Nuclear EGFR-Induced DNA-PK Activation. Am. J. Cancer Res. 2021, 11, 2838–2852. [Google Scholar] [PubMed]
- Ishigaki, S.; Riku, Y.; Fujioka, Y.; Endo, K.; Iwade, N.; Kawai, K.; Ishibashi, M.; Yokoi, S.; Katsuno, M.; Watanabe, H.; et al. Aberrant Interaction between FUS and SFPQ in Neurons in a Wide Range of FTLD Spectrum Diseases. Brain 2020, 143, 2398–2405. [Google Scholar] [CrossRef]
- Jaafar, L.; Li, Z.; Li, S.; Dynan, W.S. SFPQ•NONO and XLF Function Separately and Together to Promote DNA Double-Strand Break Repair via Canonical Nonhomologous End Joining. Nucleic Acids Res. 2017, 45, 1848–1859. [Google Scholar] [CrossRef]
- Krietsch, J.; Caron, M.-C.; Gagné, J.-P.; Ethier, C.; Vignard, J.; Vincent, M.; Rouleau, M.; Hendzel, M.J.; Poirier, G.G.; Masson, J.-Y. PARP Activation Regulates the RNA-Binding Protein NONO in the DNA Damage Response to DNA Double-Strand Breaks. Nucleic Acids Res. 2012, 40, 10287–10301. [Google Scholar] [CrossRef]
- Lim, Y.W.; James, D.; Huang, J.; Lee, M. The Emerging Role of the RNA-Binding Protein SFPQ in Neuronal Function and Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 7151. [Google Scholar] [CrossRef]
- Udayakumar, D.; Dynan, W.S. Characterization of DNA Binding and Pairing Activities Associated with the Native SFPQ·NONO DNA Repair Protein Complex. Biochem. Biophys. Res. Commun. 2015, 463, 473–478. [Google Scholar] [CrossRef]
- Guerrero, E.N.; Wang, H.; Mitra, J.; Hegde, P.M.; Stowell, S.E.; Liachko, N.F.; Kraemer, B.C.; Garruto, R.M.; Rao, K.S.; Hegde, M.L. TDP-43/FUS in Motor Neuron Disease: Complexity and Challenges. Prog. Neurobiol. 2016, 145–146, 78–97. [Google Scholar] [CrossRef]
- Baechtold, H.; Kuroda, M.; Sok, J.; Ron, D.; Lopez, B.S.; Akhmedov, A.T. Human 75-kDa DNA-Pairing Protein Is Identical to the Pro-Oncoprotein TLS/FUS and Is Able to Promote D-Loop Formation. J. Biol. Chem. 1999, 274, 34337–34342. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS Causes DNA Ligation Defects to Inhibit Oxidative Damage Repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.A.; Huang, E.J.; Tsai, L.-H. Interaction of FUS and HDAC1 Regulates DNA Damage Response and Repair in Neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.Q.; Gómez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 Dependent Recruitment of the Amyotrophic Lateral Sclerosis-Associated Protein FUS/TLS to Sites of Oxidative DNA Damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrov, R.; Dotchev, A.; Poser, I.; Krastev, D.; Georgiev, G.; Panova, G.; Babukov, Y.; Danovski, G.; Dyankova, T.; Hubatsch, L.; et al. Protein Dynamics in Complex DNA Lesions. Mol. Cell 2018, 69, 1046–1061.e5. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Chitta, R.K.; High, A.A.; Taylor, J.P. Global Analysis of TDP-43 Interacting Proteins Reveals Strong Association with RNA Splicing and Translation Machinery. J. Proteome Res. 2010, 9, 1104–1120. [Google Scholar] [CrossRef]
- Neven, J.; Issayama, L.K.; Dewachter, I.; Wilson, D.M. Genomic Stress and Impaired DNA Repair in Alzheimer Disease. DNA Repair 2024, 139, 103678. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Human Neurological Syndrome | Mutated Gene | Rodent Models | References |
|---|---|---|---|
| Nucleotide excision repair | |||
| Cockayne syndrome: progressive neurodegeneration | XPB | Xpa and XpbXPCS double mutants: CS-like symptoms, including neurological defects | [15,143] |
| ERCC6/CSB | Csb mutants mimicking human CS1AN allele: minor neurologic abnormalities | [15,144] | |
| Xeroderma Pigmentosum (XP): part of XP patients develops neurological symptoms including microcephaly, mental retardation, cerebellar ataxia and peripheral neuropathy | XPA-XPG | Xpa−/−; Csb−/− or Xpc−/−; Csb−/− double mutants: CS- and XP-like symptoms including ataxia, motor dysfunction, reduced cerebellar neurogenesis, and neurodegeneration | [145,146] |
| Base excision repair/Single-strand break repair | |||
| Spinocerebellar Ataxia with Axonal Neuropathy (SCAN1): cerebellar atrophy | TDP1 | Tdp1−/− mutant: progressive cerebellar atrophy | [15,147] |
| Biallelic mutations in human XRCC1: ocular motor apraxia, axonal neuropathy, and progressive cerebellar ataxia | XRCC1 | Xrcc1−/− mutant: embryonic lethality, but double Parp1−/−_Xrcc1−/− mutant: reduced loss of cerebellar neurons and ataxia i | [148,149] |
| Ataxia with oculomotor apraxia 4 (AOA4), microcephaly with seizures (MCSZ) | PNKP | Pnkp: sensitivity of the myelin-producing oligodendrocytes to PNKP loss and DNA damage accumulation | [150] |
| Alzheimer disease | POLB | Polβ knockdown models in an AD mouse: an increase in synaptic problems as observed in AD patients | [151] |
| Ligase 3 (no human syndrome) | LIG 3 | Lig3Nes-cre conditional inactivation in mouse NS: mtDNA loss leading to ataxia | [18,152] |
| Double strand break repair | |||
| ATR-Seckel Syndrome: microcephaly, dwarfism | ATR | AtrS/S mutant mimicking ATR-Seckel Syndrome: microcephaly | [15,153] |
| LIG4 Syndrome: microcephaly | LIG4 | Lig4 mutant: p53 dependent apoptosis of post-mitotic neurons | [154] |
| Ataxia-telangiectasia (A-T): progressive cerebellar ataxia that develops into severe motor dysfunction | ATM | AtmL2262P/L2262P mutant rats: neuroinflammation and neurodegeneration (hind-limb paralysis) | [155] |
| Atm−/− mice: microglia activation and mild cerebellar degeneration | [156,157] | ||
| ATM−/− mice: aberrant astrocytic morphology and alterations of vasculature both in cerebellum and the visual system; reduced myelin basic protein immunoreactivity and signs of inflammation in ATM-deficient cerebella and optic nerve | [158] | ||
| Atm−/− mice: lose the ability to induce apoptosis in differentiating neuronal cells, but not in proliferating precursor neuroblasts, in response to DNA damage induced by ionizing radiation | [159,160] | ||
| Name | Associated Neurological Disorder | Associated DNA Repair Pathway | Subcellular Localization/MLO | References |
|---|---|---|---|---|
| FUS | ALS, FTLD | NHEJ | Nucleus Paraspeckle Nucleolus Cajal body Cytoplasm Stress granule Transport granule | [178,179,193,212,214,215,216,217,218] |
| TDP-43 | ALS, FTLD, AD, PD, HD | NHEJ MMR | Nucleus Paraspeckle Nucleolus Cajal body Cytoplasm Stress granule Transport granule | [109,138,139,176,179,201,207,219,220,221,222,223,224] |
| C9ORF72 | ALS, FTLD HD, PD | BER (SSBR) | Nucleus Cytoplasm (co-aggregates with TDP-43) | [170,225,226,227,228,229,230,231,232] |
| Aβ | AD | NHEJ | Nucleus Cytoplasm Cell surface | [184,185,213,233,234,235,236,237,238,239,240,241] |
| αSyn | PD | NHEJ | Nucleus Colocalized with γH2AX or PAR foci Cytoplasm Presynaptic terminals | [183,206,209,242,243,244] |
| Tau | AD, ALS, FTLD | NHEJ | Nucleus Cytoplasm Axon, Dendrite, Cell membrane | [182,245,246,247,248,249,250,251,252,253] |
| Htt | HD | BER | Nucleus Cytoplasm | [129,254,255,256,257,258,259,260,261] |
| NONO/SFPQ | AD, PD, DLB, FTLD | NHEJ | Nucleus Paraspeckle | [262,263,264,265,266,267,268,269] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khodyreva, S.N.; Dyrkheeva, N.S.; Lavrik, O.I. Proteins Associated with Neurodegenerative Diseases: Link to DNA Repair. Biomedicines 2024, 12, 2808. https://doi.org/10.3390/biomedicines12122808
Khodyreva SN, Dyrkheeva NS, Lavrik OI. Proteins Associated with Neurodegenerative Diseases: Link to DNA Repair. Biomedicines. 2024; 12(12):2808. https://doi.org/10.3390/biomedicines12122808
Chicago/Turabian StyleKhodyreva, Svetlana N., Nadezhda S. Dyrkheeva, and Olga I. Lavrik. 2024. "Proteins Associated with Neurodegenerative Diseases: Link to DNA Repair" Biomedicines 12, no. 12: 2808. https://doi.org/10.3390/biomedicines12122808
APA StyleKhodyreva, S. N., Dyrkheeva, N. S., & Lavrik, O. I. (2024). Proteins Associated with Neurodegenerative Diseases: Link to DNA Repair. Biomedicines, 12(12), 2808. https://doi.org/10.3390/biomedicines12122808