

Mitochondrial Dysfunction in Atrial Fibrillation: The Need for a Strong Pharmacological Approach

 ,
,  and
and 
Abstract

1. Introduction
2. Role of the Mitochondria in Systemic Disease
3. Mitochondrial Diseases Correlated with Atrial Fibrillation
4. Acquired Mitochondrial Dysfunction
5. Cellular Alterations in the Heart and Electrogenesis of Atrial Fibrillation
ATP Homeostasis, ROS Production, and Atrial Fibrillation
6. Drugs with Mitochondrial Effects
6.1. Oral Hypoglycaemic Agents
6.2. Hypolipidemic Drugs
6.3. Others
7. Nutraceutical with Mitochondrial Effects
8. Experimental Drugs with Mitochondrial Effects
9. Effect of Anticoagulant Drugs on Mitochondrial Function
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, R.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Klopstock, T.; Priglinger, C.; Yilmaz, A.; Kornblum, C.; Distelmaier, F.; Prokisch, H. Mitochondrial Disorders. Dtsch. Aerzteblatt Online 2021, 118, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Yamazoe, M.; Sasano, T.; Ihara, K.; Takahashi, K.; Nakamura, W.; Takahashi, N.; Komuro, H.; Hamada, S.; Furukawa, T. Sparsely methylated mitochondrial cell free DNA released from cardiomyocytes contributes to systemic inflammatory response accompanied by atrial fibrillation. Sci. Rep. 2021, 11, 5837. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, T.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced Oxidative Stress and its Role in Diabetes Mellitus Related Cardiovascular Diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef]
- Rao, Y.; Chen, J.; Guo, Y.; Ji, T.; Xie, P. Rivaroxaban ameliorates angiotensin II-induced cardiac remodeling by attenuating TXNIP/Trx2 interaction in KKAy mice. Thromb. Res. 2020, 193, 45–52. [Google Scholar] [CrossRef]
- Qu, K.; Yan, F.; Qin, X.; Zhang, K.; He, W.; Dong, M.; Wu, G. Mitochondrial dysfunction in vascular endothelial cells and its role in atherosclerosis. Front. Physiol. 2022, 13, 1084604. [Google Scholar] [CrossRef]
- Popoiu, T.-A.; Dudek, J.; Maack, C.; Bertero, E. Cardiac Involvement in Mitochondrial Disorders. Curr. Heart Fail. Rep. 2023, 20, 76–87. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Ng, Y.S.; Turnbull, D.M. Mitochondrial disease: Genetics and management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef]
- Davis, R.L.; Liang, C.; Sue, C.M. Mitochondrial diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 125–141. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassègue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. Nox2-Induced Production of Mitochondrial Superoxide in Angiotensin II-Mediated Endothelial Oxidative Stress and Hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef]
- Yang, K.-C.; Bonini, M.G.; Dudley, S.C. Mitochondria and arrhythmias. Free Radic. Biol. Med. 2014, 71, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Mason, F.E.; Pronto, J.R.D.; Alhussini, K.; Maack, C.; Voigt, N. Cellular and mitochondrial mechanisms of atrial fibrillation. Basic Res. Cardiol. 2020, 115, 72. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Jiang, Y.; Chen, Z.B.; Rhee, J.-W.; Deng, Y.; Wang, Z.V. Mitochondrial Dysfunction in Cardiac Arrhythmias. Cells 2023, 12, 679. [Google Scholar] [CrossRef] [PubMed]
- Black, N.; Mohammad, F.; Saraf, K.; Morris, G. Endothelial function and atrial fibrillation: A missing piece of the puzzle? J. Cardiovasc. Electrophysiol. 2022, 33, 109–116. [Google Scholar] [CrossRef]
- Khan, A.A.; Thomas, G.N.; Lip, G.Y.H.; Shantsila, A. Endothelial function in patients with atrial fibrillation. Ann. Med. 2020, 52, 1–11. [Google Scholar] [CrossRef]
- Qin, S.; Boidin, M.; Buckley, B.J.R.; Lip, G.Y.H.; Thijssen, D.H.J. Endothelial dysfunction and vascular maladaptation in atrial fibrillation. Eur. J. Clin. Investig. 2021, 51, e13477. [Google Scholar] [CrossRef]
- Guazzi, M.; Arena, R. Endothelial dysfunction and pathophysiological correlates in atrial fibrillation. Heart 2009, 95, 102–106. [Google Scholar] [CrossRef]
- Balint, B.; Jaremek, V.; Thorburn, V.; Whitehead, S.N.; Sposato, L.A. Left atrial microvascular endothelial dysfunction, myocardial inflammation and fibrosis after selective insular cortex ischemic stroke. Int. J. Cardiol. 2019, 292, 148–155. [Google Scholar] [CrossRef]
- Muszyński, P.; Bonda, T.A. Mitochondrial Dysfunction in Atrial Fibrillation—Mechanisms and Pharmacological Interventions. J. Clin. Med. 2021, 10, 2385. [Google Scholar] [CrossRef]
- Caturano, A.; D’Angelo, M.; Mormone, A.; Russo, V.; Mollica, M.P.; Salvatore, T.; Galiero, R.; Rinaldi, L.; Vetrano, E.; Marfella, R.; et al. Oxidative Stress in Type 2 Diabetes: Impacts from Pathogenesis to Lifestyle Modifications. Curr. Issues Mol. Biol. 2023, 45, 6651–6666. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, T.; Watanabe, M.; Yokota, T.; Tsuda, M.; Handa, H.; Koya, J.; Nishino, K.; Tatsuta, D.; Natsui, H.; Kadosaka, T.; et al. Empagliflozin suppresses mitochondrial reactive oxygen species generation and mitigates the inducibility of atrial fibrillation in diabetic rats. Front. Cardiovasc. Med. 2023, 10, 1005408. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Y.; Han, X.; Wang, X.; Qu, C.; Liu, X.; Yang, B. Dapagliflozin attenuates the vulnerability to atrial fibrillation in rats with lipopolysaccharide-induced myocardial injury. Int. Immunopharmacol. 2023, 125, 111038. [Google Scholar] [CrossRef]
- Myasoedova, V.A.; Bozzi, M.; Valerio, V.; Moschetta, D.; Massaiu, I.; Rusconi, V.; Di Napoli, D.; Ciccarelli, M.; Parisi, V.; Agostoni, P.; et al. Anti-Inflammation and Anti-Oxidation: The Key to Unlocking the Cardiovascular Potential of SGLT2 Inhibitors and GLP1 Receptor Agonists. Antioxidants 2023, 13, 16. [Google Scholar] [CrossRef]
- Nuamnaichati, N.; Mangmool, S.; Chattipakorn, N.; Parichatikanond, W. Stimulation of GLP-1 Receptor Inhibits Methylglyoxal-Induced Mitochondrial Dysfunctions in H9c2 Cardiomyoblasts: Potential Role of Epac/PI3K/Akt Pathway. Front. Pharmacol. 2020, 11, 805. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, J.; Diao, S.; Zhang, G.; Xiao, M.; Chang, D. GLP-1 receptor agonist liraglutide protects cardiomyocytes from IL-1β-induced metabolic disturbance and mitochondrial dysfunction. Chem. Biol. Interact. 2020, 332, 109252. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, L.; Feng, B.; He, N.; Zhang, Y.; Ye, H. Protective effects of glucagon-like peptide-1 on cardiac remodeling by inhibiting oxidative stress through mammalian target of rapamycin complex 1/p70 ribosomal protein S6 kinase pathway in diabetes mellitus. J. Diabetes Investig. 2020, 11, 39–51. [Google Scholar] [CrossRef]
- Wei, J.; Wang, R.; Ye, H.; Wang, Y.; Wang, L.; Zhang, X. Effects of GLP-1 receptor agonists on arrhythmias and its subtypes in patients with type 2 diabetes: A systematic review and meta-analysis. Front. Endocrinol. 2022, 13, 910256. [Google Scholar] [CrossRef]
- Fauchier, G.; Bisson, A.; Bodin, A.; Herbert, J.; Angoulvant, D.; Ducluzeau, P.H.; Lip, G.Y.H.; Fauchier, L. Glucose-lowering drug use and new-onset atrial fibrillation in patients with diabetes mellitus. Diabetologia 2021, 64, 2602–2605. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, J.; Wang, C.; Zhang, Q.; Xu, Y.; Liu, H.; Xiang, X.; Ma, J. DPP-4 inhibitor anagliptin protects against hypoxia-induced cytotoxicity in cardiac H9C2 cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3823–3831. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Zhao, Y.; Jiang, N.; Qiu, J.; Yang, Y.; Li, J.; Liang, X.; Wang, X.; Tse, G.; et al. Alogliptin, a Dipeptidyl Peptidase-4 Inhibitor, Alleviates Atrial Remodeling and Improves Mitochondrial Function and Biogenesis in Diabetic Rabbits. J. Am. Hear. Assoc. 2017, 6, e005945. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, T.; Niwano, S.; Niwano, H.; Yoshizawa, T.; Nakamura, H.; Fukaya, H.; Fujiishi, T.; Ishizue, N.; Satoh, A.; Kishihara, J.; et al. Linagliptin prevents atrial electrical and structural remodeling in a canine model of atrial fibrillation. Heart Vessels 2018, 33, 1258–1265. [Google Scholar] [CrossRef]
- Patoulias, D.I.; Boulmpou, A.; Teperikidis, E.; Katsimardou, A.; Siskos, F.; Doumas, M.; Papadopoulos, C.E.; Vassilikos, V. Cardiovascular efficacy and safety of dipeptidyl peptidase-4 inhibitors: A meta-analysis of cardiovascular outcome trials. World J. Cardiol. 2021, 13, 585–592. [Google Scholar] [CrossRef]
- Chan, Y.-H.; Chao, T.F.; Chen, S.W.; Lee, H.F.; Li, P.R.; Chen, W.M.; Yeh, Y.H.; Kuo, C.T.; See, L.C.; Lip, G.Y.H. The risk of incident atrial fibrillation in patients with type 2 diabetes treated with sodium glucose cotransporter-2 inhibitors, glucagon-like peptide-1 receptor agonists, and dipeptidyl peptidase-4 inhibitors: A nationwide cohort study. Cardiovasc. Diabetol. 2022, 21, 118. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Yeh, Y.-H.; Chan, Y.-H.; Liu, J.-R.; Chang, S.-H.; Lee, H.-F.; Wu, L.-S.; Yen, K.-C.; Kuo, C.-T.; See, L.-C. Dipeptidyl peptidase-4 inhibitor decreases the risk of atrial fibrillation in patients with type 2 diabetes: A nationwide cohort study in Taiwan. Cardiovasc. Diabetol. 2017, 16, 159. [Google Scholar] [CrossRef]
- Chang, S.H.; Wu, L.S.; Chiou, M.J.; Liu, J.R.; Yu, K.H.; Kuo, C.F.; Wen, M.S.; Chen, W.J.; Yeh, Y.H.; See, L.C. Association of metformin with lower atrial fibrillation risk among patients with type 2 diabetes mellitus: A population-based dynamic cohort and in vitro studies. Cardiovasc. Diabetol. 2014, 13, 123. [Google Scholar] [CrossRef]
- Sun, D.; Yang, F. Metformin improves cardiac function in mice with heart failure after myocardial infarction by regulating mitochondrial energy metabolism. Biochem. Biophys. Res. Commun. 2017, 486, 329–335. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Korantzopoulos, P.; Letsas, K.P.; Tse, G.; Gong, M.; Meng, L.; Li, G.; Liu, T. Thiazolidinedione use and atrial fibrillation in diabetic patients: A meta-analysis. BMC Cardiovasc. Disord. 2017, 17, 96. [Google Scholar] [CrossRef]
- Liu, C.; Liu, R.; Fu, H.; Li, J.; Wang, X.; Cheng, L.; Korantzopoulos, P.; Tse, G.; Li, G.; Liu, T. Pioglitazone attenuates atrial remodeling and vulnerability to atrial fibrillation in alloxan-induced diabetic rabbits. Cardiovasc. Ther. 2017, 35, e12284. [Google Scholar] [CrossRef] [PubMed]
- Pallisgaard, J.L.; Brooks, M.M.; Chaitman, B.R.; Boothroyd, D.B.; Perez, M.; Hlatky, M.A. Thiazolidinediones and Risk of Atrial Fibrillation Among Patients with Diabetes and Coronary Disease. Am. J. Med. 2018, 131, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Hu, W.; Song, Z.; Liu, X.; Zhang, D. PPARγ agonist use and recurrence of atrial fibrillation after successful electrical cardioversion. Hell. J. Cardiol. 2017, 58, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H.; Chung, W.-J.; Li, C.-Y.; Tsai, T.-H.; Lee, C.-H.; Hsueh, S.-K.; Wu, C.-C.; Cheng, C.-I. Statins reduce new-onset atrial fibrillation after acute myocardial infarction: A nationwide study. Medicine 2020, 99, e18517. [Google Scholar] [CrossRef]
- Alves-Cabratosa, L.; García-Gil, M.; Comas-Cufí, M.; Ponjoan, A.; Martí-Lluch, R.; Parramon, D.; Blanch, J.; Elosua-Bayes, M.; Ramos, R. Statins and new-onset atrial fibrillation in a cohort of patients with hypertension. Analysis of electronic health records, 2006–2015. PLoS ONE 2017, 12, e0186972. [Google Scholar] [CrossRef]
- Fang, W.; Li, H.; Zhang, H.; Jiang, S. The role of statin therapy in the prevention of atrial fibrillation: A meta-analysis of randomized controlled trials. Br. J. Clin. Pharmacol. 2012, 74, 744–756. [Google Scholar] [CrossRef]
- da S, A.; de França-e-Silva, A.L.G.; de Oliveira, J.M.; da Silva, D.M. Developing Pharmacological Therapies for Atrial Fibrillation Targeting Mitochondrial Dysfunction and Oxidative Stress: A Scoping Review. Int. J. Mol. Sci. 2023, 25, 535. [Google Scholar] [CrossRef]
- Shi, W. Effects of trimetazidine on mitochondrial respiratory function, biosynthesis, and fission/fusion in rats with acute myocardial ischemia. Anatol. J. Cardiol. 2017, 18, 175–181. [Google Scholar] [CrossRef]
- Li, Z.; Chaolan, L.; Chengcheng, W.; Xi, H.; Yingying, W.; Jiaqiu, L.; Wei, H. GW28-e0789 Trimetazidine decreases inducibility and duration of atrial fibrillation in a dog model of congestive heart failure. J. Am. Coll. Cardiol. 2017, 70, C29. [Google Scholar] [CrossRef]
- Ratte, A.; Wiedmann, F.; Kraft, M.; Katus, H.A.; Schmidt, C. Antiarrhythmic Properties of Ranolazine: Inhibition of Atrial Fibrillation Associated TASK-1 Potassium Channels. Front. Pharmacol. 2019, 10, 1367. [Google Scholar] [CrossRef]
- Xu, D.; Murakoshi, N.; Tajiri, K.; Duo, F.; Okabe, Y.; Murakata, Y.; Yuan, Z.; Li, S.; Aonuma, K.; Song, Z.; et al. Xanthine oxidase inhibitor febuxostat reduces atrial fibrillation susceptibility by inhibition of oxidized CaMKII in Dahl salt-sensitive rats. Clin. Sci. 2021, 135, 2409–2422. [Google Scholar] [CrossRef] [PubMed]
- Korantzopoulos, P.; Letsas, K.P.; Liu, T. Xanthine Oxidase and Uric Acid in Atrial Fibrillation. Front. Physiol. 2012, 3, 150. [Google Scholar] [CrossRef] [PubMed]
- Raizner, A.E.; Quiñones, M.A. Coenzyme Q10 for Patients With Cardiovascular Disease. J. Am. Coll. Cardiol. 2021, 77, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Kebbati, A.H.; Zhang, Y.; Tang, Y.; Okello, E.; Huang, C. Effect of Coenzyme Q10 on the Incidence of Atrial Fibrillation in Patients with Heart Failure. J. Investig. Med. 2015, 63, 735–739. [Google Scholar] [CrossRef]
- Xu, D.; Murakoshi, N.; Igarashi, M.; Hirayama, A.; Ito, Y.; Seo, Y.; Tada, H.; Aonuma, K. PPAR-γ Activator Pioglitazone Prevents Age-Related Atrial Fibrillation Susceptibility by Improving Antioxidant Capacity and Reducing Apoptosis in a Rat Model. J. Cardiovasc. Electrophysiol. 2012, 23, 209–217. [Google Scholar] [CrossRef]
- Williams, E.A.; Russo, V.; Ceraso, S.; Gupta, D.; Barrett-Jolley, R. Anti-arrhythmic properties of non-antiarrhythmic medications. Pharmacol. Res. 2020, 156, 104762. [Google Scholar] [CrossRef]
- Safaeian, L.; Mirian, M.; Bahrizadeh, S. Evolocumab, a PCSK9 inhibitor, protects human endothelial cells against H2O2-induced oxidative stress. Arch. Physiol. Biochem. 2022, 128, 1681–1686. [Google Scholar] [CrossRef]
- Yang, J.; Ma, X.; Niu, D.; Sun, Y.; Chai, X.; Deng, Y.; Wang, J.; Dong, J. PCSK9 inhibitors suppress oxidative stress and inflammation in atherosclerotic development by promoting macrophage autophagy. Am. J. Transl. Res. 2023, 15, 5129–5144. [Google Scholar]
- D’Onofrio, N.; Prattichizzo, F.; Marfella, R.; Sardu, C.; Martino, E.; Scisciola, L.; Marfella, L.; La Grotta, R.; Frigé, C.; Paolisso, G.; et al. SIRT3 mediates the effects of PCSK9 inhibitors on inflammation, autophagy, and oxidative stress in endothelial cells. Theranostics 2023, 13, 531–542. [Google Scholar] [CrossRef]
- Silla, A.; Fogacci, F.; Punzo, A.; Hrelia, S.; Simoni, P.; Caliceti, C.; Cicero, A.F.G. Treatment with PCSK9 Inhibitor Evolocumab Improves Vascular Oxidative Stress and Arterial Stiffness in Hypercholesterolemic Patients with High Cardiovascular Risk. Antioxidants 2023, 12, 578. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, R.; Wang, X.; Li, J.; Yuan, M.; Liu, E.; Liu, T.; Li, G. Attenuation of atrial remodeling by aliskiren via affecting oxidative stress, inflammation and PI3K/Akt signaling pathway. Cardiovasc. Drugs Ther. 2021, 35, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Andelova, K.; Bacova, B.S.; Sykora, M.; Hlivak, P.; Barancik, M.; Tribulova, N. Mechanisms Underlying Antiarrhythmic Properties of Cardioprotective Agents Impacting Inflammation and Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 1416. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Testai, L.; Colletti, A.; Cicero, A.F.G. Coenzyme Q10: Clinical Applications in Cardiovascular Diseases. Antioxidants 2020, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Balan, A.I.; Halațiu, V.B.; Scridon, A. Oxidative Stress, Inflammation, and Mitochondrial Dysfunction: A Link between Obesity and Atrial Fibrillation. Antioxidants 2024, 13, 117. [Google Scholar] [CrossRef]
- Pool, L.; Wijdeveld, L.F.J.M.; de Groot, N.M.S.; Brundel, B.J.J.M. The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery. Int. J. Mol. Sci. 2021, 22, 8463. [Google Scholar] [CrossRef]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Weaver, W.D.; Cohen, B.H. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology 2018, 90, e1212–e1221. [Google Scholar] [CrossRef]
- Karaa, A.; Bertini, E.; Carelli, V.; Cohen, B.H.; Enns, G.M.; Falk, M.J.; Goldstein, A.; Gorman, G.S.; Haas, R.; Hirano, M.; et al. Efficacy and Safety of Elamipretide in Individuals With Primary Mitochondrial Myopathy. Neurology 2023, 101, e238–e252. [Google Scholar] [CrossRef]
- Chatfield, K.C.; Sparagna, G.C.; Chau, S.; Phillips, E.K.; Ambardekar, A.V.; Aftab, M.; Mitchell, M.B.; Sucharov, C.C.; Miyamoto, S.D.; Stauffer, B.L. Elamipretide Improves Mitochondrial Function in the Failing Human Heart. JACC Basic Transl. Sci. 2019, 4, 147–157. [Google Scholar] [CrossRef]
- Butler, J.; Khan, M.S.; Anker, S.D.; Fonarow, G.C.; Kim, R.J.; Nodari, S.; O’Connor, C.M.; Pieske, B.; Pieske-Kraigher, E.; Sabbah, H.N.; et al. Effects of Elamipretide on Left Ventricular Function in Patients With Heart Failure With Reduced Ejection Fraction: The PROGRESS-HF Phase 2 Trial. J. Card. Fail. 2020, 26, 429–437. [Google Scholar] [CrossRef]
- Seo, K.-S.; Kim, J.-H.; Min, K.-N.; Moon, J.-A.; Roh, T.-C.; Lee, M.-J.; Lee, K.-W.; Min, J.-E.; Lee, Y.-M. KL1333, a Novel NAD+ Modulator, Improves Energy Metabolism and Mitochondrial Dysfunction in MELAS Fibroblasts. Front. Neurol. 2018, 9, 552. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, J.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef] [PubMed]
- Kleindorfer, D.O.; Towfighi, A.; Chaturvedi, S.; Cockroft, K.M.; Gutierrez, J.; Lombardi-Hill, D.; Kamel, H.; Kernan, W.N.; Kittner, S.J.; Leira, E.C.; et al. 2021 Guideline for the Prevention of Stroke in Patients With Stroke and Transient Ischemic Attack: A Guideline From the American Heart Association/American Stroke Association. Stroke 2021, 52, E364–E467. [Google Scholar] [CrossRef] [PubMed]
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenko, A.; et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: A meta-analysis of randomised trials. Lancet 2014, 383, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Falco, L.; Tessitore, V.; Ciccarelli, G.; Malvezzi, M.; D’andrea, A.; Imbalzano, E.; Golino, P.; Russo, V. Antioxidant Properties of Oral Antithrombotic Therapies in Atherosclerotic Disease and Atrial Fibrillation. Antioxidants 2023, 12, 1185. [Google Scholar] [CrossRef]
- Russo, V.; Fabiani, D. Put out the fire: The pleiotropic anti-inflammatory action of non-vitamin K oral anticoagulants. Pharmacol. Res. 2022, 182, 106335. [Google Scholar] [CrossRef]
- Goette, A.; Mollenhauer, M.; Rudolph, V.; Lamparter, M.; Meier, M.; Böhm, M. Pleiotropic effects of NOACs with focus on edoxaban: Scientific findings and potential clinical implications. Herzschrittmachertherapie Elektrophysiologie 2023, 34, 142–152. [Google Scholar] [CrossRef]
- Cate, H.T.; Guzik, T.J.; Eikelboom, J.; Spronk, H.M.H. Pleiotropic actions of factor Xa inhibition in cardiovascular prevention: Mechanistic insights and implications for anti-thrombotic treatment. Cardiovasc. Res. 2021, 117, 2030–2044. [Google Scholar] [CrossRef]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef]
- Gori, A.M.; Camilleri, E.; Bertelli, A.; Rogolino, A.; Cesari, F.; Lotti, E.; Capobianco, T.; Iannotti, W.; Giusti, B.; Marcucci, R. Pleiotropic effects of anti-thrombotic therapies: Have direct oral anticoagulants any anti-inflammatory effect? Bleeding Thromb. Vasc. Biol. 2022, 1, 34–35. [Google Scholar] [CrossRef]
- Russo, V.; Fabiani, D.; Leonardi, S.B.; Attena, E.; D’Alterio, G.; Cotticelli, C.; Rago, A.; Sarpa, S.B.; Maione, B.B.; D’Onofrio, A.; et al. Dual Pathway Inhibition with Rivaroxaban and Aspirin Reduces Inflammatory Biomarkers in Atherosclerosis. J. Cardiovasc. Pharmacol. 2023, 81, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Bețiu, A.M.; Noveanu, L.; Hâncu, I.M.; Lascu, A.; Petrescu, L.; Maack, C.; Elmér, E.; Muntean, D.M. Mitochondrial Effects of Common Cardiovascular Medications: The Good, the Bad and the Mixed. Int. J. Mol. Sci. 2022, 23, 13653. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Tsuboi, T.; Hayashi, T. An Inhibitor of Activated Blood Coagulation Factor X Shows Anti-Endothelial Senescence and Anti-Atherosclerotic Effects. J. Vasc. Res. 2019, 56, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, P.; Perzborn, E.; Hauenschild, P.; Gerdes, C.; Heitmeier, S.; Visser, M.; Summer, H.; Laux, V. Expression of pro-inflammatory genes in human endothelial cells: Comparison of rivaroxaban and dabigatran. Thromb. Res. 2016, 142, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Ueda, S.; Fukami, K.; Yamagishi, S. Advanced glycation end products potentiate citrated plasma-evoked oxidative and inflammatory reactions in endothelial cells by up-regulating protease-activated receptor-1 expression. Cardiovasc. Diabetol. 2014, 13, 60. [Google Scholar] [CrossRef]
- Bukowska, A.; Zacharias, I.; Weinert, S.; Skopp, K.; Hartmann, C.; Huth, C.; Goette, A. Coagulation factor Xa induces an inflammatory signalling by activation of protease-activated receptors in human atrial tissue. Eur. J. Pharmacol. 2013, 718, 114–123. [Google Scholar] [CrossRef]
- Zekri-Nechar, K.; Zamorano-León, J.J.; Cortina-Gredilla, M.; López-De-Andrés, A.; Jiménez-García, R.; Navarro-Cuellar, C.; López-Farré, A.; Martínez-Martínez, C.H. Mitochondrial mitophagy protection combining rivaroxaban and aspirin in high glucose-exposed human coronary artery endothelial cell. An in vitro study. Diabetes Vasc. Dis. Res. 2022, 19, 147916412211298. [Google Scholar] [CrossRef]
- Samiei, F.; Sajjadi, H.; Jamshidzadeh, A.; Seydi, E.; Pourahmad, J. Contrasting Role of Concentration in Rivaroxaban Induced Toxicity and Oxidative Stress in Isolated Kidney Mitochondria. Drug Res. 2019, 69, 523–527. [Google Scholar] [CrossRef]
- Zamorano-Leon, J.J.; de la Serna-Soto, M.; Moñux, G.; Freixer, G.; Zekri-Nechar, K.; Cabrero-Fernandez, M.; Segura, A.; Gonzalez-Cantalapiedra, A.; Serrano, J.; Farré, A.L. Factor Xa Inhibition by Rivaroxaban Modified Mitochondrial-Associated Proteins in Human Abdominal Aortic Aneurysms. Ann. Vasc. Surg. 2020, 67, 482–489. [Google Scholar] [CrossRef]
- Narita, Y.; Hamamura, K.; Kashiyama, M.; Utsumi, S.; Kakizoe, Y.; Kondo, Y.; Ishitsuka, Y.; Jono, H.; Irie, T.; Mukoyama, M.; et al. Edoxaban Exerts Antioxidant Effects Through FXa Inhibition and Direct Radical-Scavenging Activity. Int. J. Mol. Sci. 2019, 20, 4140. [Google Scholar] [CrossRef]
- Bukowska, A.; Schild, L.; Bornfleth, P.; Peter, D.; Wiese-Rischke, C.; Gardemann, A.; Isermann, B.; Walles, T.; Goette, A. Activated clotting factor X mediates mitochondrial alterations and inflammatory responses via protease-activated receptor signaling in alveolar epithelial cells. Eur. J. Pharmacol. 2020, 869, 172875. [Google Scholar] [CrossRef]
- Torramade-Moix, S.; Palomo, M.; Vera, M.; Jerez, D.; Moreno-Castaño, A.B.; Zafar, M.U.; Rovira, J.; Diekmann, F.; Garcia-Pagan, J.C.; Escolar, G.; et al. Apixaban Downregulates Endothelial Inflammatory and Prothrombotic Phenotype in an In Vitro Model of Endothelial Dysfunction in Uremia. Cardiovasc. Drugs Ther. 2021, 35, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Durmaz, S.; Kurtoğlu, T.; Rahman, F.; Tataroğlu, C.; Yılmaz, M.; Barbarus, E.; Erkan, M.H. Direct oral anticoagulant agents attenuate temporary aortic occlusion-induced renal oxidative and inflammatory responses in rats. Turk. J. Thorac. Cardiovasc. Surg. 2022, 30, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.L.; Iannucci, J.; Seeram, N.P.; Grammas, P. Inhibiting thrombin improves motor function and decreases oxidative stress in the LRRK2 transgenic Drosophila melanogaster model of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2020, 527, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Iannucci, J.; Johnson, S.L.; Majchrzak, M.; Barlock, B.J.; Akhlaghi, F.; Seeram, N.P.; Sen, A.; Grammas, P. Short-term treatment with dabigatran alters protein expression patterns in a late-stage tau-based Alzheimer’s disease mouse model. Biochem. Biophys. Rep. 2020, 24, 100862. [Google Scholar] [CrossRef]
- Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J.; Martinez, J.; Grammas, P. Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 2013, 5, 19. [Google Scholar] [CrossRef]
- Pingel, S.; Tiyerili, V.; Mueller, J.; Werner, N.; Nickenig, G.; Mueller, C. Experimental research Thrombin inhibition by dabigatran attenuates atherosclerosis in ApoE deficient mice. Arch. Med Sci. 2014, 1, 154–160. [Google Scholar] [CrossRef]
- Kadoglou, N.P.E.; Moustardas, P.; Katsimpoulas, M.; Kapelouzou, A.; Kostomitsopoulos, N.; Schafer, K.; Kostakis, A.; Liapis, C.D. The Beneficial Effects of a Direct Thrombin Inhibitor, Dabigatran Etexilate, on the Development and Stability of Atherosclerotic Lesions in Apolipoprotein E-deficient Mice. Cardiovasc. Drugs Ther. 2012, 26, 367–374. [Google Scholar] [CrossRef]
- Woźniak, E.; Broncel, M.; Bukowska, B.; Gorzelak-Pabiś, P. The Protective Effect of Dabigatran and Rivaroxaban on DNA Oxidative Changes in a Model of Vascular Endothelial Damage with Oxidized Cholesterol. Int. J. Mol. Sci. 2020, 21, 1953. [Google Scholar] [CrossRef]
- Kurokawa, H.; Taninaka, A.; Shigekawa, H.; Matsui, H. Dabigatran Etexilate Induces Cytotoxicity in Rat Gastric Epithelial Cell Line via Mitochondrial Reactive Oxygen Species Production. Cells 2021, 10, 2508. [Google Scholar] [CrossRef]
- Raskob, G.E.; van Es, N.; Verhamme, P.; Carrier, M.; Di Nisio, M.; Garcia, D.; Grosso, M.A.; Kakkar, A.K.; Kovacs, M.J.; Mercuri, M.F.; et al. Edoxaban for the Treatment of Cancer-Associated Venous Thromboembolism. N. Engl. J. Med. 2018, 378, 615–624. [Google Scholar] [CrossRef]
- Russo, V.; Falco, L.; Tessitore, V.; Mauriello, A.; Catapano, D.; Napolitano, N.; Tariq, M.; Caturano, A.; Ciccarelli, G.; D’andrea, A.; et al. Anti-Inflammatory and Anticancer Effects of Anticoagulant Therapy in Patients with Malignancy. Life 2023, 13, 1888. [Google Scholar] [CrossRef]
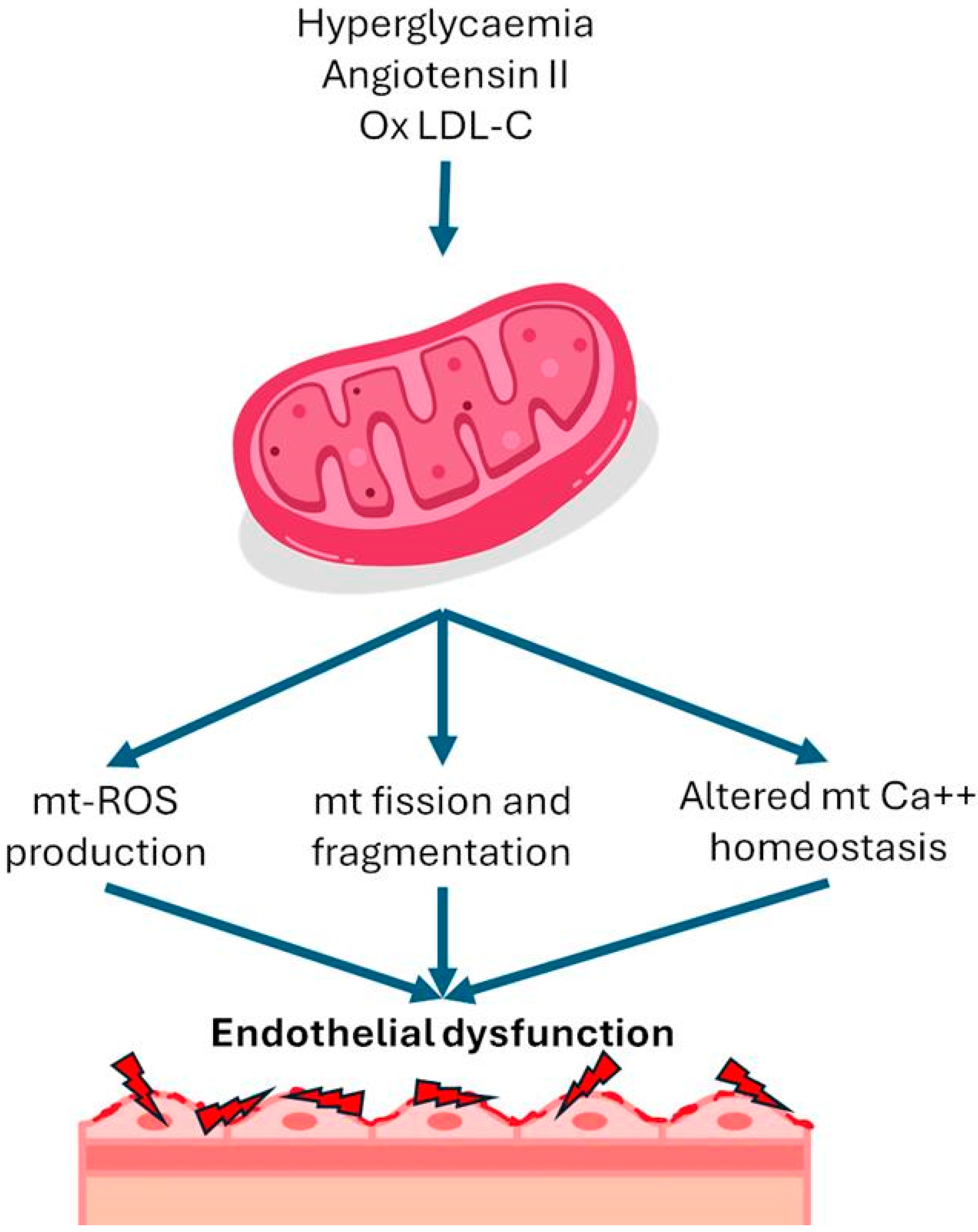
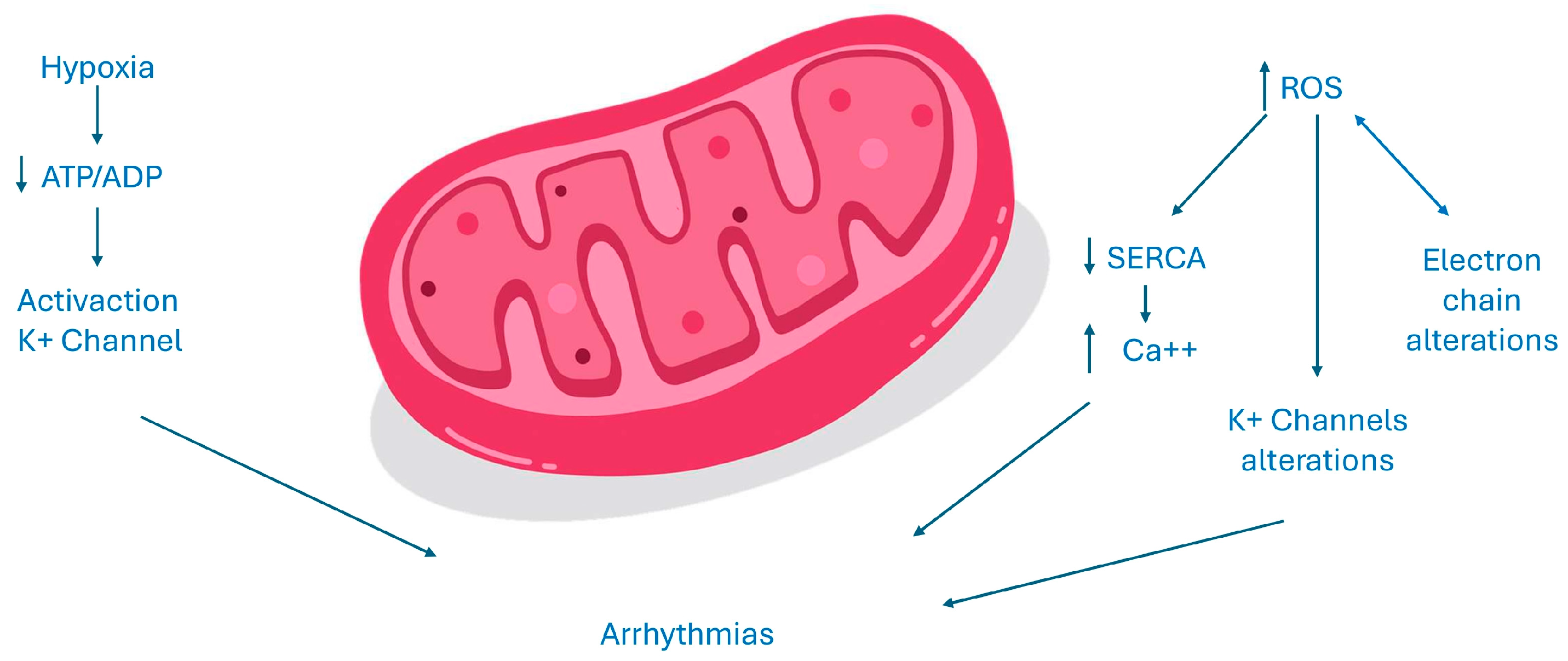

{kind=link}
{kind=link}
{kind=link}
| Risk Factors of Mitochondrial Disease |
|---|
| Smoke [3] |
| Hyperglicemia [4] |
| Fatty foods [3] |
| Sedentariety [3] |
| Alcohol [3] |
| Angiotensin II [5] |
| Use of drugs [3] |
| Dyslipidemia [6] |
| Syndrome | Causative Genes | Inheritance Pattern | Clinical Manifestations | Onset |
|---|---|---|---|---|
| Leigh syndrome | More than 80 genes in mitochondrial (MtDNA) and nuclear DNA (nDNA), including SURF1 | AR (mainly) | Seizures, encephalopathy, failure to thrive, dysphagia, cardiac involvement (HCM or DCM; valvular disease, arrhythmia, conduction defect) | Childhood |
| Sengers syndrome | Acylglycerol kinase AGK (nDNA) | AR | Cataracts, HCM, skeletal myopathy, lactic acidosis | Childhood/adulthood |
| Kearns–Sayre syndrome (KSS) | MtDNA deletion | Maternal inheritance pattern | Neurological involvement (ataxia, dementia), diabetes mellitus, cardiac conduction disorders (possible onset with sudden death), pigmentary retinopathy | Adulthood |
| Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) syndrome | MtDNA mutations (m.3243A>Gin MT-TL1, and other pathogenetic variants in MT-TL1) or MT-TV and MT-TQ | Maternal inheritance pattern | Ataxia, seizures, stroke-like episodes, myopathy, lactis acidosis, HCM, LV noncompactation, pre-excitation, atrioventricular block deafness. | Adulthood |
| Leber hereditary optic neuropathy (LHON) | Mutations in Mt-DNA m.11778G>A (MT-ND4), m.14484T>C (MT-ND6) and m.3460G>A (MT-ND1) | AR | Visual loss, cardiac involvement (pre-excitation) | Adulthood |
| Medication | Main Effect on Mithocondrial Function and AF |
|---|---|
| SGLT-2 inhibitors [22,23,24,25,26,27] | ↓ ROS production; restoration of mitochondrial membrane potential; ↑ mitochondrial biogenesis acting on PGC-1, NRF-1, Mfn-1, and AMPK; regulation of intracellulare electrolyte balance; ↓ in myocardial remodeling and fibrosis acting on TGF-beta/smad and NRF2/ARE; ↓ AF inducibility and in AF incidence |
| GLP1R antagonists [27,28,29,30,31] | ↓ ROS production and ↑ in ROS scavengers’ mechanisms; antiapoptotic effects acting on cAMP/Epac/PI3K/Akt pathway; ↓ in myocardial remodeling and fibrosis; ↓ AF inducibility in animal models, contrasting data on humans |
| DDP-4 inhibitors [32,33,34,35,36,37,38] | ↓ mitochondrial ROS production; ↓ mitochondrial membrane depolarization; ↑ mitochondrial biogenesis acting on PGC-1 /NRF1/Tfam; ↓ AF inducibility in animal models, contrasting data on humans |
| Metformin [39,40] | ↑ mitochondrial oxygen consumption and activity of complexes I, II, and IV; ↓ atrial remodeling by activating the AMPK/PGC-1/PPAR; ↓ of AF incidence by 19% |
| Thiazolidinediones [41,42,43,44] | ↓ oxidative stress; ↓ mitochondrial apoptotic signaling acting on PPAR; ↓ atrial remodeling; ↑ ion channel function (ICa and INa); ↓ AF inducibility in animal models, contrasting data on humans |
| Statins [45,46,47] | ↓ oxidative stress through ↓ Rho/ROCK pathways, ↑ PI(3)K/Akt pathway, and ↓ NAD(P)H oxidase activity; ↓incidence of AF by 19% |
| Fibrates [48] | ↑ mitochondrial function acting on PPAR/PGC-1; ↓ atrial remodeling and inducibility of AF prolonging atrial refractory period |
| Omega 3 fatty acids [48] | ↓ ROS production; regulation of ion channels and cardiac electrical activity |
| Trimetazidine [49,50] | ↓ miotochondrial ROS production by activate complex I and ETC; ↑ in mitochondrial biogenesis acting on PPAR/PGC-1α; improvement on mitochondrial fusion/fission dynamics acting on Mfn-1/Drp1/Opa-1; ↓ atrial remodeling; ↓ AF inducibility and duration in ischemic conditions |
| Ranolazine [51] | ↓ mitochondrial ROS production due to inhibition of fatty acid oxidation; antiarrhythmic proprieties due to action on sodium and potassium channels |
| Carvedilol [48] | Block on alfa1 and beta1 adrenergic receptors; antioxidative proprieties |
| ACE-I, ARB, and AT1R blocker [48] | ↓ROS production by XO and NADPH oxidase, induced by AngII; atabilization of cellular electrical proprieties blocking of the NF-κB action on SCN5A |
| Febuxostat and Allopurinol [52,53] | ↓of oxidative stress inhibiting XO; ↓of AF susceptibility inhibiting ox-Ca2+-calmodulin-dependent protein-kinase type-II (CaMKII) |
| Ubiquinone (CoQ10) [54,55] | Cofactor is involved in electron transport within the respiratory chain; anti-inflammatory and anti-oxidant activity. |
| Medication | Main Effect on Mithocondrial Function and AF |
|---|---|
| Ubiquinone (CoQ10) [54,55] | Cofactor involved in electron transport within the respiratory chain; anti-inflammatory and anti-oxidant activity |
| Vitamin C and E [65] | Anti-inflammatory and anti-oxidant activity; ↓ post-surgical AF and AF recurrence after electrical cardioversion |
| N-acetyl cysteine [65] | ↓ risk of AF by ↑ the density of L-type calcium current |
| L-glutamine [66] | ↓ ROS production and stabilize the microtubule network through heightened heat-shock protein (HSP) expression. |
| Costunolide [48] | ↑ mitochondrial function and ↓ in ROS production; anti-inflammatory and anti-fibrotic properties |
| Andrographolide [48] | ↑ mitochondrial function and ↓ in ROS production; anti-inflammatory proprieties through regulation of calcium homeostasis genes |
| Medication | Mechanism | Effect on Mitochondrial Function |
|---|---|---|
| Rivaroxaban [5,81,84,87,88,89,99] | Factor Xa inhibitor | ↓ ROS production; restoration of mitochondrial membrane potential; ↑ mitophagy; ↑ citrate synthase; ↑ cytochrome C oxidase |
| Edoxaban [77,90,101] | Factor Xa inhibitor | ↓ ROS production; ↑ mitochondrial oxigen consumption; ↑ ATP production; ↓ atrial remodeling |
| Apixaban [92,102] | Factor Xa inhibitor | ↓ ROS production |
| Dabigatran [100] | Thrombin inhibitor | ↓ ROS production; ↓ ROS-induced DNA strand breakage; ↓ SOD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauriello, A.; Correra, A.; Molinari, R.; Del Vecchio, G.E.; Tessitore, V.; D’Andrea, A.; Russo, V. Mitochondrial Dysfunction in Atrial Fibrillation: The Need for a Strong Pharmacological Approach. Biomedicines 2024, 12, 2720. https://doi.org/10.3390/biomedicines12122720
Mauriello A, Correra A, Molinari R, Del Vecchio GE, Tessitore V, D’Andrea A, Russo V. Mitochondrial Dysfunction in Atrial Fibrillation: The Need for a Strong Pharmacological Approach. Biomedicines. 2024; 12(12):2720. https://doi.org/10.3390/biomedicines12122720
Chicago/Turabian StyleMauriello, Alfredo, Adriana Correra, Riccardo Molinari, Gerardo Elia Del Vecchio, Viviana Tessitore, Antonello D’Andrea, and Vincenzo Russo. 2024. "Mitochondrial Dysfunction in Atrial Fibrillation: The Need for a Strong Pharmacological Approach" Biomedicines 12, no. 12: 2720. https://doi.org/10.3390/biomedicines12122720
APA StyleMauriello, A., Correra, A., Molinari, R., Del Vecchio, G. E., Tessitore, V., D’Andrea, A., & Russo, V. (2024). Mitochondrial Dysfunction in Atrial Fibrillation: The Need for a Strong Pharmacological Approach. Biomedicines, 12(12), 2720. https://doi.org/10.3390/biomedicines12122720