
Levo-Stepholidine as a Potential Cognitive Enhancer: Insights into Executive Function and Memory Improvements



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
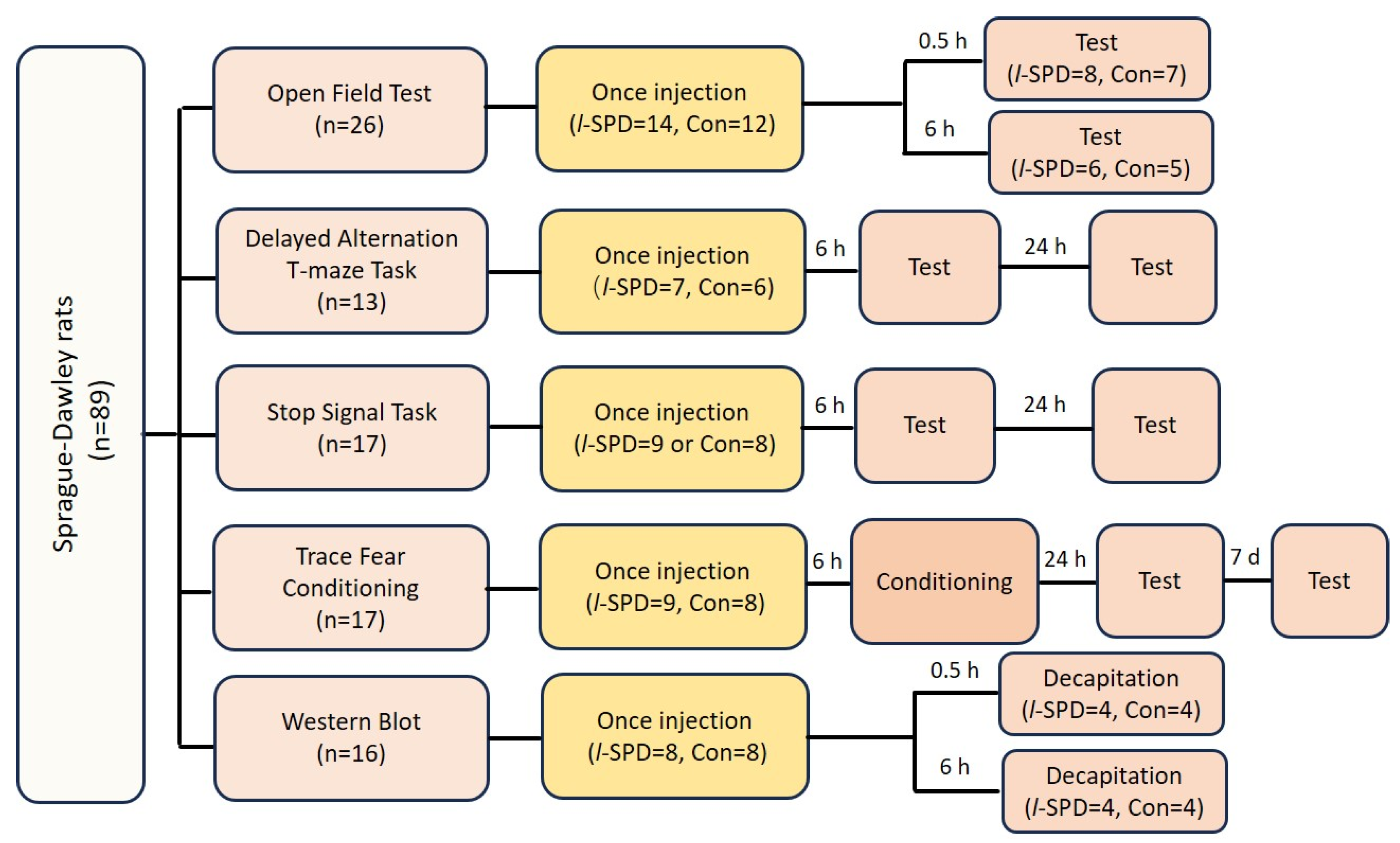
2.1. Subjects
2.2. Behavioral Experiments
2.3. Open Field Test
2.4. The Delayed Alternative Task in T-Maze
2.5. The Stop-Signal Task
2.6. Trace Fear Conditioning
2.7. Western Blot
2.8. Chemicals
2.9. Data Analysis
3. Results
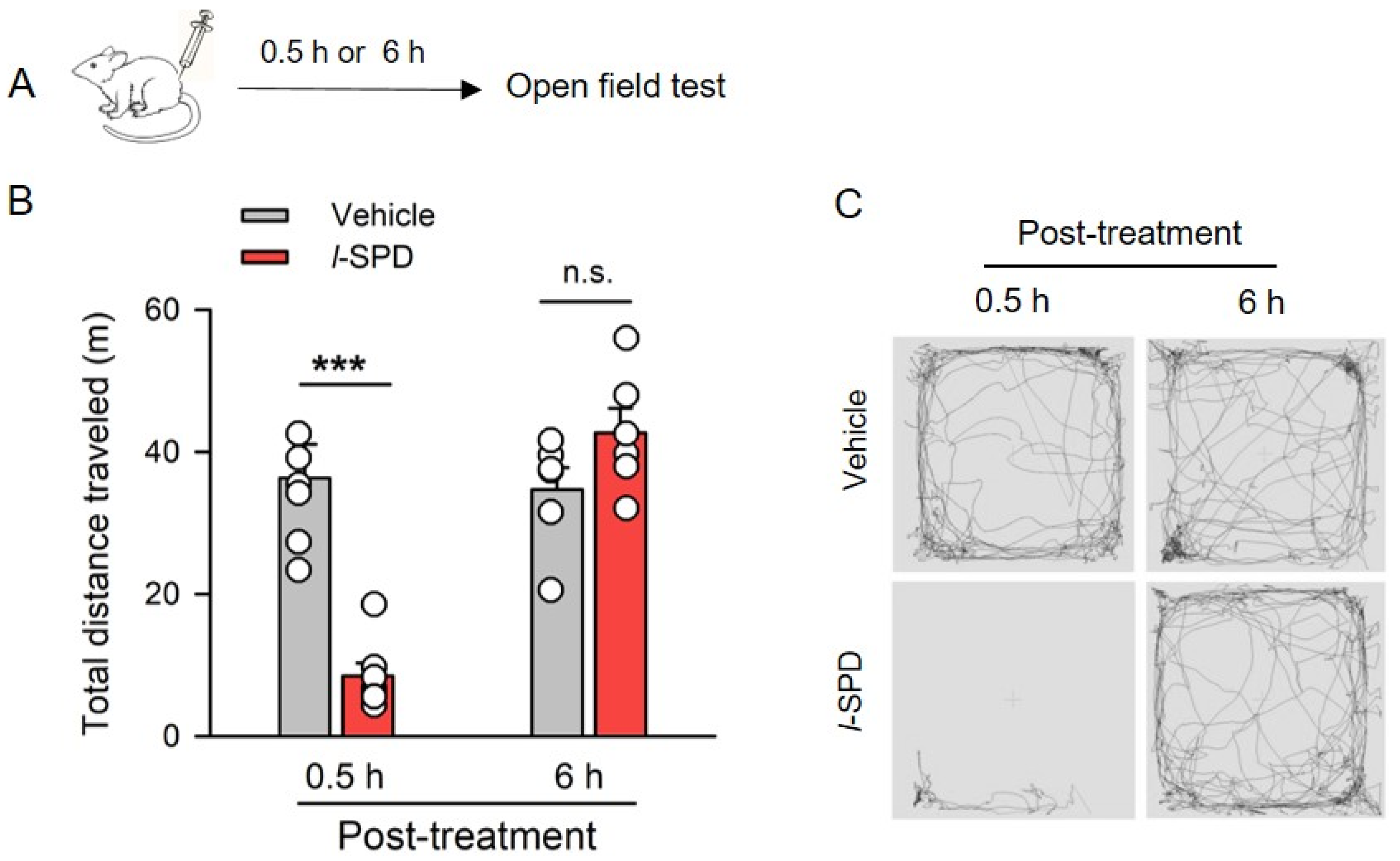
3.1. Effect of l-SPD on Spontaneous Activity of Rats
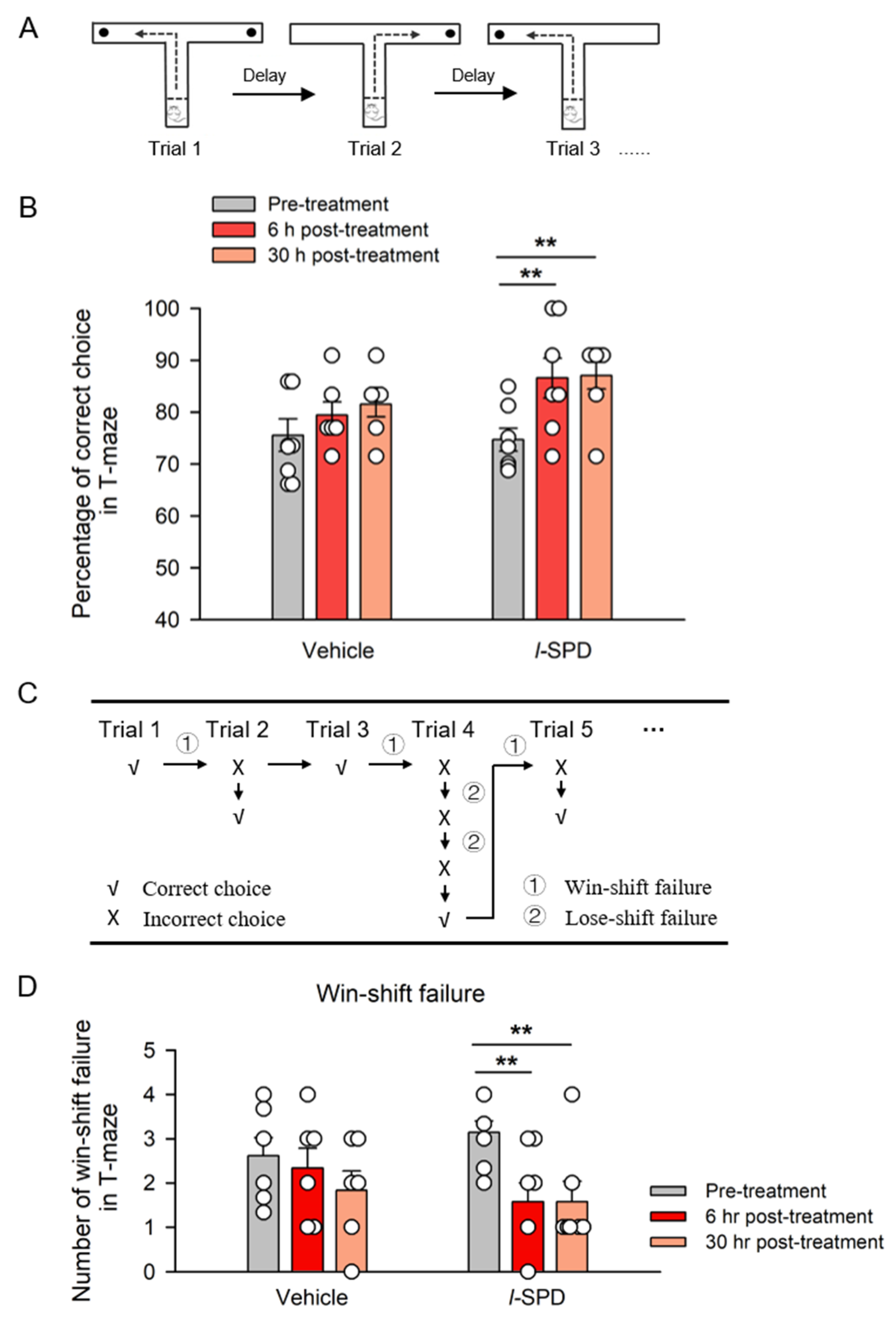
3.2. L-SPD Enhances the Working Memory in Delayed Alternative T-Maze Task
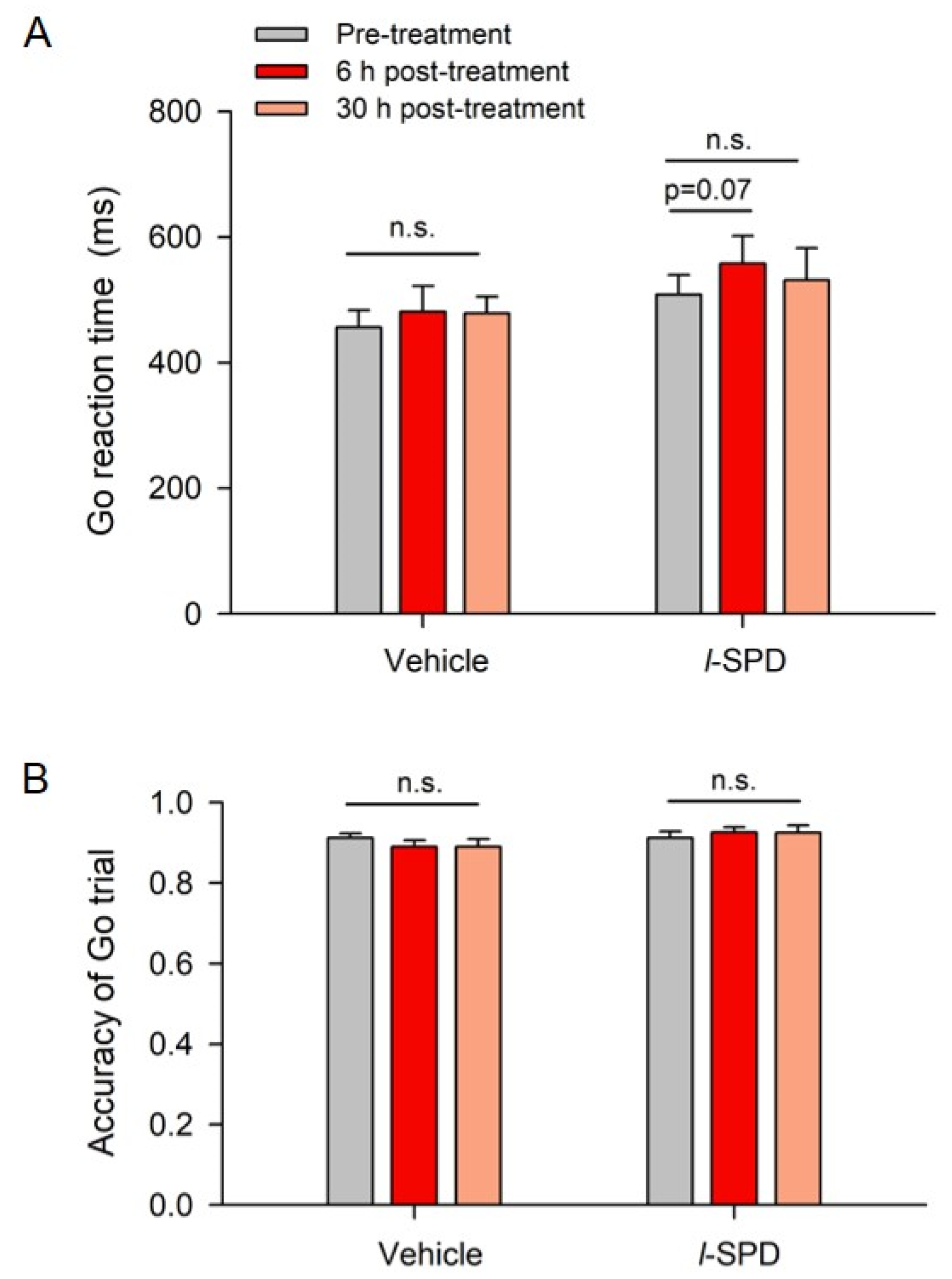
3.3. l-SPD Enhances Response Inhibition in the Stop-Signal Task
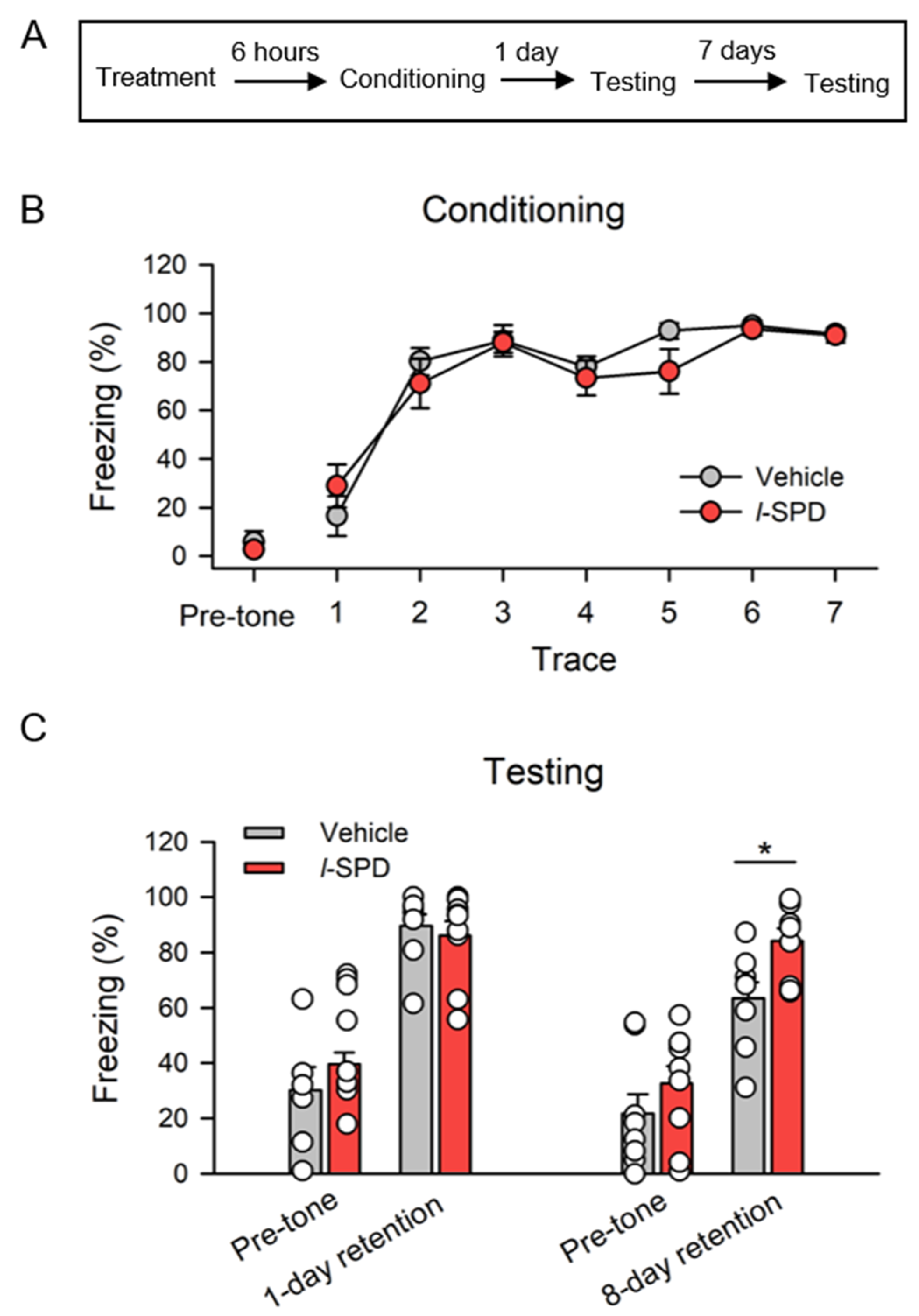
3.4. l-SPD Enhances Long-Term Memory Retention
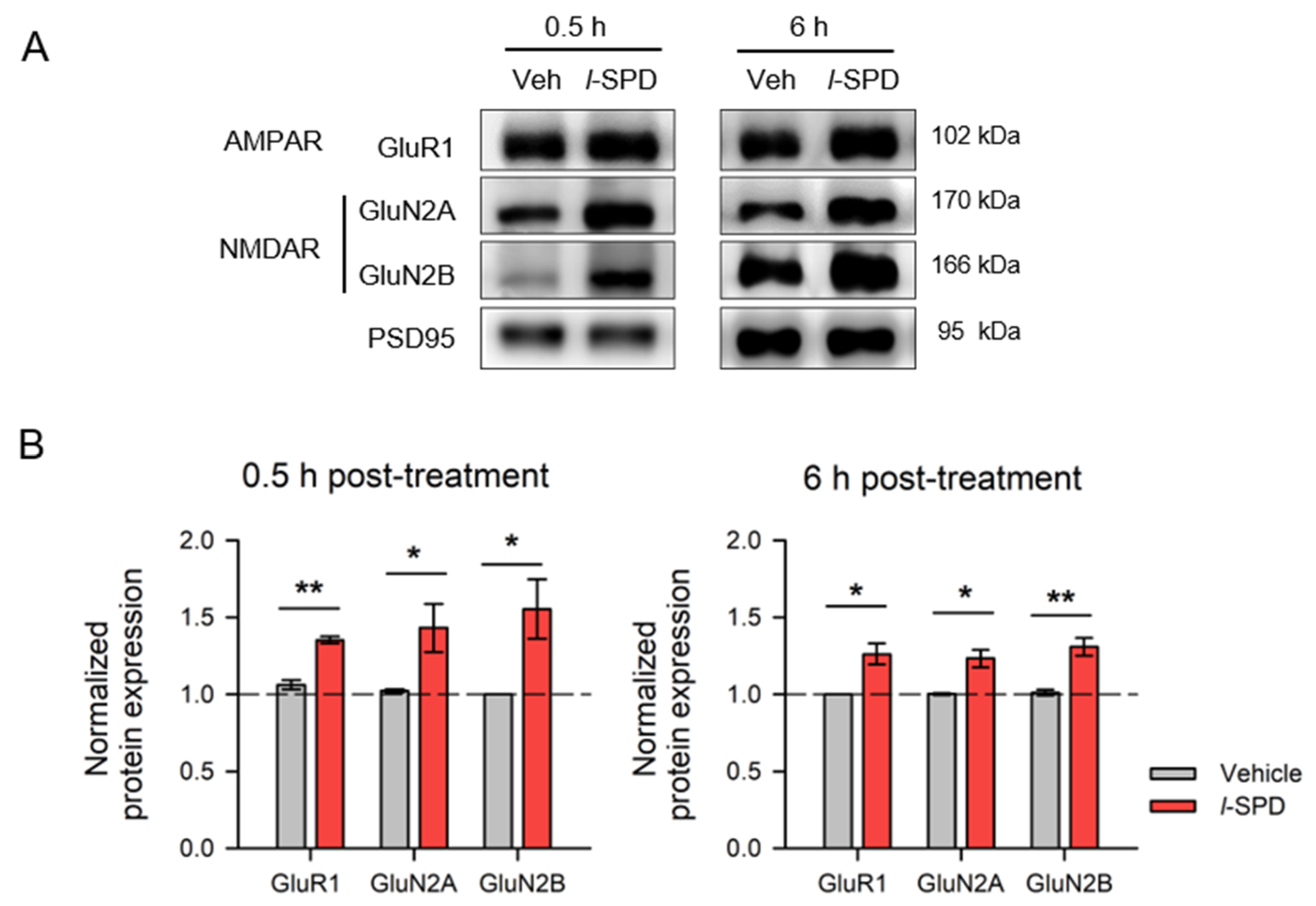
3.5. l-SPD Upregulates NMDA and AMPA Receptor Expression in PFC
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shahrokh, N.C.; Hales, R.E.; Phillips, K.A.; Yudofsky, S.C. The Language of Mental Health: A Glossary of Psychiatric Terms; American Psychiatric Publishing: Arlington, VA, USA, 2011. [Google Scholar]
- Harvey, P.D.; Bosia, M.; Cavallaro, R.; Howes, O.D.; Kahn, R.S.; Leucht, S.; Müller, D.R.; Penadés, R.; Vita, A. Cognitive dysfunction in schizophrenia: An expert group paper on the current state of the art. Schizophr. Res. Cogn. 2022, 29, 100249. [Google Scholar] [CrossRef] [PubMed]
- Sabe, M.; Pillinger, T.; Kaiser, S.; Chen, C.; Taipale, H.; Tanskanen, A.; Tiihonen, J.; Leucht, S.; Correll, C.U.; Solmi, M. Half a century of research on antipsychotics and schizophrenia: A scientometric study of hotspots, nodes, bursts, and trends. Neurosci. Biobehav. Rev. 2022, 136, 104608. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.S.; Keefe, R.S. Schizophrenia is a cognitive illness: Time for a change in focus. JAMA Psychiatry 2013, 70, 1107–1112. [Google Scholar] [CrossRef]
- Vita, A.; Gaebel, W.; Mucci, A.; Sachs, G.; Barlati, S.; Giordano, G.M.; Nibbio, G.; Nordentoft, M.; Wykes, T.; Galderisi, S. European Psychiatric Association guidance on treatment of cognitive impairment in schizophrenia. Eur. Psychiatry 2022, 65, e57. [Google Scholar] [CrossRef]
- Green, M.F.; Kern, R.S.; Braff, D.L.; Mintz, J. Neurocognitive deficits and functional outcome in schizophrenia: Are we measuring the “right stuff”? Schizophr. Bull. 2000, 26, 119–136. [Google Scholar] [CrossRef]
- Falkai, P.; Schmitt, A. The need to develop personalized interventions to improve cognition in schizophrenia. World Psychiatry 2019, 18, 170. [Google Scholar] [CrossRef]
- Meier, M.H.; Caspi, A.; Reichenberg, A.; Keefe, R.S.; Fisher, H.L.; Harrington, H.; Houts, R.; Poulton, R.; Moffitt, T.E. Neuropsychological decline in schizophrenia from the premorbid to the postonset period: Evidence from a population-representative longitudinal study. Am. J. Psychiatry 2014, 171, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Reichenberg, A.; Harvey, P.D. Neuropsychological impairments in schizophrenia: Integration of performance-based and brain imaging findings. Psychol. Bull. 2007, 133, 833–858. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.; Moffitt, T.E.; Caspi, A.; Murray, R.M.; Harrington, H.; Poulton, R. Neuropsychological performance at the age of 13 years and adult schizophreniform disorder: Prospective birth cohort study. Br. J. Psychiatry 2006, 189, 463–464. [Google Scholar] [CrossRef]
- El-Mallakh, P.; Findlay, J. Strategies to improve medication adherence in patients with schizophrenia: The role of support services. Neuropsychiatr. Dis. Treat. 2015, 11, 1077–1090. [Google Scholar] [CrossRef]
- Miller, E.K.; Cohen, J.D. An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci. 2001, 24, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Reimann, Z.; Miller, J.R.; Dahle, K.M.; Hooper, A.P.; Young, A.M.; Goates, M.C.; Magnusson, B.M.; Crandall, A. Executive functions and health behaviors associated with the leading causes of death in the United States: A systematic review. J. Health Psychol. 2020, 25, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Miyake, A.; Friedman, N.P.; Emerson, M.J.; Witzki, A.H.; Howerter, A.; Wager, T.D. The unity and diversity of executive functions and their contributions to complex “Frontal Lobe” tasks: A latent variable analysis. Cogn. Psychol. 2000, 41, 49–100. [Google Scholar] [CrossRef]
- Baddeley, A. Working Memory: The Interface between Memory and Cognition. J. Cogn. Neurosci. 1992, 4, 281–288. [Google Scholar] [CrossRef]
- Forbes, N.F.; Carrick, L.A.; McIntosh, A.M.; Lawrie, S.M. Working memory in schizophrenia: A meta-analysis. Psychol. Med. 2009, 39, 889–905. [Google Scholar] [CrossRef]
- Lett, T.A.; Voineskos, A.N.; Kennedy, J.L.; Levine, B.; Daskalakis, Z.J. Treating working memory deficits in schizophrenia: A review of the neurobiology. Biol. Psychiatry 2014, 75, 361–370. [Google Scholar] [CrossRef]
- Collins, A.G.; Brown, J.K.; Gold, J.M.; Waltz, J.A.; Frank, M.J. Working memory contributions to reinforcement learning impairments in schizophrenia. J. Neurosci. 2014, 34, 13747–13756. [Google Scholar] [CrossRef]
- Gray, B.E.; Hahn, B.; Robinson, B.; Harvey, A.; Leonard, C.J.; Luck, S.J.; Gold, J.M. Relationships between divided attention and working memory impairment in people with schizophrenia. Schizophr. Bull. 2014, 40, 1462–1471. [Google Scholar] [CrossRef]
- Silver, H.; Feldman, P.; Bilker, W.; Gur, R.C. Working memory deficit as a core neuropsychological dysfunction in schizophrenia. Am. J. Psychiatry 2003, 160, 1809–1816. [Google Scholar] [CrossRef]
- Goldman-Rakic, P.S. Development of cortical circuitry and cognitive function. Child Dev. 1987, 58, 601–622. [Google Scholar] [CrossRef]
- Tritsch, N.X.; Sabatini, B.L. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 2012, 76, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Sawaguchi, T.; Goldman-Rakic, P.S. The role of D1-dopamine receptor in working memory: Local injections of dopamine antagonists into the prefrontal cortex of rhesus monkeys performing an oculomotor delayed-response task. J. Neurophysiol. 1994, 71, 515–528. [Google Scholar] [CrossRef]
- Williams, S.M.; Goldman-Rakic, P.S. Widespread origin of the primate mesofrontal dopamine system. Cereb. Cortex 1998, 8, 321–345. [Google Scholar] [CrossRef] [PubMed]
- Seamans, J.K.; Floresco, S.B.; Phillips, A.G. D1 receptor modulation of hippocampal-prefrontal cortical circuits integrating spatial memory with executive functions in the rat. J. Neurosci. 1998, 18, 1613–1621. [Google Scholar] [CrossRef]
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 2016, 17, 524–532. [Google Scholar] [CrossRef]
- Volk, L.; Chiu, S.-L.; Sharma, K.; Huganir, R.L. Glutamate synapses in human cognitive disorders. Annu. Rev. Neurosci. 2015, 38, 127–149. [Google Scholar] [CrossRef]
- Laruelle, M.; Kegeles, L.S.; Abi-Dargham, A. Glutamate, dopamine, and schizophrenia: From pathophysiology to treatment. Ann. N. Y. Acad. Sci. 2003, 1003, 138–158. [Google Scholar] [CrossRef]
- Li, C.T.; Yang, K.C.; Lin, W.C. Glutamatergic Dysfunction and Glutamatergic Compounds for Major Psychiatric Disorders: Evidence from Clinical Neuroimaging Studies. Front. Psychiatry 2018, 9, 767. [Google Scholar] [CrossRef]
- Sun, X.; Zhao, Y.; Wolf, M.E. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J. Neurosci. 2005, 25, 7342–7351. [Google Scholar] [CrossRef]
- Gao, C.; Wolf, M.E. Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J. Neurochem. 2008, 106, 2489–2501. [Google Scholar] [CrossRef]
- Natesan, S.; Reckless, G.E.; Barlow, K.B.L.; Odontiadis, J.; Nobrega, J.N.; Baker, G.B.; George, S.R.; Mamo, D.; Kapur, S. The antipsychotic potential of l-stepholidine--a naturally occurring dopamine receptor D1 agonist and D2 antagonist. Psychopharmacology 2008, 199, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.Z. [Progress in studies of the pharmacology of l-tetrahydropalmatine and l-stepholidine]. Yao Xue Xue Bao 1987, 22, 472–480. [Google Scholar] [PubMed]
- Sun, Y.; Dai, J.; Hu, Z.; Du, F.; Niu, W.; Wang, F.; Liu, F.; Jin, G.; Li, C. Oral bioavailability and brain penetration of (-)-stepholidine, a tetrahydroprotoberberine agonist at dopamine D(1) and antagonist at D(2) receptors, in rats. Br. J. Pharmacol. 2009, 158, 1302–1312. [Google Scholar] [CrossRef]
- Jin, G.Z.; Zhu, Z.T.; Fu, Y. (-)-Stepholidine: A potential novel antipsychotic drug with dual D1 receptor agonist and D2 receptor antagonist actions. Trends Pharmacol. Sci. 2002, 23, 4–7. [Google Scholar] [CrossRef]
- Abi-Dargham, A. Do we still believe in the dopamine hypothesis? New data bring new evidence. Int. J. Neuropsychopharmacol. 2004, 7 (Suppl. 1), S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.-R.; Sun, N.; Lei, L.; Li, X.-Y.; Yao, B.; Sun, K.; Hu, R.; Zhang, X.; Shi, X.-D.; Gao, C. L-Stepholidine rescues memory deficit and synaptic plasticity in models of Alzheimer’s disease via activating dopamine D1 receptor/PKA signaling pathway. Cell Death Dis. 2015, 6, e1965. [Google Scholar] [CrossRef]
- Ellenbroek, B.A.; Zhang, X.X.; Jin, G.Z. Effects of (-)stepholidine in animal models for schizophrenia. Acta Pharmacol. Sin. 2006, 27, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Guo, F.; Ma, Y.; Song, Y.; Lin, R.; Shen, F.-Y.; Jin, G.-Z.; Li, Y.; Liu, Z.-Q. Activation of D1R/PKA/mTOR signaling cascade in medial prefrontal cortex underlying the antidepressant effects of l-SPD. Sci. Rep. 2017, 7, 3809. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Gong, X.; Ru, Q.; Xiong, Q.; Chen, L.; Si, Y.; Xiao, H.; Li, C. The Neuroprotective Effect of L-Stepholidine on Methamphetamine-Induced Memory Deficits in Mice. Neurotox. Res. 2019, 36, 376–386. [Google Scholar] [CrossRef]
- Yue, K.; Ma, B.; Chen, L.; Tian, X.; Ru, Q.; Gan, Y.; Wang, D.; Jin, G.; Li, C. L-Stepholidine, a naturally occurring dopamine D1 receptor agonist and D2 receptor antagonist, attenuates heroin self-administration and cue-induced reinstatement in rats. Neuroreport 2014, 25, 7–11. [Google Scholar] [CrossRef]
- Goldman-Rakic, P.S. Cellular basis of working memory. Neuron 1995, 14, 477–485. [Google Scholar] [CrossRef]
- Diamond, A. Executive functions. Annu. Rev. Psychol. 2013, 64, 135–168. [Google Scholar] [CrossRef]
- Fuster, J.M.; Bauer, R.H.; Jervey, J.P. Functional interactions between inferotemporal and prefrontal cortex in a cognitive task. Brain Res. 1985, 330, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-H.; Liu, S.-S.; Yi, F.; Zhuo, M.; Li, B.-M. Delay-dependent impairment of spatial working memory with inhibition of NR2B-containing NMDA receptors in hippocampal CA1 region of rats. Mol. Brain 2013, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Bari, A.; Robbins, T.W. Inhibition and impulsivity: Behavioral and neural basis of response control. Prog. Neurobiol. 2013, 108, 44–79. [Google Scholar] [CrossRef]
- Zhang, D.D.; Zhang, Y.Q.; Zhang, X.H. Prefrontal AMPA receptors are involved in the effect of methylphenidate on response inhibition in rats. Acta Pharmacol. Sin. 2018, 39, 607–615. [Google Scholar] [CrossRef]
- Logan, G.D.; Cowan, W.B.; Davis, K.A. On the ability to inhibit simple and choice reaction time responses: A model and a method. J. Exp. Psychol. Hum. Percept. Perform. 1984, 10, 276–291. [Google Scholar] [CrossRef]
- Dias, R.; Robbins, T.W.; Roberts, A.C. Dissociable forms of inhibitory control within prefrontal cortex with an analog of the Wisconsin Card Sort Test: Restriction to novel situations and independence from “on-line” processing. J. Neurosci. 1997, 17, 9285–9297. [Google Scholar] [CrossRef]
- Runyan, J.D.; Moore, A.N.; Dash, P.K. A role for prefrontal cortex in memory storage for trace fear conditioning. J. Neurosci. 2004, 24, 1288–1295. [Google Scholar] [CrossRef]
- Tao, X.-D.; Liu, Z.-R.; Zhang, Y.-Q.; Zhang, X.-H. Connexin43 hemichannels contribute to working memory and excitatory synaptic transmission of pyramidal neurons in the prefrontal cortex of rats. Life Sci. 2021, 286, 120049. [Google Scholar] [CrossRef]
- Bari, A.; Mar, A.C.; Theobald, D.E.; Elands, S.A.; Oganya, K.C.N.A.; Eagle, D.M.; Robbins, T.W. Prefrontal and monoaminergic contributions to stop-signal task performance in rats. J. Neurosci. 2011, 31, 9254–9263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.C.; Sun, Y.Y.; Cai, W.; He, X.T.; Yi, F.; Li, B.M.; Zhang, X.H. Activation of beta2-adrenoceptor enhances synaptic potentiation and behavioral memory via cAMP-PKA signaling in the medial prefrontal cortex of rats. Learn. Mem. 2013, 20, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-Y.; Cai, W.; Yu, J.; Liu, S.-S.; Zhuo, M.; Li, B.-M.; Zhang, X.-H. Surface expression of hippocampal NMDA GluN2B receptors regulated by fear conditioning determines its contribution to memory consolidation in adult rats. Sci. Rep. 2016, 6, 30743. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Graziane, N.M.; Wang, H.; Zhong, P.; Wang, Q.; Liu, W.; Hayashi-Takagi, A.; Korth, C.; Sawa, A.; Brandon, N.J.; et al. Regulation of N-methyl-D-aspartate receptors by disrupted-in-schizophrenia-1. Biol. Psychiatry 2014, 75, 414–424. [Google Scholar] [CrossRef]
- Lisman, J.E.; Fellous, J.M.; Wang, X.J. A role for NMDA-receptor channels in working memory. Nat. Neurosci. 1998, 1, 273–275. [Google Scholar] [CrossRef]
- Mo, J.; Zhang, H.; Yu, L.-P.; Sun, P.-H.; Jin, G.-Z.; Zhen, X. L-stepholidine reduced L-DOPA-induced dyskinesia in 6-OHDA-lesioned rat model of Parkinson’s disease. Neurobiol. Aging 2010, 31, 926–936. [Google Scholar] [CrossRef]
- Hicks, C.; Huang, P.; Ramos, L.; Nayak, S.U.; Caro, Y.; Reitz, A.B.; Smith, G.R.; Lee, D.Y.-W.; Rawls, S.M.; Liu-Chen, L.-Y. Dopamine D1-Like Receptor Agonist and D2-Like Receptor Antagonist (-)-Stepholidine Reduces Reinstatement of Drug-Seeking Behavior for 3,4-Methylenedioxypyrovalerone (MDPV) in Rats. ACS Chem. Neurosci. 2018, 9, 1327–1337. [Google Scholar] [CrossRef]
- Ma, B.; Yue, K.; Chen, L.; Tian, X.; Ru, Q.; Gan, Y.; Wang, D.; Jin, G.; Li, C. L-stepholidine, a natural dopamine receptor D1 agonist and D2 antagonist, inhibits heroin-induced reinstatement. Neurosci. Lett. 2014, 559, 67–71. [Google Scholar] [CrossRef]
- Fedulov, V.; Rex, C.S.; Simmons, D.A.; Palmer, L.; Gall, C.M.; Lynch, G. Evidence that long-term potentiation occurs within individual hippocampal synapses during learning. J. Neurosci. 2007, 27, 8031–8039. [Google Scholar] [CrossRef]
- Hlinak, Z.; Krejci, I. Spontaneous alternation behaviour in rats: Kynurenic acid attenuated deficits induced by MK-801. Behav. Brain Res. 2006, 168, 144–149. [Google Scholar] [CrossRef]
- Braun, U.; Harneit, A.; Pergola, G.; Menara, T.; Schäfer, A.; Betzel, R.F.; Zang, Z.; Schweiger, J.I.; Zhang, X.; Schwarz, K.; et al. Brain network dynamics during working memory are modulated by dopamine and diminished in schizophrenia. Nat. Commun. 2021, 12, 3478. [Google Scholar] [CrossRef] [PubMed]
- Ott, T.; Nieder, A. Dopamine D2 Receptors Enhance Population Dynamics in Primate Prefrontal Working Memory Circuits. Cereb. Cortex 2017, 27, 4423–4435. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Bies, R.R.; Stroup, S.T.; Keefe, R.S.E.; Rajji, T.K.; Suzuki, T.; Mamo, D.C.; Pollock, B.G.; Watanabe, K.; Mimura, M.; et al. Dopamine D2 receptor occupancy and cognition in schizophrenia: Analysis of the CATIE data. Schizophr. Bull. 2013, 39, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y.; Suhara, T.; Suzuki, K.; Kobayashi, K.; Inoue, O.; Terasaki, O.; Someya, Y.; Sassa, T.; Sudo, Y.; Matsushima, E.; et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 1997, 385, 634–636. [Google Scholar] [CrossRef]
- Abi-Dargham, A.; Mawlawi, O.; Lombardo, I.; Gil, R.; Martinez, D.; Huang, Y.; Hwang, D.-R.; Keilp, J.; Kochan, L.; Van Heertum, R.; et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J. Neurosci. 2002, 22, 3708–3719. [Google Scholar] [CrossRef]
- Vijayraghavan, S.; Wang, M.; Birnbaum, S.G.; Williams, G.V.; Arnsten, A.F.T. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat. Neurosci. 2007, 10, 376–384. [Google Scholar] [CrossRef]
- Reneaux, M.; Gupta, R. Prefronto-cortical dopamine D1 receptor sensitivity can critically influence working memory maintenance during delayed response tasks. PLoS ONE 2018, 13, e0198136. [Google Scholar] [CrossRef]
- Yang, Y.; Lee, S.-M.; Imamura, F.; Gowda, K.; Amin, S.; Mailman, R.B. D1 dopamine receptors intrinsic activity and functional selectivity affect working memory in prefrontal cortex. Mol. Psychiatry 2021, 26, 645–655. [Google Scholar] [CrossRef]
- Yang, Y.; Kocher, S.D.; Lewis, M.M.; Mailman, R.B. Dose-Dependent Regulation on Prefrontal Neuronal Working Memory by Dopamine D(1) Agonists: Evidence of Receptor Functional Selectivity-Related Mechanisms. Front. Neurosci. 2022, 16, 898051. [Google Scholar] [CrossRef]
- Cimino, J.X.; Zhou, M.; Waxmonsky, J.; Mailman, R.B.; Yang, Y. Characterization of behavioral changes in T-maze alternation from dopamine D(1) agonists with different receptor coupling mechanisms. Psychopharmacology 2023, 240, 2187–2199. [Google Scholar] [CrossRef]
- Eagle, D.M.; Wong, J.C.K.; Allan, M.E.; Mar, A.C.; Theobald, D.E.; Robbins, T.W. Contrasting roles for dopamine D1 and D2 receptor subtypes in the dorsomedial striatum but not the nucleus accumbens core during behavioral inhibition in the stop-signal task in rats. J. Neurosci. 2011, 31, 7349–7356. [Google Scholar] [CrossRef]
- Dong, Z.J.; Jin, G.Z.; Huang, H.Y. Augmentation of dopamine release by (-)-stepholidine from rabbit and rat caudate slices. Zhongguo Yao Li Xue Bao 1995, 16, 497–501. [Google Scholar]
- Gao, M.; Chu, H.Y.; Jin, G.Z.; Zhang, Z.J.; Wu, J.; Zhen, X.C. l-Stepholidine-induced excitation of dopamine neurons in rat ventral tegmental area is associated with its 5-HT(1A) receptor partial agonistic activity. Synapse 2011, 65, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, Y.; Wang, C.-J.; Gamo, N.J.; Jin, L.E.; Mazer, J.A.; Morrison, J.H.; Wang, X.-J.; Arnsten, A.F. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron 2013, 77, 736–749. [Google Scholar] [CrossRef]
- Yuen, E.Y.; Wei, J.; Liu, W.; Zhong, P.; Li, X.; Yan, Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron 2012, 73, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Bitanihirwe, B.; Lim, M.; Kelley, J.; Kaneko, T.; Woo, T. Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry 2009, 9, 71. [Google Scholar] [CrossRef]
- Tang, Y.-P.; Shimizu, E.; Dube, G.R.; Rampon, C.; Kerchner, G.A.; Zhuo, M.; Liu, G.; Tsien, J.Z. Genetic enhancement of learning and memory in mice. Nature 1999, 401, 63–69. [Google Scholar] [CrossRef]
- Runyan, J.D.; Dash, P.K. Intra-medial prefrontal administration of SCH-23390 attenuates ERK phosphorylation and long-term memory for trace fear conditioning in rats. Neurobiol. Learn. Mem. 2004, 82, 65–70. [Google Scholar] [CrossRef]
- Gilmartin, M.R.; Helmstetter, F.J. Trace and contextual fear conditioning require neural activity and NMDA receptor-dependent transmission in the medial prefrontal cortex. Learn. Mem. 2010, 17, 289–296. [Google Scholar] [CrossRef]
- Ho, C.-S.; Chen, H.-J.; Chiu, N.-C.; Shen, E.-Y.; Lue, H.-C. Short-term sulpiride treatment of children and adolescents with Tourette syndrome or chronic tic disorder. J. Formos. Med. Assoc. 2009, 108, 788–793. [Google Scholar] [CrossRef]
- Gallo, E.F.; Salling, M.C.; Feng, B.; A Morón, J.; Harrison, N.L.; Javitch, J.A.; Kellendonk, C. Upregulation of dopamine D2 receptors in the nucleus accumbens indirect pathway increases locomotion but does not reduce alcohol consumption. Neuropsychopharmacology 2015, 40, 1609–1618. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Zhu, X.; Liang, Y.; Zhang, Y.; Zheng, P.; Zhang, X. Levo-Stepholidine as a Potential Cognitive Enhancer: Insights into Executive Function and Memory Improvements. Biomedicines 2024, 12, 2680. https://doi.org/10.3390/biomedicines12122680
Hu Z, Zhu X, Liang Y, Zhang Y, Zheng P, Zhang X. Levo-Stepholidine as a Potential Cognitive Enhancer: Insights into Executive Function and Memory Improvements. Biomedicines. 2024; 12(12):2680. https://doi.org/10.3390/biomedicines12122680
Chicago/Turabian StyleHu, Zhengwei, Xueqing Zhu, Yirui Liang, Yuqiu Zhang, Ping Zheng, and Xuehan Zhang. 2024. "Levo-Stepholidine as a Potential Cognitive Enhancer: Insights into Executive Function and Memory Improvements" Biomedicines 12, no. 12: 2680. https://doi.org/10.3390/biomedicines12122680
APA StyleHu, Z., Zhu, X., Liang, Y., Zhang, Y., Zheng, P., & Zhang, X. (2024). Levo-Stepholidine as a Potential Cognitive Enhancer: Insights into Executive Function and Memory Improvements. Biomedicines, 12(12), 2680. https://doi.org/10.3390/biomedicines12122680