
Heme Oxygenase-1 Inhibition Modulates Autophagy and Augments Arsenic Trioxide Cytotoxicity in Pancreatic Cancer Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Western Blot
2.2. Flow Cytometry for Apoptosis
2.3. Non-Radioactive Cell Proliferation Assay (MTT)
2.4. Confocal Microscopy
2.5. ROS Detection
2.6. Lysosomal Staining
2.7. Transfection
2.8. Autophagy
2.9. Glutathione (GSH) Analysis
2.10. Statistical Analysis
3. Results
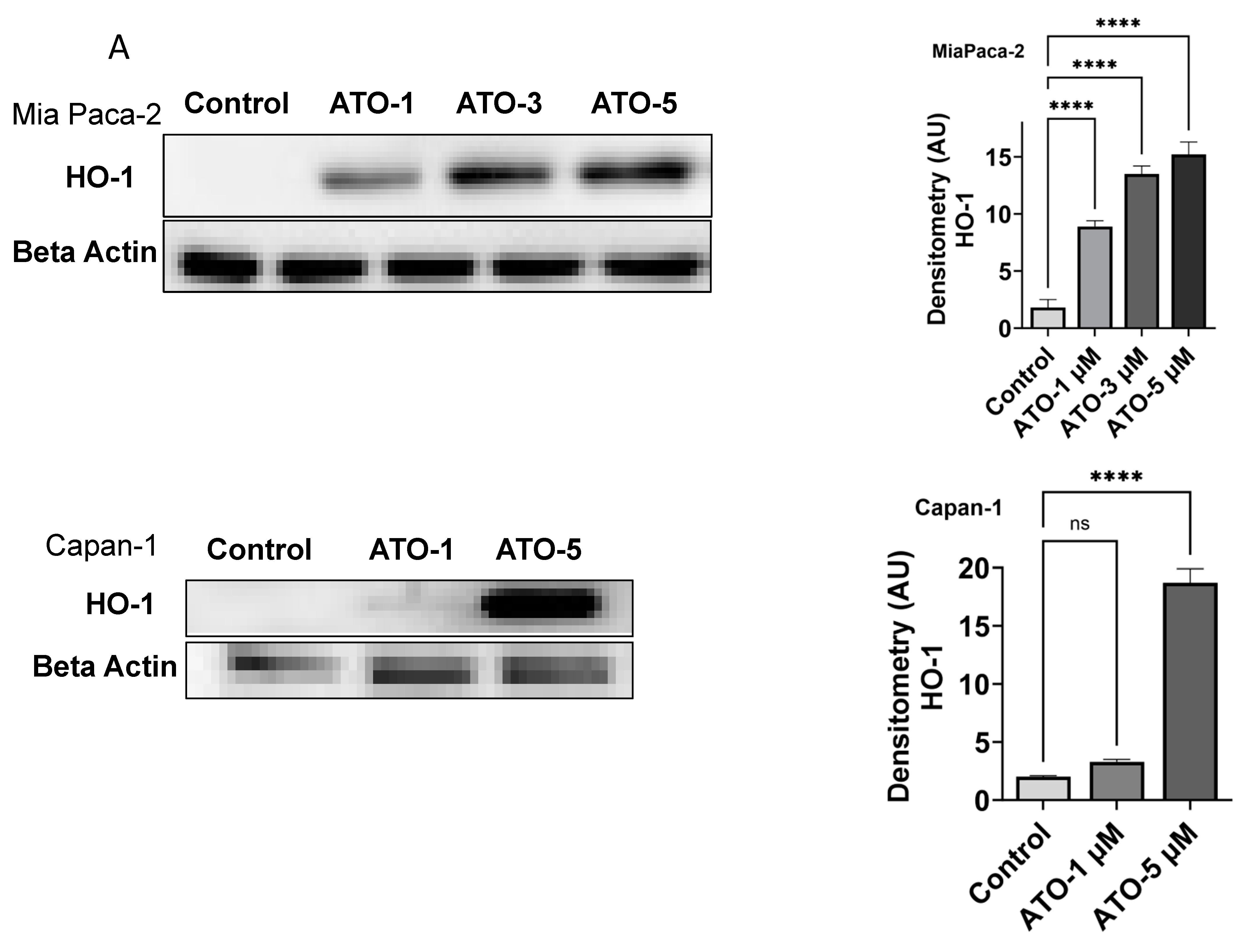
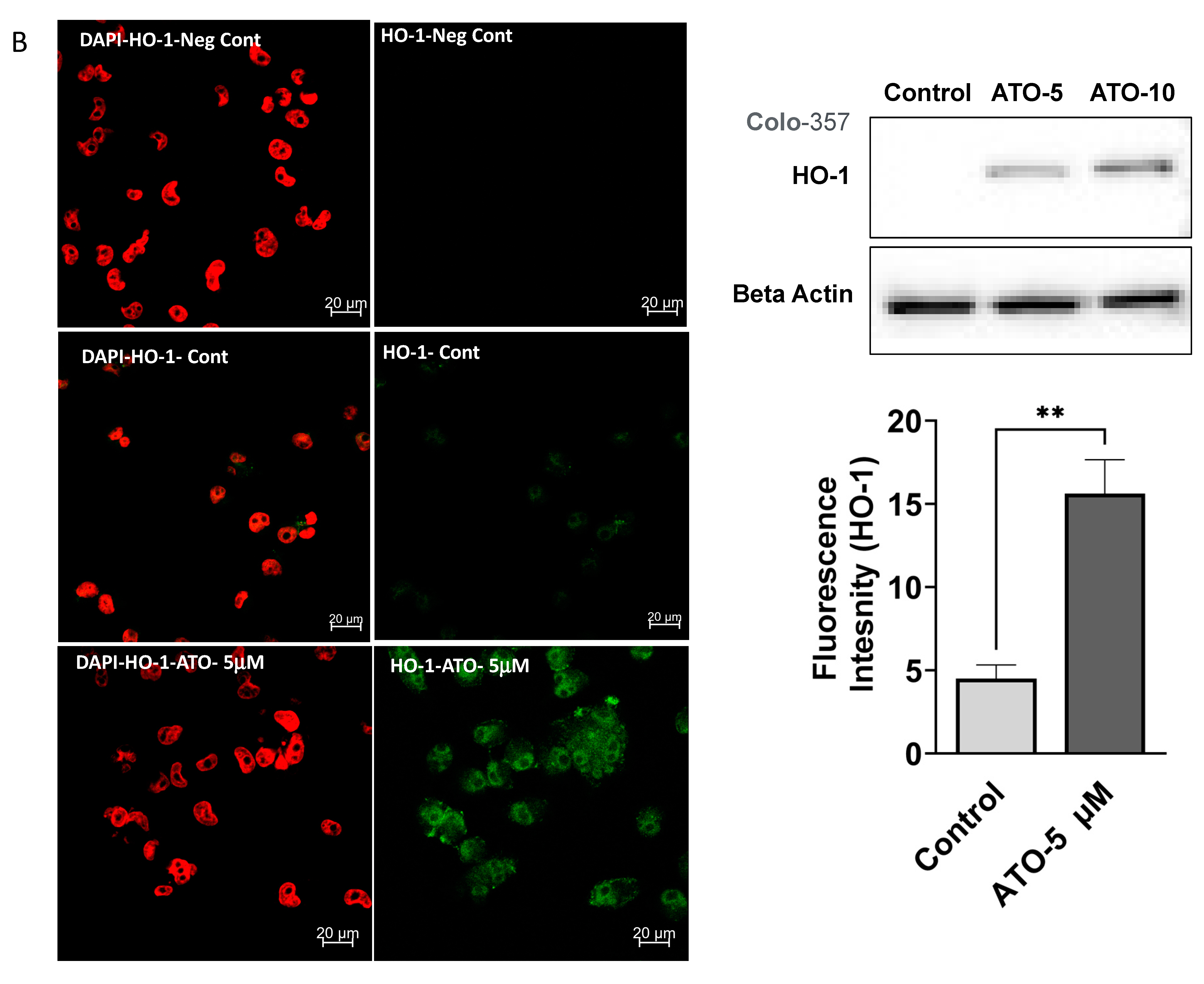
3.1. ATO Treatment Induces HO-1 Expression in PDAC Cells
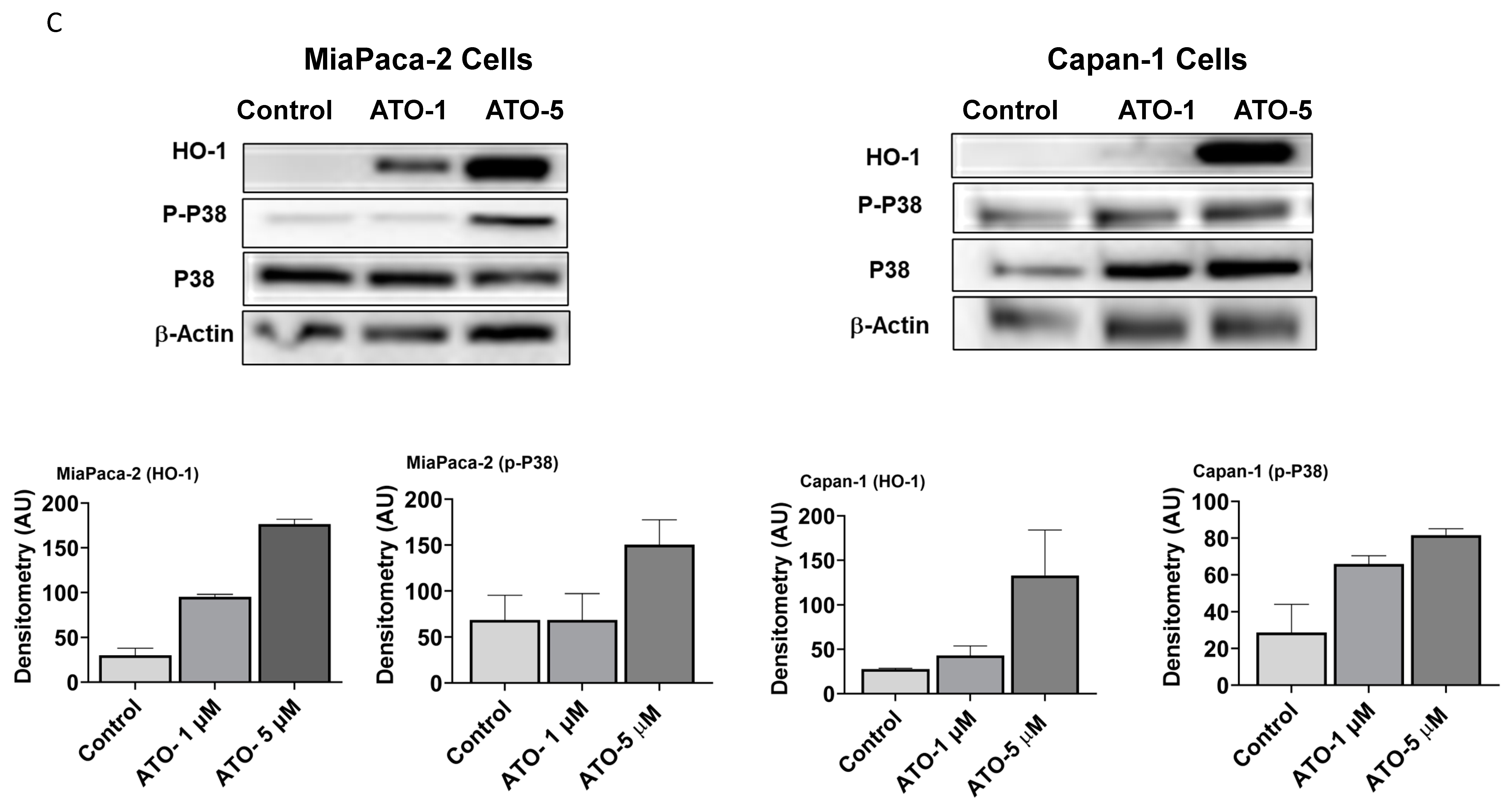
3.2. ATO Induces Phosphorylation of p38 MAPK Pathway in Human PDAC Cells
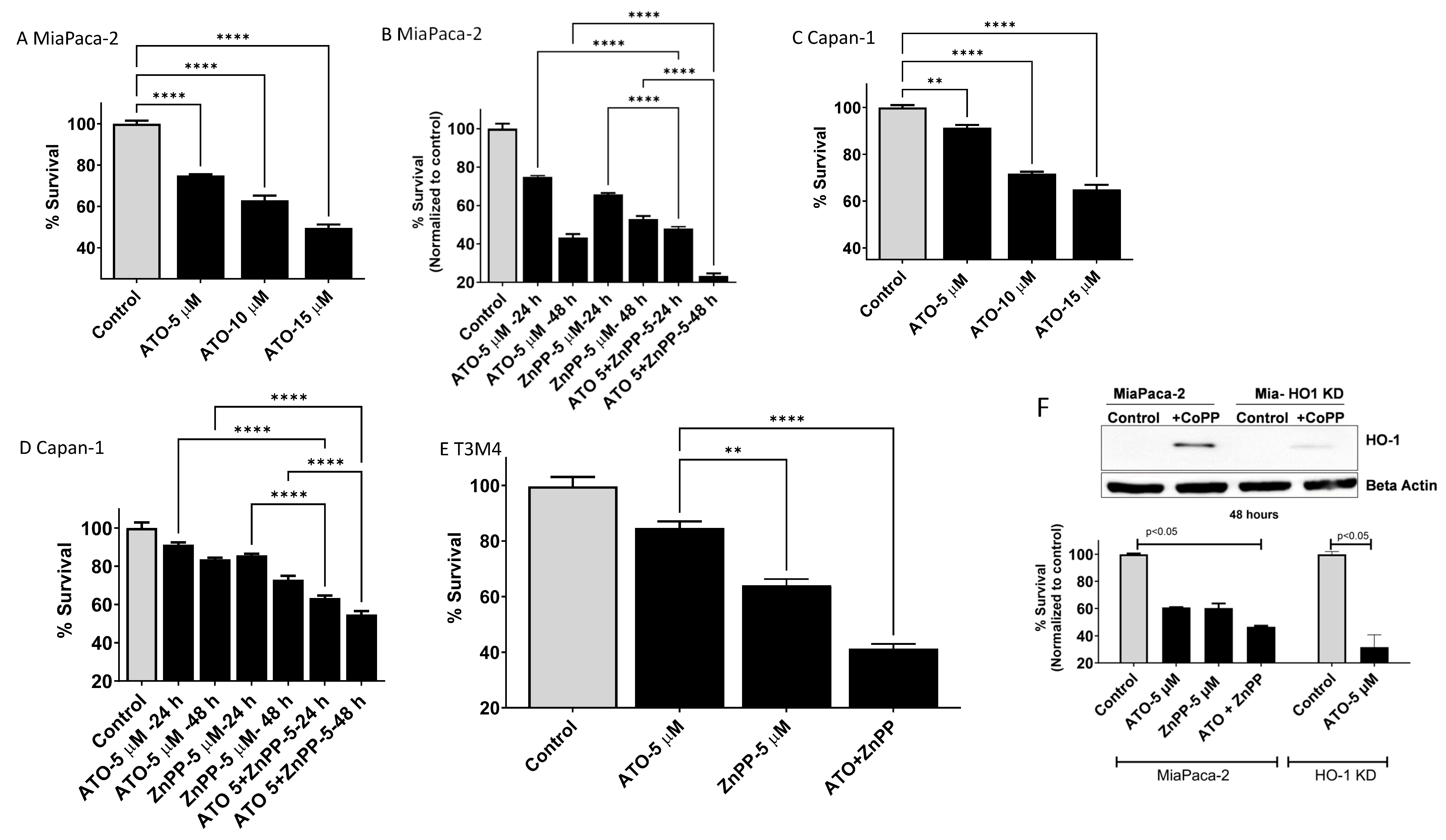
3.3. HO-1 Inhibition Enhanced ATO-Mediated Cell Killing in PDAC Cells
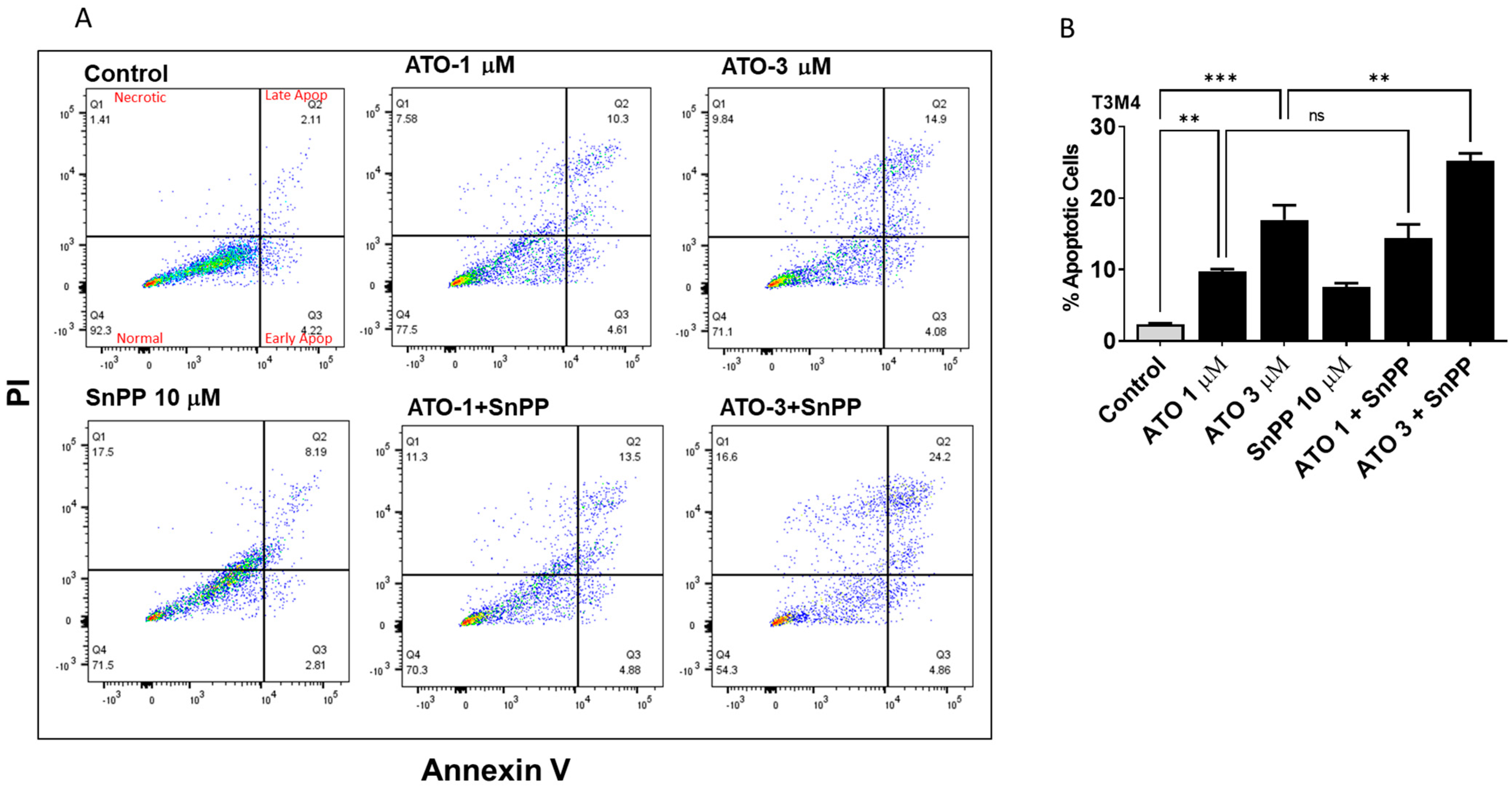
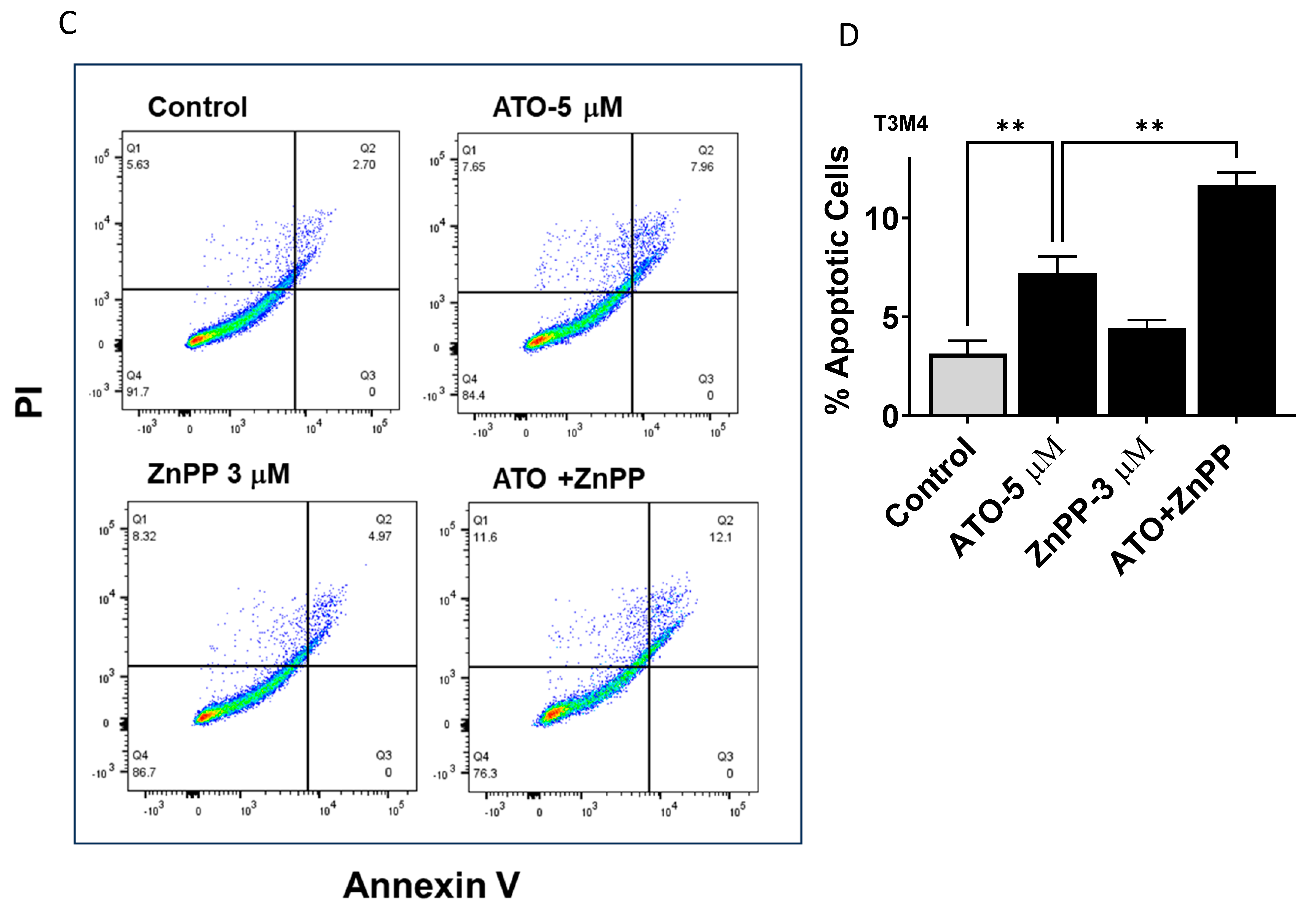
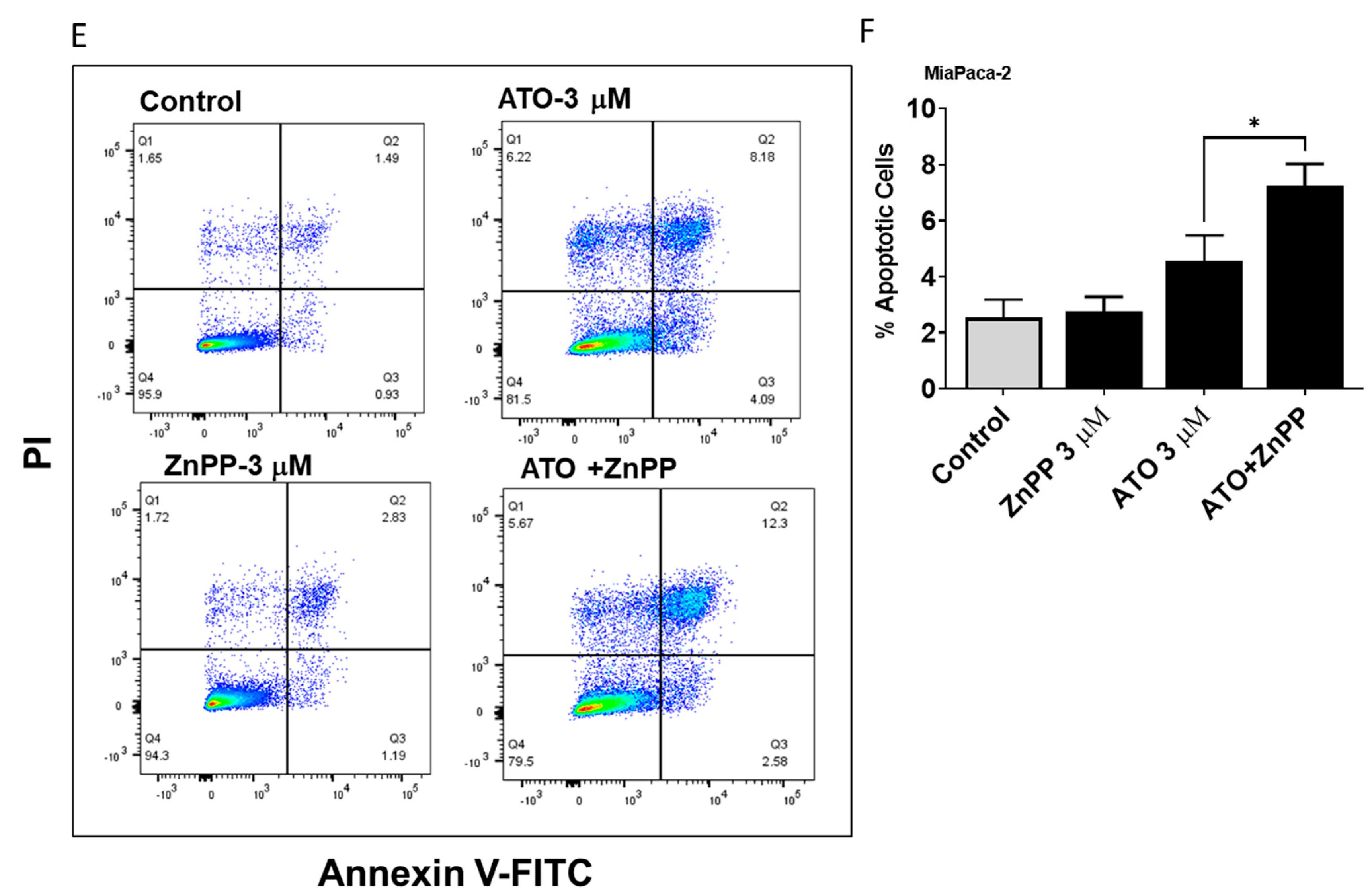
3.4. HO-1 Inhibition Enhanced ATO Induced Apoptosis in PDAC Cells
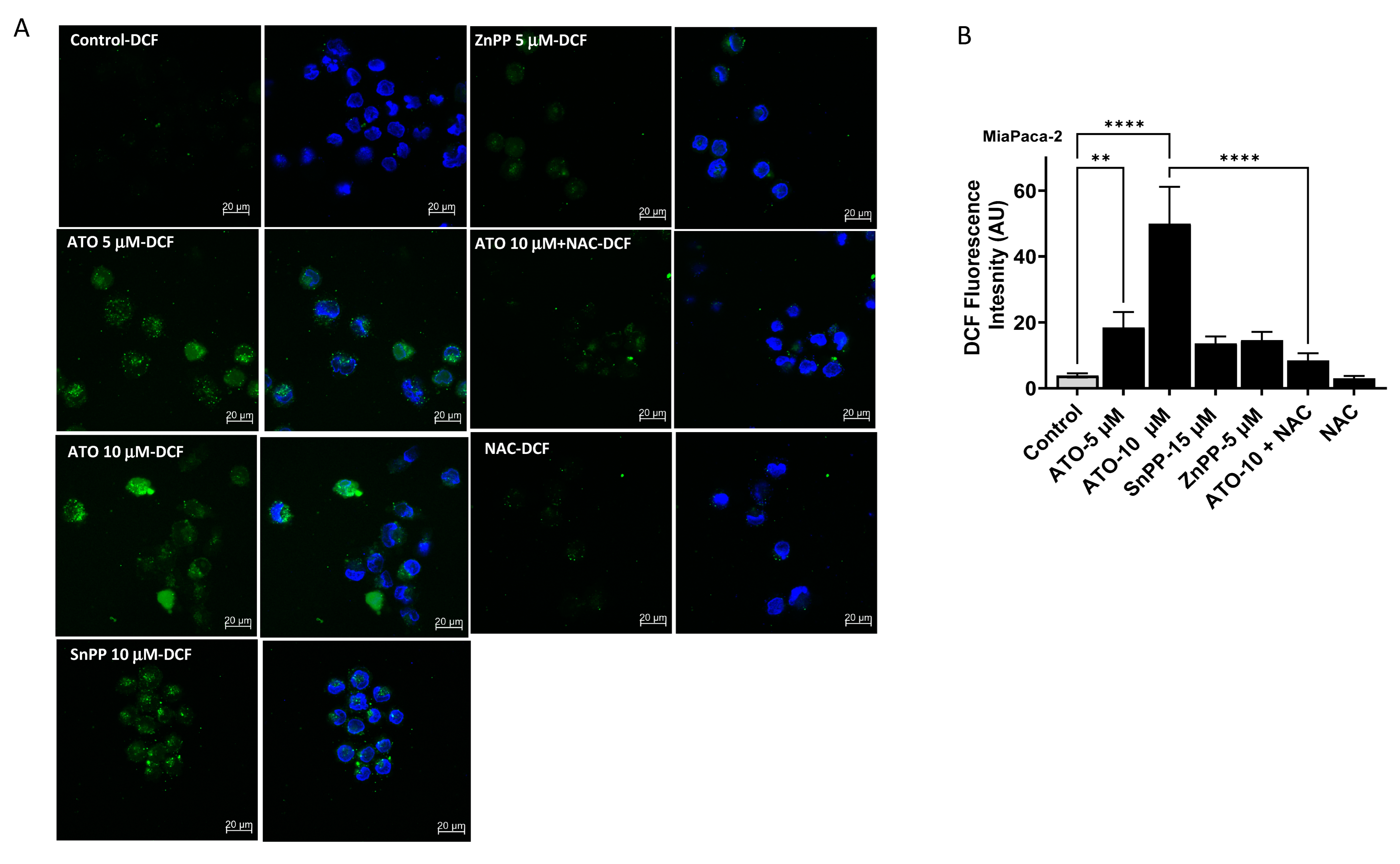
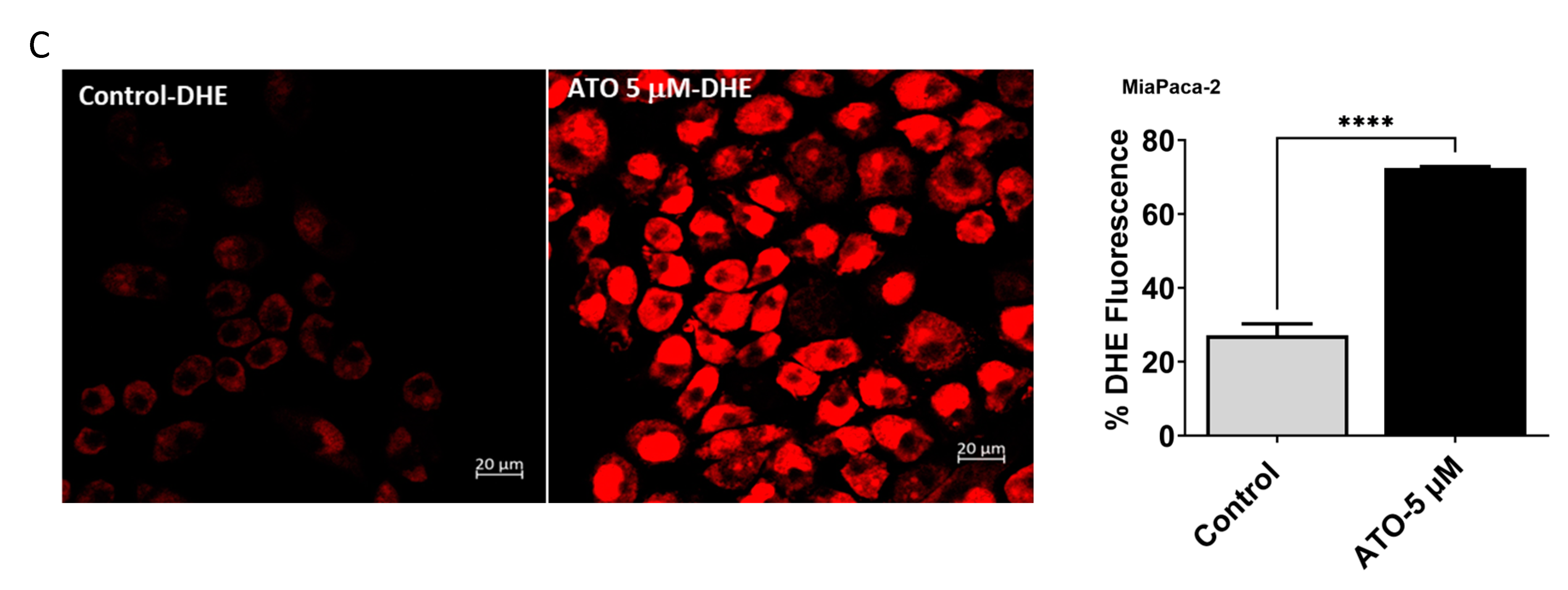
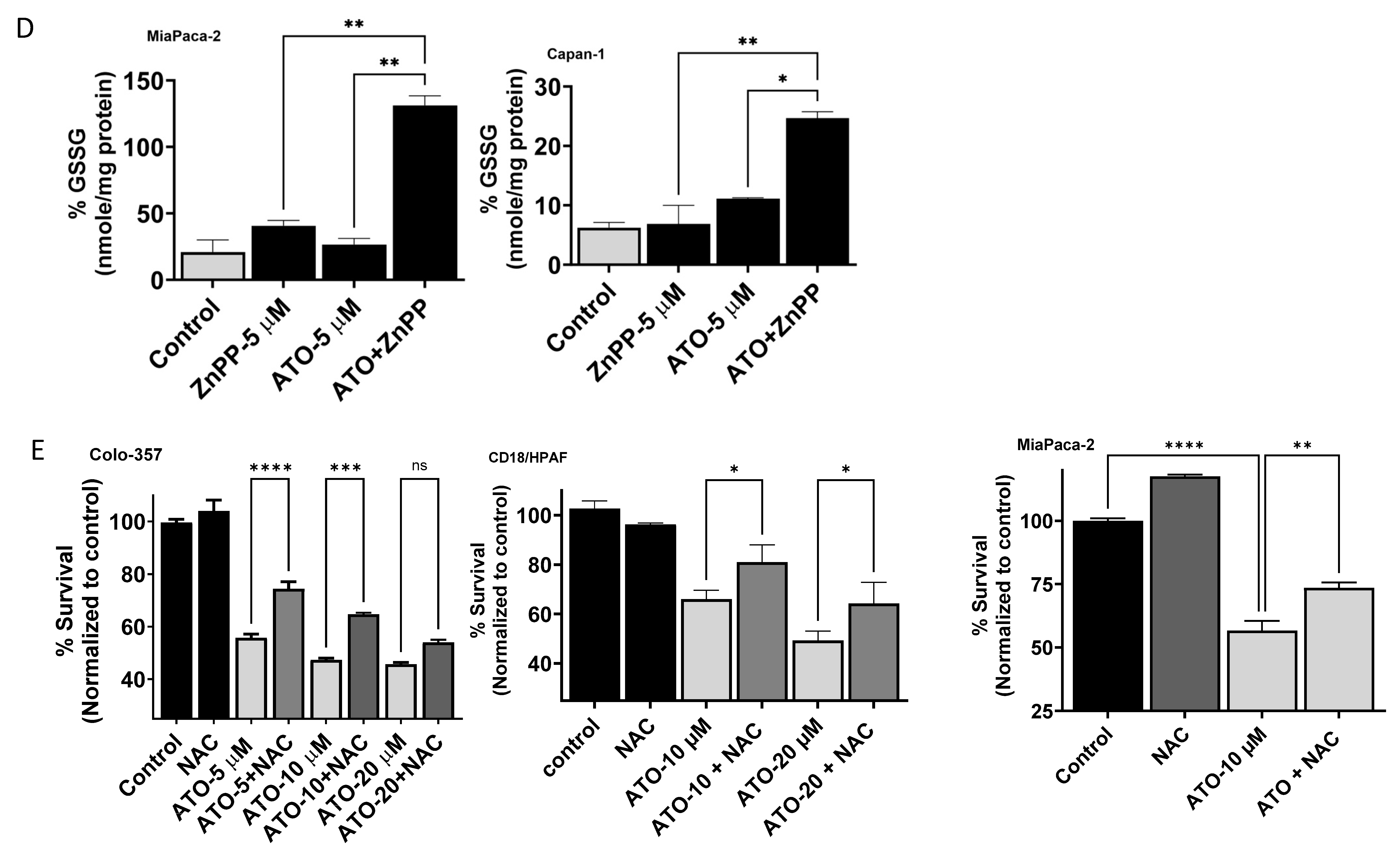
3.5. ATO/HO-1 Inhibition Combination Stimulates ROS Generation and Disrupts ROS Scavenging in PDAC Cells
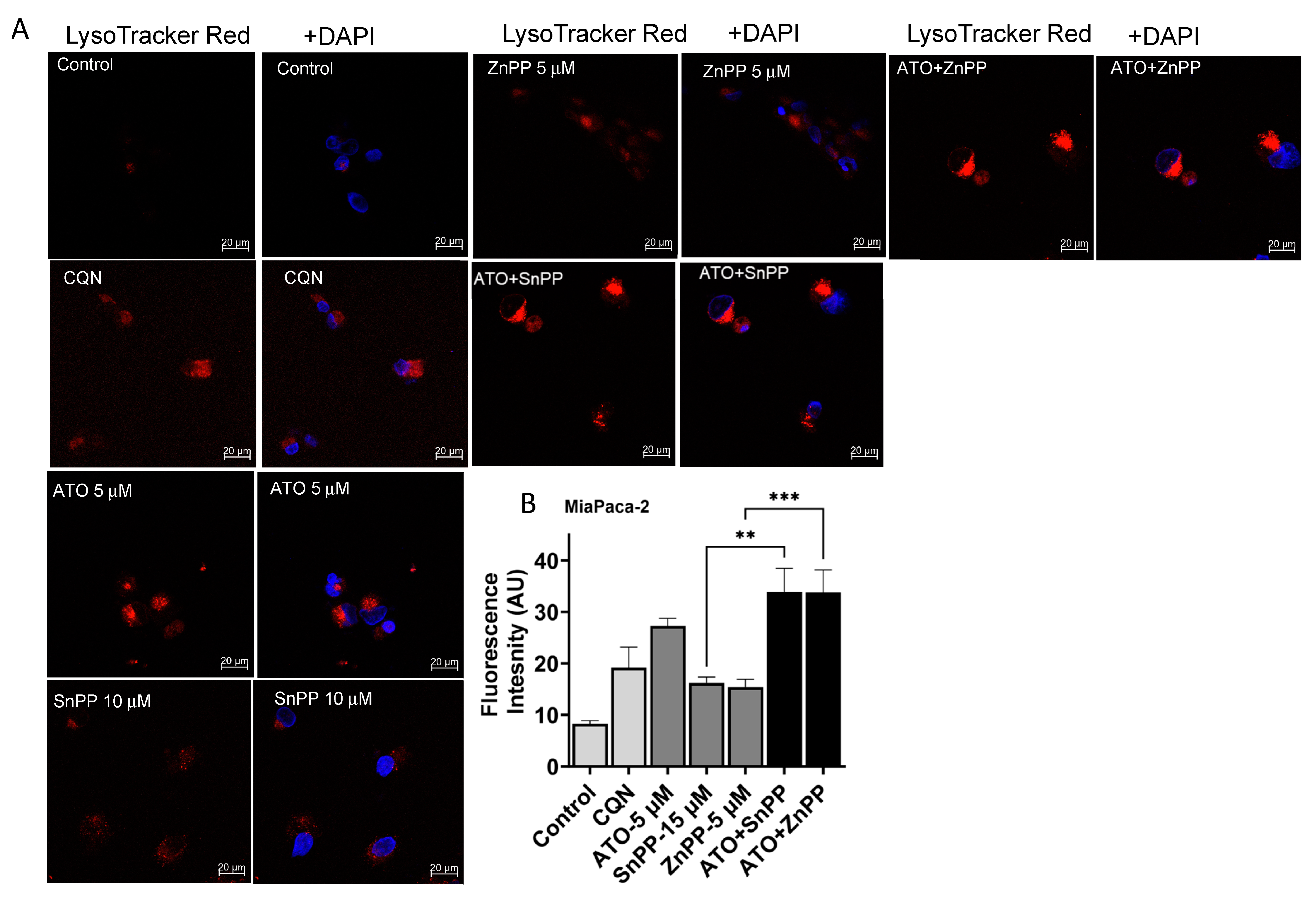
3.6. ATO and HO-1 Inhibitors Increased Lysosomal Staining in PDAC Cells
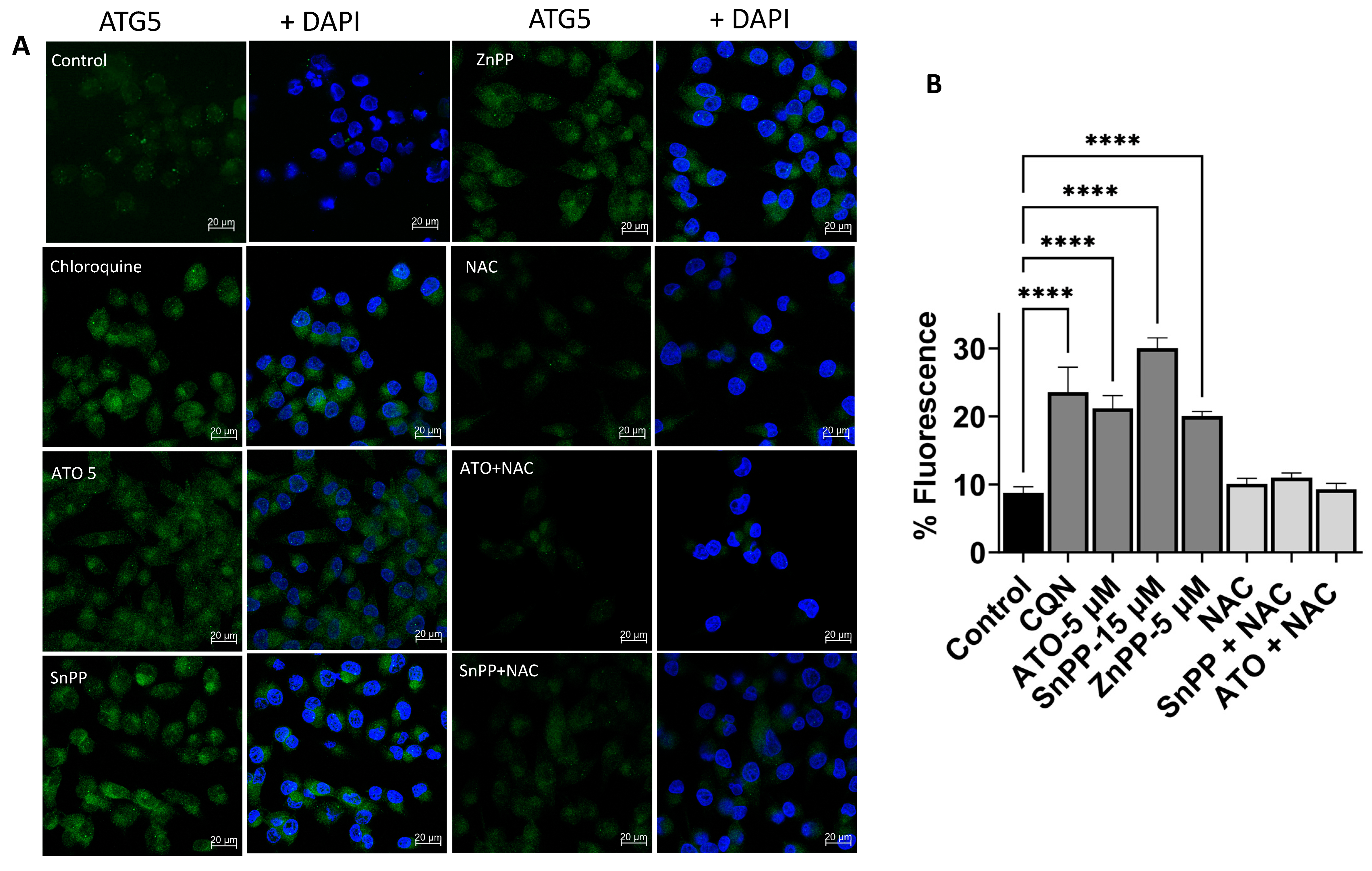
3.7. HO-1 Inhibition and ATO Regulate Autophagy through ROS-Mediated Mechanism
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Kong, D.; Sarkar, F.H. Pancreatic cancer: Understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Antman, K.H. Introduction: The history of arsenic trioxide in cancer therapy. Oncologist 2001, 6 (Suppl. S2), 1–2. [Google Scholar] [CrossRef] [PubMed]
- Vuky, J.; Yu, R.; Schwartz, L.; Motzer, R.J. Phase II trial of arsenic trioxide in patients with metastatic renal cell carcinoma. Investig. New Drugs 2002, 20, 327–330. [Google Scholar] [CrossRef]
- Bael, T.E.; Peterson, B.L.; Gollob, J.A. Phase II trial of arsenic trioxide and ascorbic acid with temozolomide in patients with metastatic melanoma with or without central nervous system metastases. Melanoma Res. 2008, 18, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.T.; Yao, Q.J.; Meng, Y.L.; Li, H.L.; Zhang, H.; Luo, J.P.; Guo, C.Y.; Geng, X. Arsenic trioxide intravenous infusion combined with transcatheter arterial chemoembolization for the treatment of hepatocellular carcinoma with pulmonary metastasis: Long-term outcome analysis. J. Gastroenterol. Hepatol. 2017, 32, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Aklilu, M.; Nattam, S.; Vokes, E.E. Arsenic trioxide in patients with adenocarcinoma of the pancreas refractory to gemcitabine: A phase II trial of the University of Chicago Phase II Consortium. Am. J. Clin. Oncol. 2008, 31, 553–556. [Google Scholar] [CrossRef]
- Li, X.; Ding, X.; Adrian, T.E. Arsenic trioxide inhibits proliferation and induces apoptosis in pancreatic cancer cells. Anticancer Res. 2002, 22, 2205–2213. [Google Scholar]
- Subbarayan, P.R.; Lima, M.; Ardalan, B. Arsenic trioxide/ascorbic acid therapy in patients with refractory metastatic colorectal carcinoma: A clinical experience. Acta Oncol. 2007, 46, 557–561. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme Oxygenase-1/Carbon Monoxide: From Basic Science to Therapeutic Applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Sunamura, M.; Duda, D.G.; Ghattas, M.H.; Lozonschi, L.; Motoi, F.; Yamauchi, J.; Matsuno, S.; Shibahara, S.; Abraham, N.G. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis 2003, 6, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Berberat, P.O.; Dambrauskas, Z.; Gulbinas, A.; Giese, T.; Giese, N.; Kunzli, B.; Autschbach, F.; Meuer, S.; Buchler, M.W.; Friess, H. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin. Cancer Res. 2005, 11, 3790–3798. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.M.; Dafferner, A.J.; O’connell, K.A.; Mehla, K.; Britigan, B.E.; Hollingsworth, M.A.; Abdalla, M.Y. Heme Oxygenase-1 Inhibition Potentiates the Effects of Nab-Paclitaxel-Gemcitabine and Modulates the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 2264. [Google Scholar] [CrossRef]
- Abdalla, M.Y.; Ahmad, I.M.; Rachagani, S.; Banerjee, K.; Thompson, C.M.; Maurer, H.C.; Olive, K.P.; Bailey, K.L.; Britigan, B.E.; Kumar, S. Enhancing responsiveness of pancreatic cancer cells to gemcitabine treatment under hypoxia by heme oxygenase-1 inhibition. Transl. Res. 2019, 207, 56–69. [Google Scholar] [CrossRef]
- Nuhn, P.; Kunzli, B.M.; Hennig, R.; Mitkus, T.; Ramanauskas, T.; Nobiling, R.; Meuer, S.C.; Friess, H.; Berberat, P.O. Heme oxygenase-1 and its metabolites affect pancreatic tumor growth in vivo. Mol. Cancer 2009, 8, 37. [Google Scholar] [CrossRef]
- Choi, B.M.; Pae, H.O.; Jeong, Y.R.; Oh, G.S.; Jun, C.D.; Kim, B.R.; Kim, Y.M.; Chung, H.T. Overexpression of heme oxygenase (HO)-1 renders Jurkat T cells resistant to fas-mediated apoptosis: Involvement of iron released by HO-1. Free. Radic. Biol. Med. 2004, 36, 858–871. [Google Scholar] [CrossRef]
- Morse, D.; Lin, L.; Choi, A.M.; Ryter, S.W. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radic. Biol. Med. 2009, 47, 1–12. [Google Scholar] [CrossRef]
- Chen, N.; Wu, L.; Yuan, H.; Wang, J. ROS/Autophagy/Nrf2 Pathway Mediated Low-Dose Radiation Induced Radio-Resistance in Human Lung Adenocarcinoma A549 Cell. Int. J. Biol. Sci. 2015, 11, 833–844. [Google Scholar] [CrossRef]
- Qian, W.; Liu, J.; Jin, J.; Ni, W.; Xu, W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk. Res. 2007, 31, 329–339. [Google Scholar] [CrossRef]
- Prawan, A.; Kundu, J.K.; Surh, Y.J. Molecular basis of heme oxygenase-1 induction: Implications for chemoprevention and chemoprotection. Antioxid. Redox Signal. 2005, 7, 1688–1703. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, X.; Zhou, Y.; Chen, F.X.; Wang, H.; Li, K.; Fan, H.; Tang, X.; Jiang, G.; Zhang, J. Synergy between arsenic trioxide and JQ1 on autophagy in pancreatic cancer. Oncogene 2019, 38, 7249–7265. [Google Scholar] [CrossRef] [PubMed]
- Baysan, A.; Yel, L.; Gollapudi, S.; Su, H.; Gupta, S. Arsenic trioxide induces apoptosis via the mitochondrial pathway by upregulating the expression of Bax and Bim in human B cells. Int. J. Oncol. 2007, 30, 313–318. [Google Scholar] [PubMed]
- Partridge, M.A.; Huang, S.X.; Hernandez-Rosa, E.; Davidson, M.M.; Hei, T.K. Arsenic induced mitochondrial DNA damage and altered mitochondrial oxidative function: Implications for genotoxic mechanisms in mammalian cells. Cancer Res. 2007, 67, 5239–5247. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007, 6, 304–312. [Google Scholar] [CrossRef]
- Jing, Y.; Wang, L.; Xia, L.; Chen, G.Q.; Chen, Z.; Miller, W.H.; Waxman, S. Combined effect of all-trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia cells in vitro and in vivo. Blood 2001, 97, 264–269. [Google Scholar] [CrossRef]
- Han, J.B.; Sang, F.; Chang, J.J.; Hua, Y.Q.; Shi, W.D.; Tang, L.H.; Liu, L.M. Arsenic trioxide inhibits viability of pancreatic cancer stem cells in culture and in a xenograft model via binding to SHH-Gli. OncoTargets Ther. 2013, 6, 1129–1138. [Google Scholar] [CrossRef]
- Alaluf, E.; Vokaer, B.; Detavernier, A.; Azouz, A.; Splittgerber, M.; Carrette, A.; Boon, L.; Libert, F.; Soares, M.; Le Moine, A.; et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight 2020, 5, e133929. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Chu, W.; Li, C.; Qu, X.; Zhao, D.; Wang, X.; Yu, X.; Cai, F.; Liang, H.; Zhang, Y.; Zhao, X.; et al. Arsenic-induced interstitial myocardial fibrosis reveals a new insight into drug-induced long QT syndrome. Cardiovasc. Res. 2012, 96, 90–98. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, J.; Zhao, J.; Ma, N.; Kim, S.W.; Qiao, S.; Ma, X. Autophagy: The Last Defense against Cellular Nutritional Stress. Adv. Nutr. 2018, 9, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Chen, S.Y.; Li, T.J.; Lin, T.W.; Chen, C.C.; Yen, G.C. 4-Acetylantroquinonol B enhances cell death and inhibits autophagy by downregulating the PI3K/Akt/MDR1 pathway in gemcitabine-resistant pancreatic cancer cells. Oncol Lett. 2022, 23, 128. [Google Scholar] [CrossRef]
- Clark, C.A.; Gupta, H.B.; Curiel, T.J. Tumor cell-intrinsic CD274/PD-L1: A novel metabolic balancing act with clinical potential. Autophagy 2017, 13, 987–988. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Mainz, L.; Sarhan, M.; Roth, S.; Sauer, U.; Maurus, K.; Hartmann, E.M.; Seibert, H.D.; Rosenwald, A.; Diefenbacher, M.E.; Rosenfeldt, M.T. Autophagy Blockage Reduces the Incidence of Pancreatic Ductal Adenocarcinoma in the Context of Mutant Trp53. Front. Cell Dev. Biol. 2022, 10, 785252. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Hashimoto, D.; Blauer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur. J. Cancer 2014, 50, 1382–1390. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef]
- Kardeh, S.; Ashkani-Esfahani, S.; Alizadeh, A.M. Paradoxical action of reactive oxygen species in creation and therapy of cancer. Eur. J. Pharmacol. 2014, 735, 150–168. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.T.; Pu, Z.J.; Liu, L.X.; Li, G.P.; Feng, J.L.; Zhu, H.C.; Wu, L.F. Inhibition of autophagy enhances adenosineinduced apoptosis in human hepatoblastoma HepG2 cells. Oncol. Rep. 2019, 41, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Li, X.; Fan, Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1alpha and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010, 70, 5942–5952. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hu, C.; Cao, H.; Wu, X.; Wang, R.; Lu, H.; Li, H.; Chen, H. MicroRNA-29c Increases the Chemosensitivity of Pancreatic Cancer Cells by Inhibiting USP22 Mediated Autophagy. Cell. Physiol. Biochem. 2018, 47, 747–758. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, I.M.; Dafferner, A.J.; Salloom, R.J.; Abdalla, M.Y. Heme Oxygenase-1 Inhibition Modulates Autophagy and Augments Arsenic Trioxide Cytotoxicity in Pancreatic Cancer Cells. Biomedicines 2023, 11, 2580. https://doi.org/10.3390/biomedicines11092580
Ahmad IM, Dafferner AJ, Salloom RJ, Abdalla MY. Heme Oxygenase-1 Inhibition Modulates Autophagy and Augments Arsenic Trioxide Cytotoxicity in Pancreatic Cancer Cells. Biomedicines. 2023; 11(9):2580. https://doi.org/10.3390/biomedicines11092580
Chicago/Turabian StyleAhmad, Iman M., Alicia J. Dafferner, Ramia J. Salloom, and Maher Y. Abdalla. 2023. "Heme Oxygenase-1 Inhibition Modulates Autophagy and Augments Arsenic Trioxide Cytotoxicity in Pancreatic Cancer Cells" Biomedicines 11, no. 9: 2580. https://doi.org/10.3390/biomedicines11092580
APA StyleAhmad, I. M., Dafferner, A. J., Salloom, R. J., & Abdalla, M. Y. (2023). Heme Oxygenase-1 Inhibition Modulates Autophagy and Augments Arsenic Trioxide Cytotoxicity in Pancreatic Cancer Cells. Biomedicines, 11(9), 2580. https://doi.org/10.3390/biomedicines11092580