
Systematic Exploration of Functional Group Relevance for Anti-Leishmanial Activity of Anisomycin

 ,
,  and
and 
Abstract
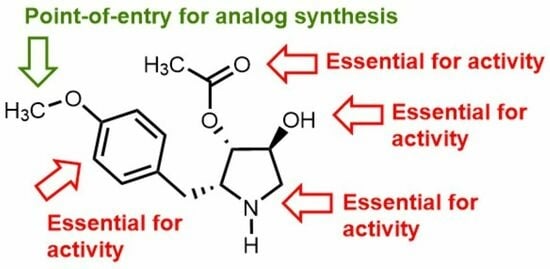
:
1. Introduction
2. Materials and Methods
2.1. In Vitro Inhibition of Translation Assays
2.2. Leishmania Cultures and Anti-Leishmanial Activity Determination
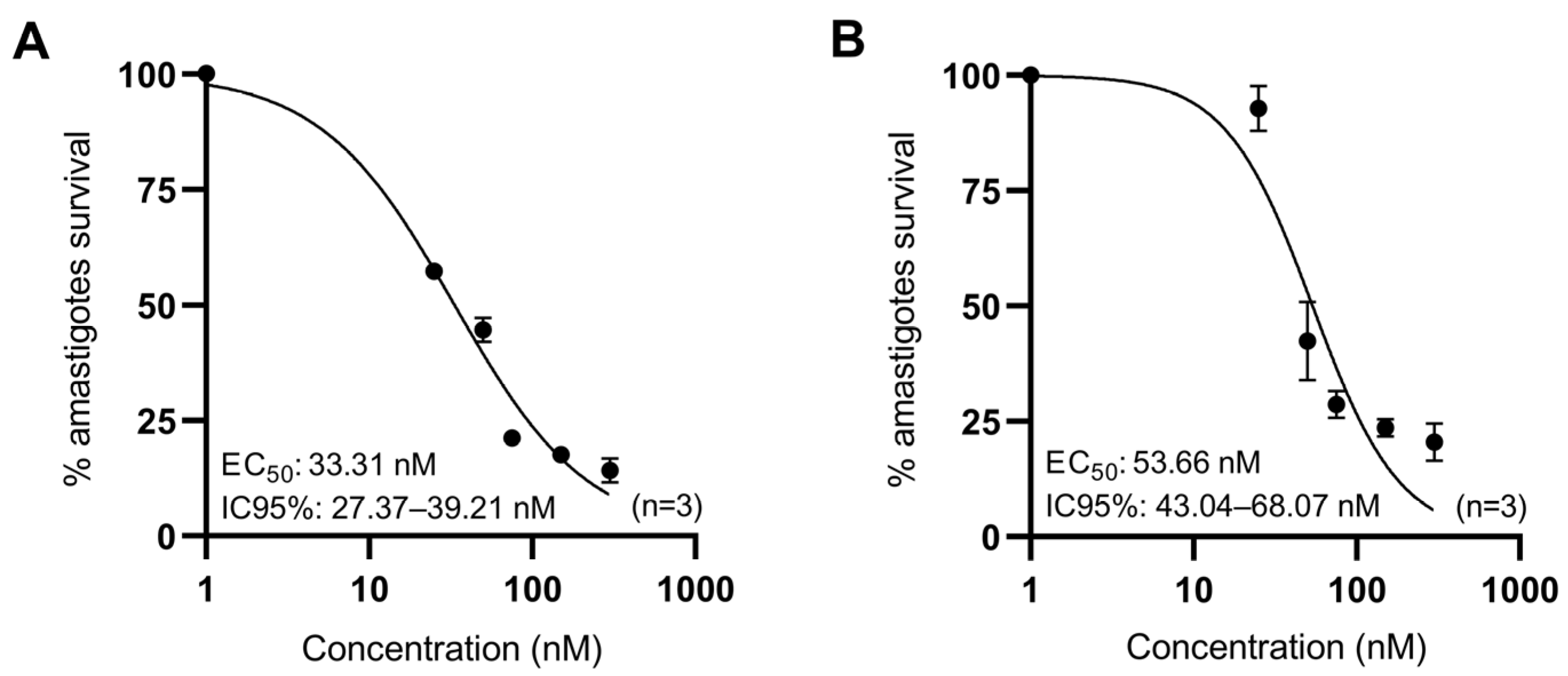
2.3. Drug Susceptibility Assays in Infected BMDM
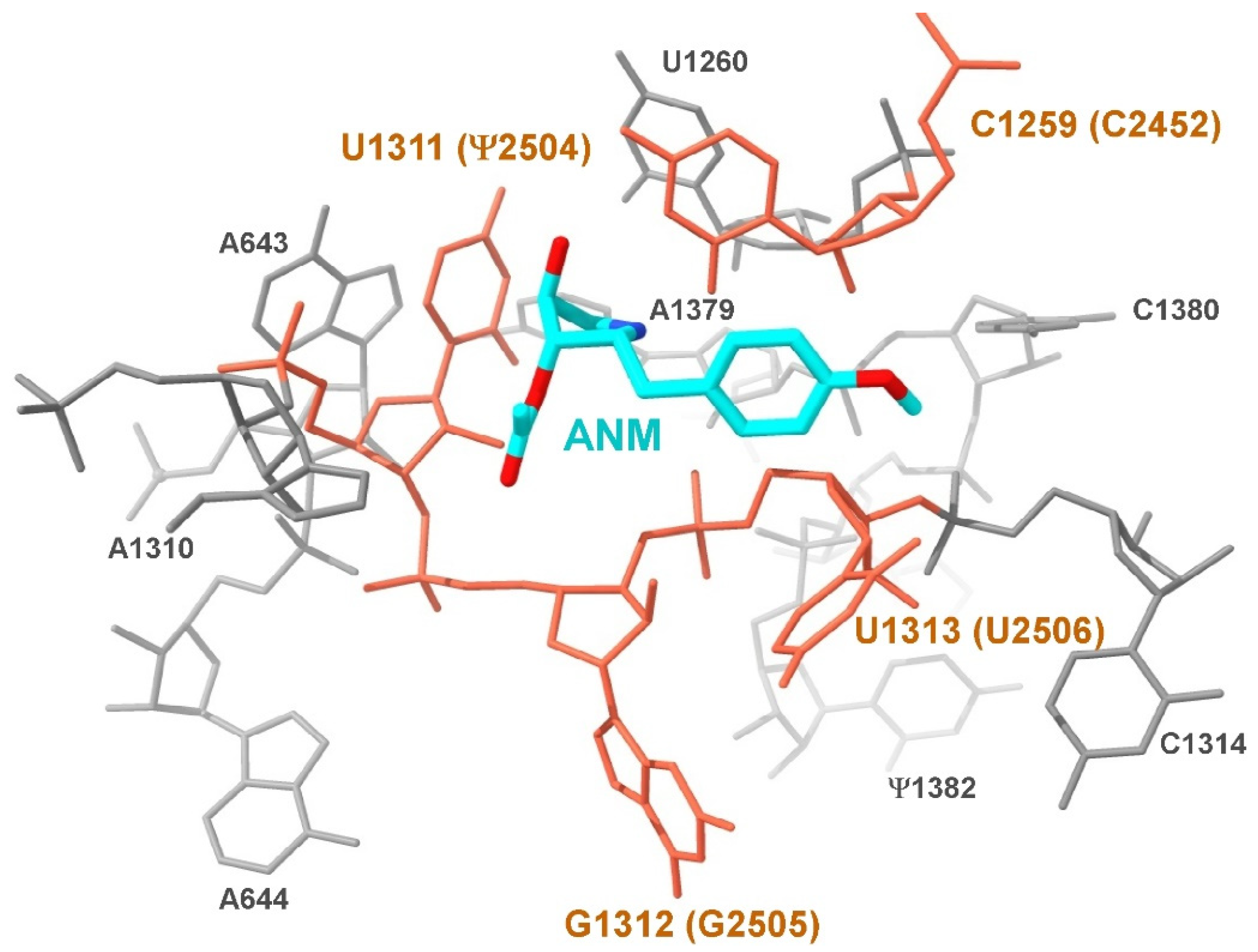
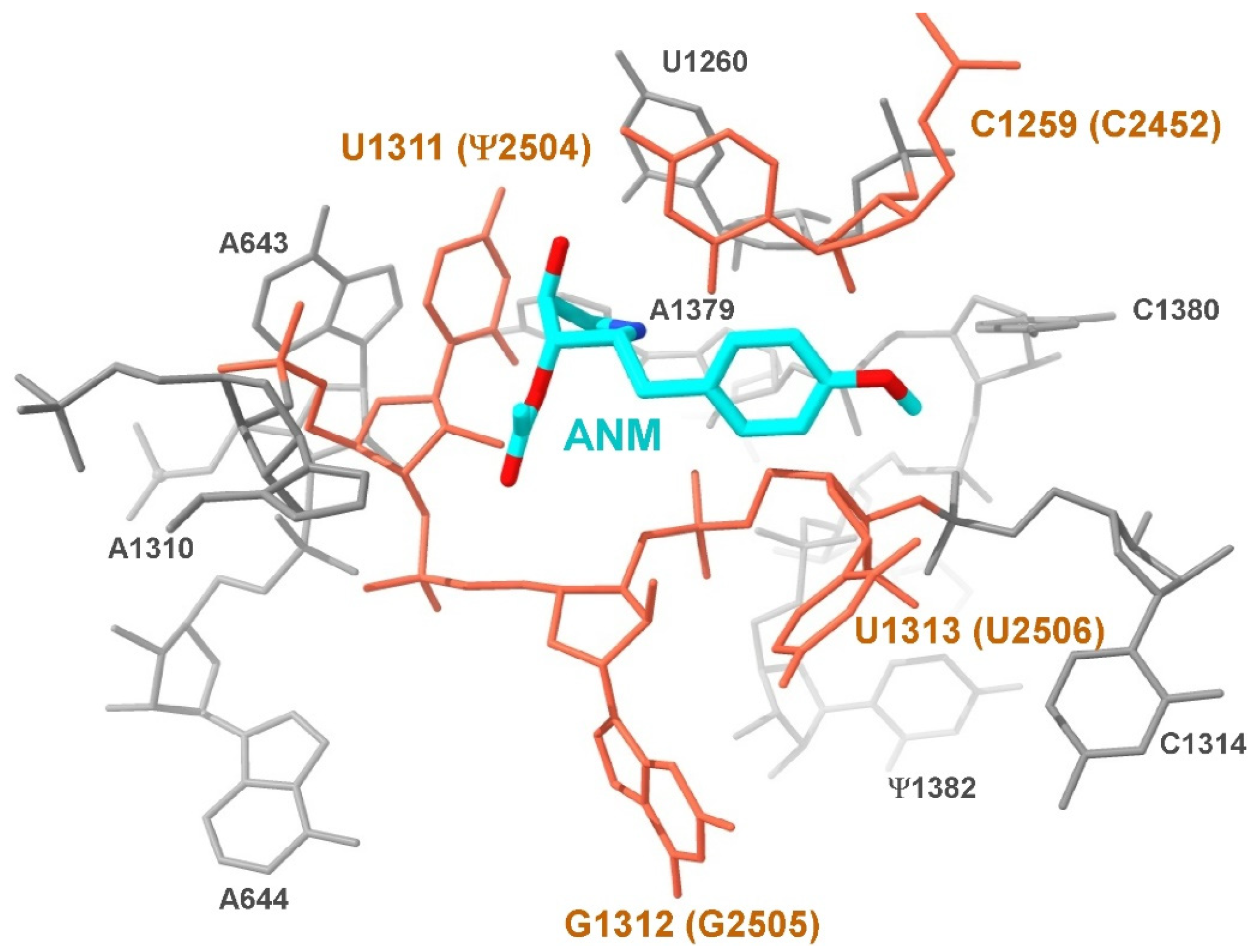
2.4. Superimposition of Ribosome Structures
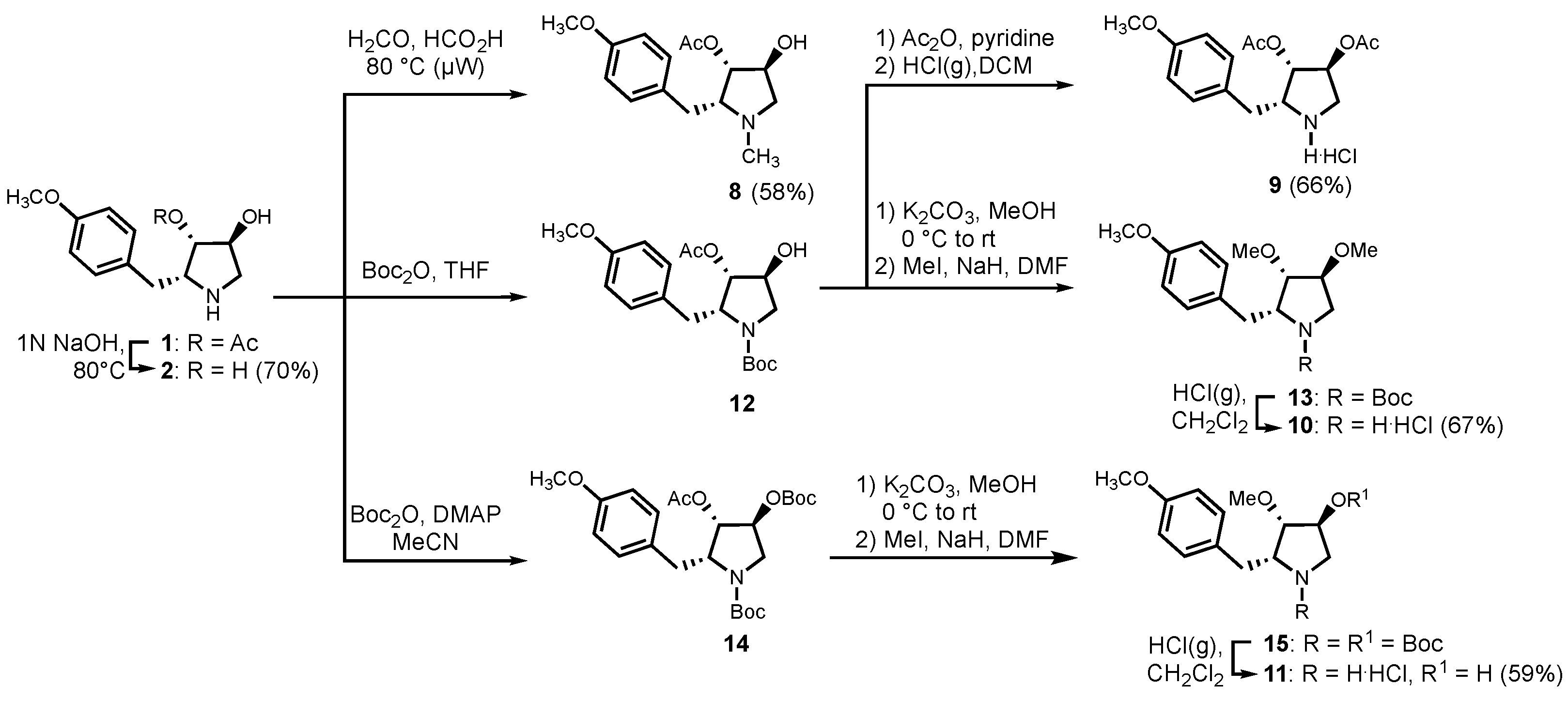
2.5. Chemistry
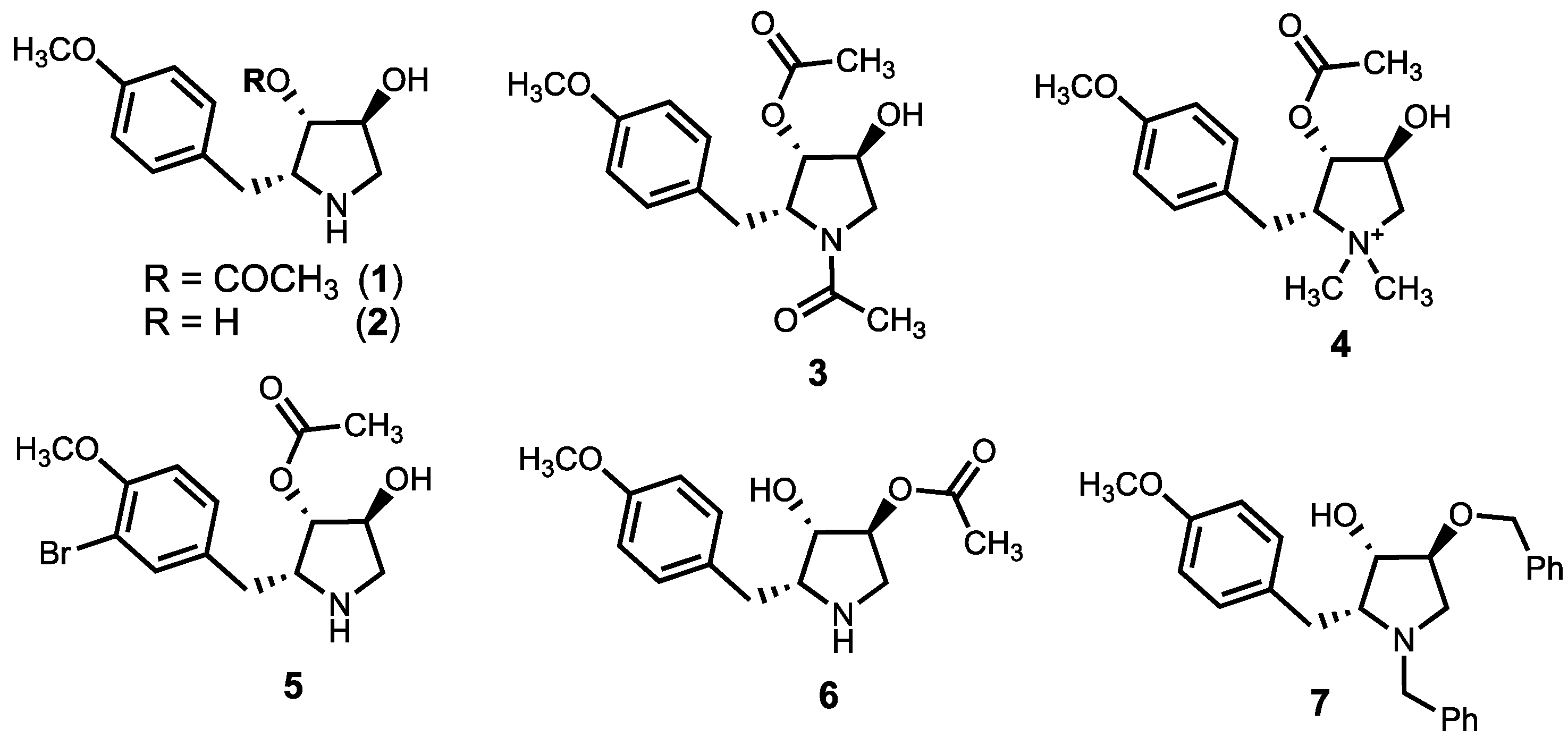
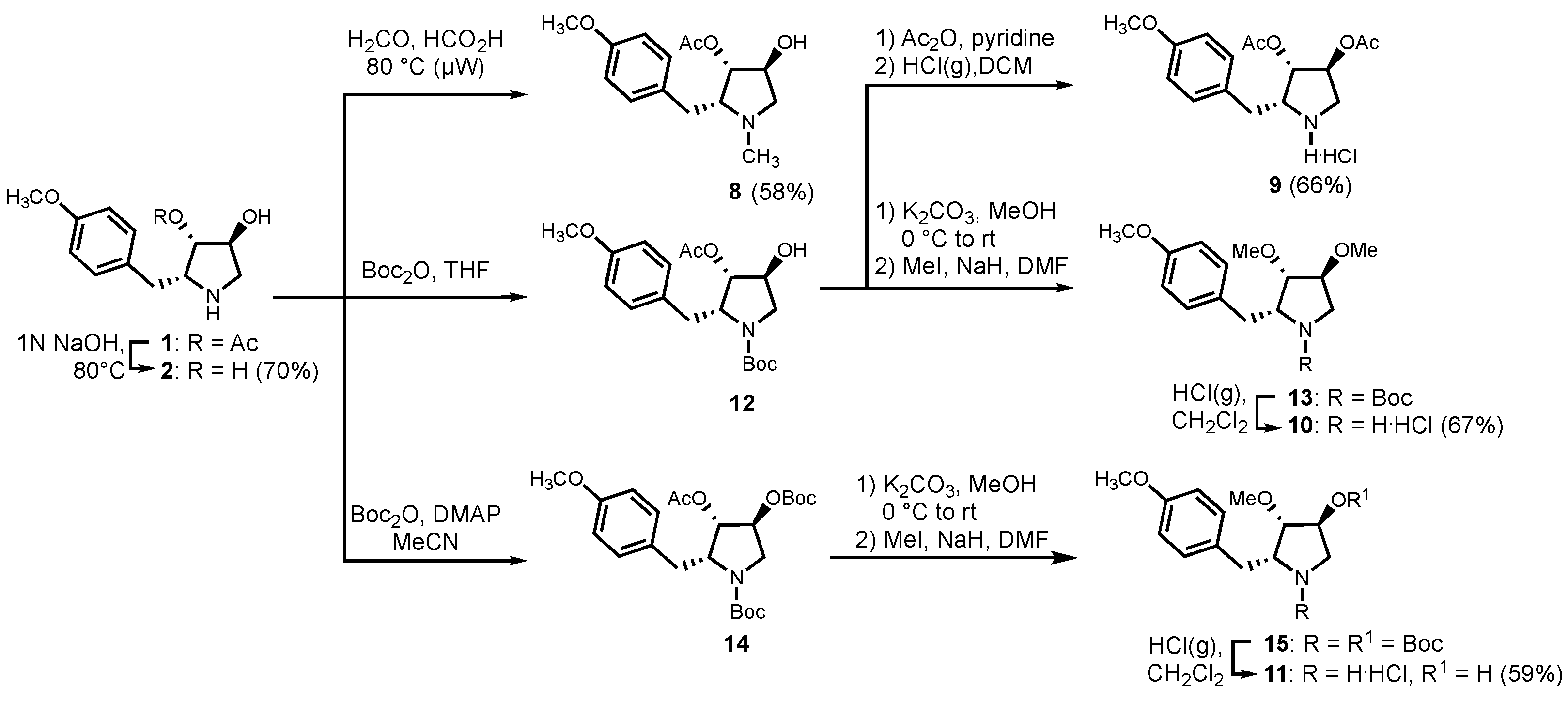
2.5.1. (2R,3S,4S)-3,4-dihydroxy-2-(4-methoxybenzyl)-pyrrolidine (2)
2.5.2. (2R,3S,4S)-1-methyl-4-hydroxy-2-(4-methoxybenzyl)-pyrrolidin-3-yl acetate (8)
2.5.3. (2R,3S,4S)-3,4-diacetoxy-2-(4-methoxybenzyl)-pyrrolidine hydrochloride (9)
2.5.4. (2R,3S,4S)-3,4-dimethoxy-2-(4-methoxybenzyl)-pyrrolidine hydrochloride (10)
2.5.5. (2R,3S,4S)-3-methoxy-4-hydroxy-2-(4-methoxybenzyl)-pyrrolidine hydrochloride (11)
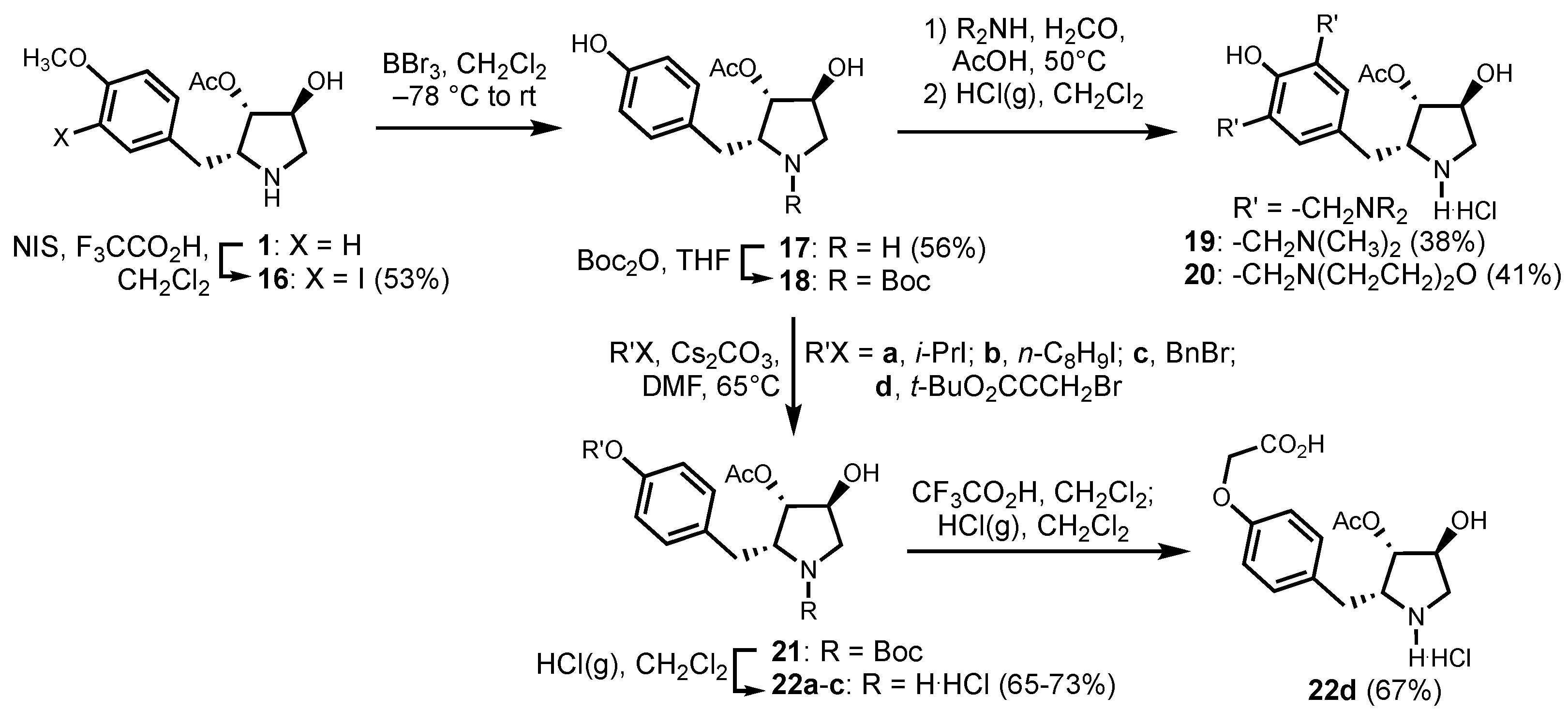
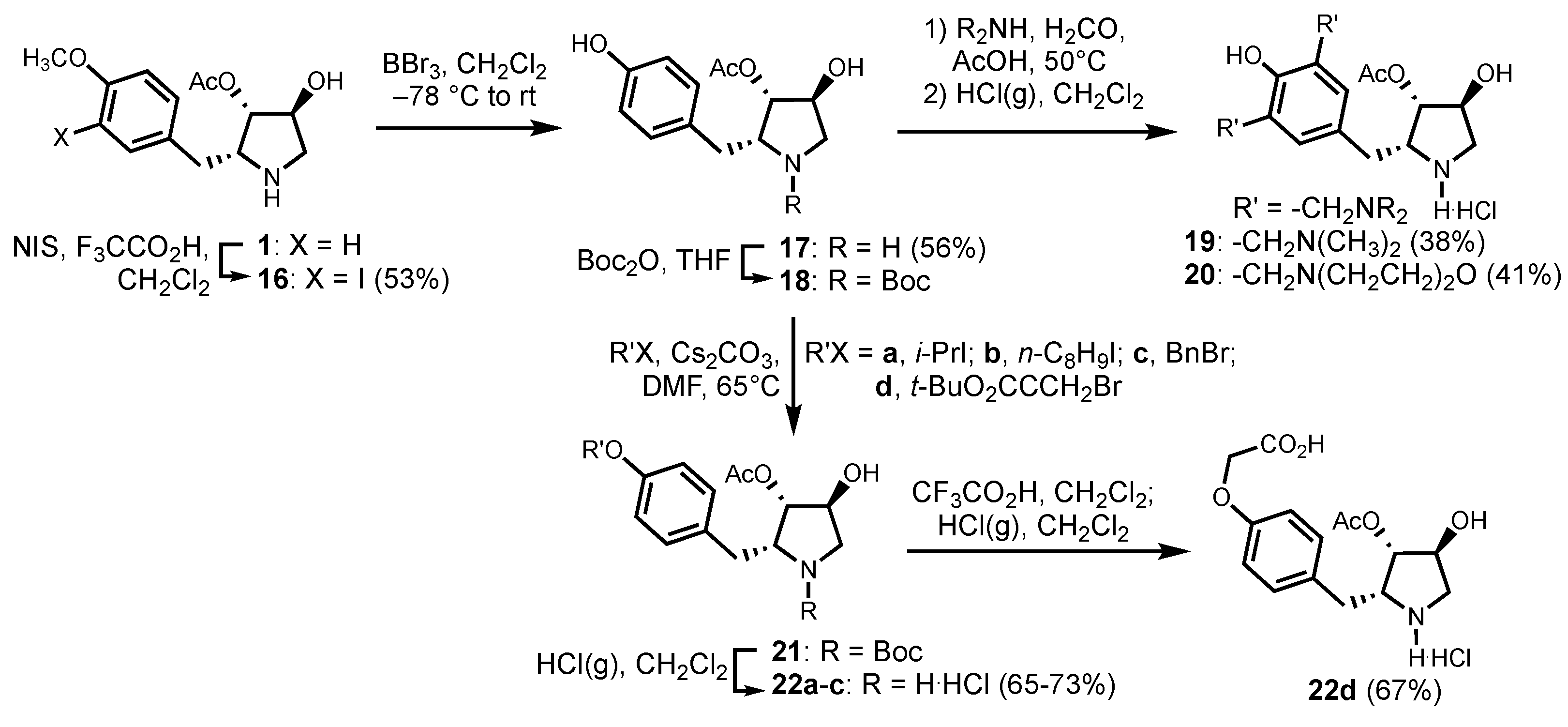
2.5.6. (2R,3S,4S)-4-hydroxy-2-(3-iodo-4-methoxybenzyl)-pyrrolidin-3-yl acetate (16)
2.5.7. (2R,3S,4S)-4-hydroxy-2-(4-hydroxybenzyl)-pyrrolidin-3-yl acetate (17)
2.5.8. (2R,3S,4S)-4-hydroxy-2-[3,5-di-(dimethylaminomethyl)-4-hydroxybenzyl]-pyrrolidin-3-yl acetate (19)
2.5.9. (2R,3S,4S)-4-hydroxy-2-[3,5-di-(morpholinomethyl)-4-hydroxybenzyl]-pyrrolidin-3-yl acetate (20)
2.5.10. (2R,3S,4S)-4-hydroxy-2-(4-iso-propoxybenzyl)-pyrrolidin-3-yl acetate (22a)
2.5.11. (2R,3S,4S)-4-hydroxy-2-(4-octyloxybenzyl)-pyrrolidin-3-yl acetate (22b)
2.5.12. (2R,3S,4S)-4-hydroxy-2-(4-benzyloxybenzyl)-pyrrolidin-3-yl acetate (22c)
2.5.13. (2R,3S,4S)-4-hydroxy-2-(4-carboxymethyloxybenzyl)-pyrrolidin-3-yl acetate (22d)
3. Results
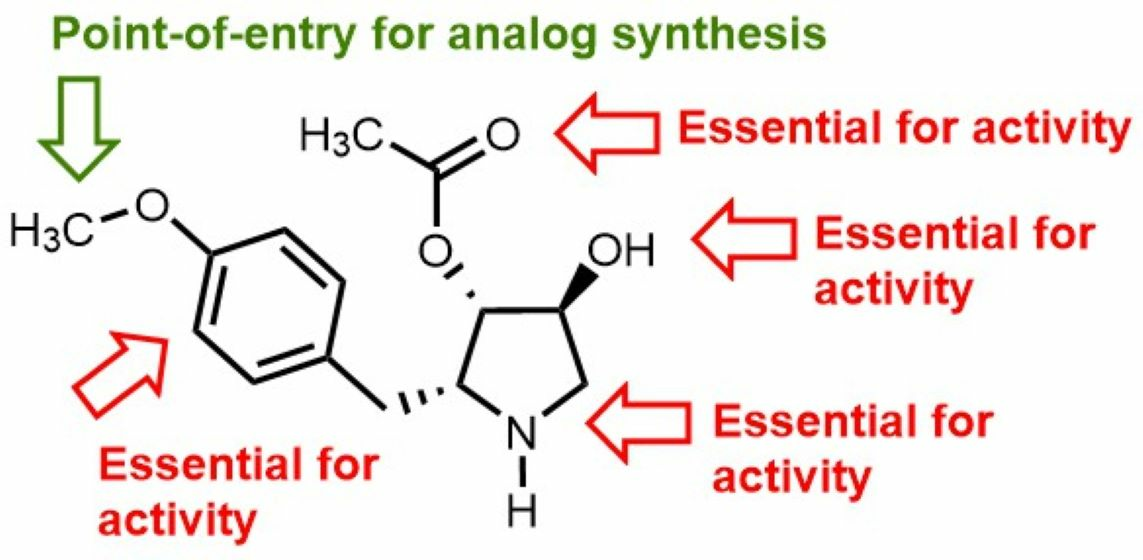
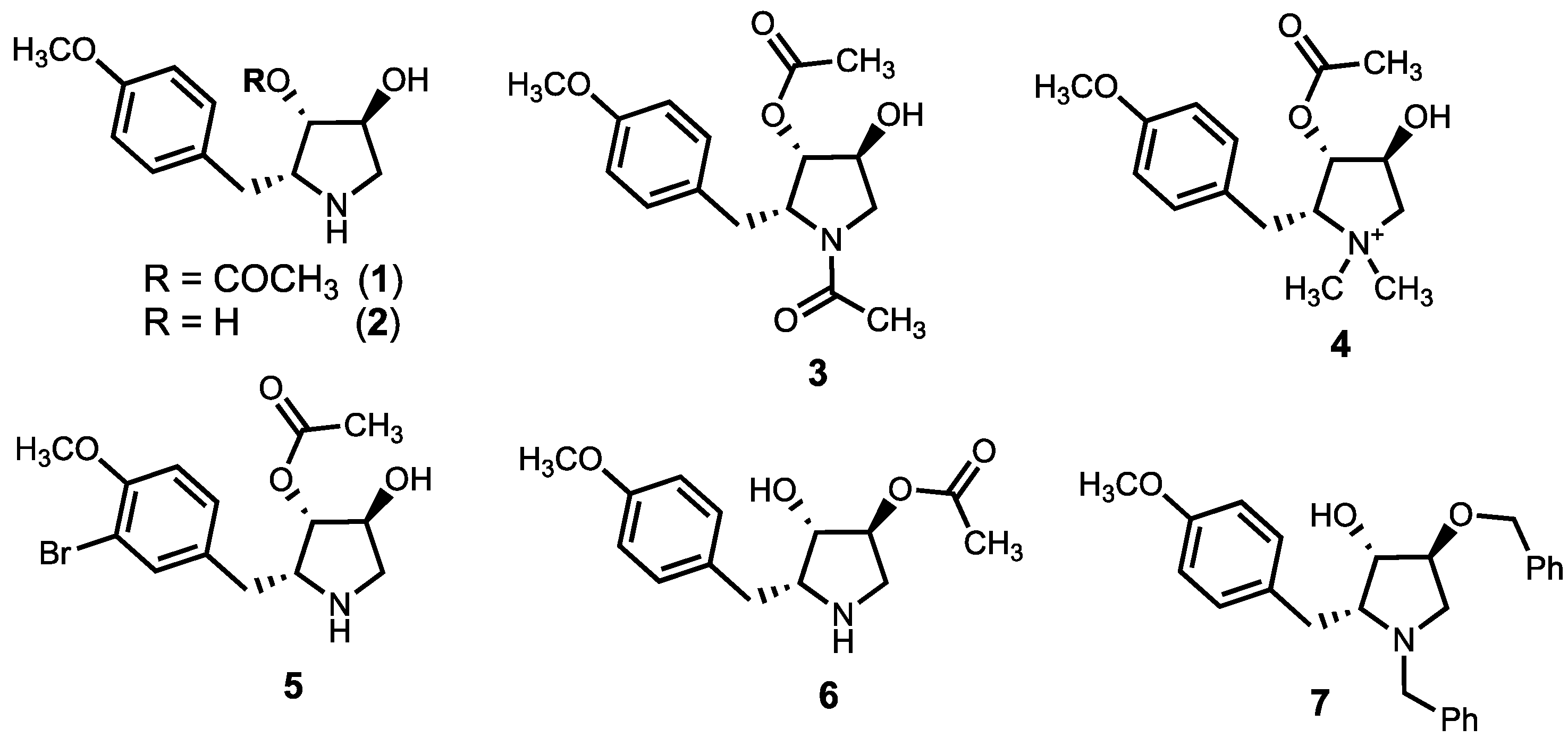
3.1. Chemistry
3.2. Anti-Leishmanial Activity of Anisomycin Derivatives
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sobin, B.A.; Tanner, F.W., Jr. Anisomycin, 1 A new anti-protozoan antibiotic. J. Am. Chem. Soc. 1954, 76, 4053. [Google Scholar] [CrossRef]
- Grollman, A.P. Inhibitors of protein biosynthesis: II. Mode of action of anisomycin. J. Biol. Chem. 1967, 242, 3226–3233. [Google Scholar] [CrossRef] [PubMed]
- Macías-Silva, M.; Vázquez-Victorio, G.; Hernández-Damián, J. Anisomycin is a multifunctional drug: More than just a tool to inhibit protein synthesis. Curr. Chem. Biol. 2010, 4, 124–132. [Google Scholar]
- de Loubresse, N.G.; Prokhorova, I.; Holtkamp, W.; Rodnina, M.V.; Yusupova, G.; Yusupov, M. Structural basis for the inhibition of the eukaryotic ribosome. Nature 2014, 513, 517–522. [Google Scholar] [CrossRef]
- Mahadevan, L.C.; Edwards, D.R. Signalling and superinduction. Nature 1991, 349, 747–748. [Google Scholar] [CrossRef]
- Cano, E.; Mahadevan, L.C. Parallel signal processing among mammalian MAPKs. Trends Biochem. Sci. 1995, 20, 117–122. [Google Scholar] [CrossRef]
- Barros, L.F.; Young, M.; Saklatvala, J.; Baldwin, S.A. Evidence of two mechanisms for the activation of the glucose transporter GLUT1 by anisomycin: P38 (MAP kinase) activation and protein synthesis inhibition in mammalian cells. J. Physiol. 1997, 504, 517–525. [Google Scholar] [CrossRef]
- Junghae, M.; Raynes, J.G. Activation of p38 mitogen-activated protein kinase attenuates Leishmania donovani infection in macrophages. Infect. Immun. 2002, 70, 5026–5035. [Google Scholar] [CrossRef]
- Chang, C.-C.; Ou, Y.-C.; Raung, S.-L.; Chen, C.-J. Antiviral effect of dehydroepiandrosterone on Japanese encephalitis virus infection. J. Gen. Virol. 2005, 86, 2513–2523. [Google Scholar] [CrossRef]
- Quintana, V.M.; Selisko, B.; Brunetti, J.E.; Eydoux, C.; Guillemot, J.; Canard, B.; Damonte, E.B.; Julander, J.; Castilla, V. Antiviral activity of the natural alkaloid anisomycin against dengue and Zika viruses. Antivir. Res. 2020, 176, 104749. [Google Scholar] [CrossRef]
- Croons, V.; Martinet, W.; Herman, A.G.; Timmermans, J.-P.; De Meyer, G.R. The protein synthesis inhibitor anisomycin induces macrophage apoptosis in rabbit atherosclerotic plaques through p38 mitogen-activated protein kinase. J. Pharmacol. Exp. Ther. 2009, 329, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Ni, Z.; Yicheng, S.; Pan, H.; Huang, Y.; Xiong, Y.; Liu, T. Anisomycin inhibits angiogenesis in ovarian cancer by attenuating the molecular sponge effect of the lncRNA-Meg3/miR-421/PDGFRA axis. Int. J. Oncol. 2019, 55, 1296–1312. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Xing, F.; Chen, D.; Yu, Y.; Yu, C.; Di, J.; Liu, J. In vivo toxicological evaluation of Anisomycin. Toxicol. Lett. 2012, 208, 1–11. [Google Scholar] [CrossRef] [PubMed]
- El Nemr, A.; El Ashry, E. New Developments in the Synthesis of Anisomycin and Its Analogues. In Heterocycles from Carbohydrate Precursors; Springer: Berlin/Heidelberg, Germany, 2007; pp. 249–285. [Google Scholar]
- Six, P.; Goossens, J.-F.; Riquet, W.; Brel, V.; Fournier, E.; Annereau, J.-P.; Kruczynski, A.; Castillo-Aguilera, O.; Depreux, P.; Bailly, C.; et al. Hemisynthesis of Anisomycin Derivatives as Antitumor Agents. Med. Chem. 2015, 5, 183–191. [Google Scholar] [CrossRef]
- Hall, S.S.; Loebenberg, D.; Schumacher, D.P. Structure-activity relationships of synthetic antibiotic analogs of anisomycin. J. Med. Chem. 1983, 26, 469–475. [Google Scholar] [CrossRef]
- Monaghan, D.; O’Connell, E.; Cruickshank, F.L.; O’Sullivan, B.; Giles, F.J.; Hulme, A.N.; Fearnhead, H.O. Inhibition of protein synthesis and JNK activation are not required for cell death induced by anisomycin and anisomycin analogues. Biochem. Biophys. Res. Commun. 2014, 443, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.M.; Morton, S.; Ashton, K.S.; Cohen, P.; Hulme, A.N. Synthetic anisomycin analogues activating the JNK/SAPK1 and p38/SAPK2 pathways. Org. Biomol. Chem. 2004, 2, 142–149. [Google Scholar] [CrossRef]
- Shi, S.; Zhu, S.; Gerritz, S.W.; Esposito, K.; Padmanabha, R.; Li, W.; Herbst, J.J.; Wong, H.; Shu, Y.Z.; Lam, K.S. Solid-phase synthesis and anti-infective activity of a combinatorial library based on the natural product anisomycin. Bioorg. Med. Chem. Lett. 2005, 15, 4151–4154. [Google Scholar] [CrossRef]
- Ehrenkaufer, G.; Li, P.; Stebbins, E.E.; Kangussu-Marcolino, M.M.; Debnath, A.; White, C.V.; Moser, M.S.; DeRisi, J.; Gisselberg, J.; Yeh, E. Identification of anisomycin, prodigiosin and obatoclax as compounds with broad-spectrum anti-parasitic activity. PLoS Negl. Trop. Dis. 2020, 14, e0008150. [Google Scholar] [CrossRef]
- Awasthi, A.; Mathur, R.; Khan, A.; Joshi, B.N.; Jain, N.; Sawant, S.; Boppana, R.; Mitra, D.; Saha, B. CD40 signaling is impaired in L. major–infected macrophages and is rescued by a p38MAPK activator establishing a host-protective memory T cell response. J. Exp. Med. 2003, 197, 1037–1043. [Google Scholar] [CrossRef]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef] [PubMed]
- Poulaki, A.; Piperaki, E.T.; Voulgarelis, M. Effects of visceralising leishmania on the spleen, liver, and bone marrow: A pathophysiological perspective. Microorganisms 2021, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Bilgic-Temel, A.; Murrell, D.F.; Uzun, S. Cutaneous leishmaniasis: A neglected disfiguring disease for women. Int. J. Womens Dermatol. 2019, 5, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Strazzulla, A.; Cocuzza, S.; Pinzone, M.R.; Postorino, M.C.; Cosentino, S.; Serra, A.; Cacopardo, B.; Nunnari, G. Mucosal leishmaniasis: An underestimated presentation of a neglected disease. Biochem. Biophys. Res. Commun. 2013, 2013, 805108. [Google Scholar] [CrossRef] [PubMed]
- Olias-Molero, A.I.; de la Fuente, C.; Cuquerella, M.; Torrado, J.J.; Alunda, J.M. Antileishmanial drug discovery and development: Time to reset the model? Microorganisms 2021, 9, 2500. [Google Scholar] [CrossRef]
- Olivier, M.; Gregory, D.J.; Forget, G. Subversion mechanisms by which Leishmania parasites can escape the host immune response: A signaling point of view. Clin. Microbiol. Rev. 2005, 18, 293–305. [Google Scholar] [CrossRef]
- Leatherbarrow, R.J. GraFit, version 7; Erithacus Software Ltd.: East Grinstead, UK, 2010. [Google Scholar]
- Brotherton, M.-C.; Bourassa, S.; Leprohon, P.; Légaré, D.; Poirier, G.G.; Droit, A.; Ouellette, M. Proteomic and genomic analyses of antimony resistant Leishmania infantum mutant. PLoS ONE 2013, 8, e81899. [Google Scholar] [CrossRef]
- El Fadili, K.; Messier, N.; Leprohon, P.; Roy, G.; Guimond, C.; Trudel, N.; Saravia, N.G.; Papadopoulou, B.; Légaré, D.; Ouellette, M. Role of the ABC transporter MRPA (PGPA) in antimony resistance in Leishmania infantum axenic and intracellular amastigotes. Antimicrob. Agents Chemother. 2005, 49, 1988–1993. [Google Scholar] [CrossRef]
- Brotherton, M.-C.; Bourassa, S.; Légaré, D.; Poirier, G.G.; Droit, A.; Ouellette, M. Quantitative proteomic analysis of amphotericin B resistance in Leishmania infantum. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 126–132. [Google Scholar] [CrossRef]
- Fernandez-Prada, C.; Vincent, I.M.; Brotherton, M.-C.; Roberts, M.; Roy, G.; Rivas, L.; Leprohon, P.; Smith, T.K.; Ouellette, M. Different mutations in a P-type ATPase transporter in Leishmania parasites are associated with cross-resistance to two leading drugs by distinct mechanisms. PLoS Negl. Trop. Dis. 2016, 10, e0005171. [Google Scholar] [CrossRef]
- Revilla-López, G.; Rodríguez-Ropero, F.; Curcó, D.; Torras, J.; Isabel Calaza, M.; Zanuy, D.; Jiménez, A.I.; Cativiela, C.; Nussinov, R.; Alemán, C. Integrating the intrinsic conformational preferences of noncoded α-amino acids modified at the peptide bond into the noncoded amino acids database. Proteins 2011, 79, 1841–1852. [Google Scholar] [CrossRef]
- Douanne, N.; Wagner, V.; Roy, G.; Leprohon, P.; Ouellette, M.; Fernandez-Prada, C. MRPA-independent mechanisms of antimony resistance in Leishmania infantum. Int. J. Parasitol. Drugs Drug Resist. 2020, 13, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Eliaz, D.; Doniger, T.; Tkacz, I.D.; Biswas, V.K.; Gupta, S.K.; Kolev, N.G.; Unger, R.; Ullu, E.; Tschudi, C.; Michaeli, S. Genome-wide analysis of small nucleolar RNAs of Leishmania major reveals a rich repertoire of RNAs involved in modification and processing of rRNA. RNA Biol. 2015, 12, 1222–1255. [Google Scholar] [CrossRef]
- Nickell, L.G.; Pennington, F.C.; Solomons, I.A. Parasiticidal agent and process for producing same. U.S. Patent No. 2,935,444, 3 May 1960. [Google Scholar]
- Tokuda, M.; Fujita, H.; Miyamoto, T.; Suginome, H. New total synthesis of (+)-N-methylanisomycin by anodic cyclization of δ-alkenylamine. Tetrahedron 1993, 49, 2413–2426. [Google Scholar] [CrossRef]
- Eschweiler, W. Ersatz von an Stickstoff gebundenen Wasserstoffatomen durch die Methylgruppe mit Hülfe von Formaldehyd. Berichte Dtsch. Chem. Ges. 1905, 38, 880–882. [Google Scholar] [CrossRef]
- Basel, Y.; Hassner, A. Di-tert-butyl dicarbonate and 4-(dimethylamino) pyridine revisited. Their reactions with amines and alcohols1. J. Org. Chem. 2000, 65, 6368–6380. [Google Scholar] [CrossRef]
- McOmie, J.; Watts, M.; West, D. Demethylation of aryl methyl ethers by boron tribromide. Tetrahedron 1968, 24, 2289–2292. [Google Scholar] [CrossRef]
- Shalev, M.; Rozenberg, H.; Smolkin, B.; Nasereddin, A.; Kopelyanskiy, D.; Belakhov, V.; Schrepfer, T.; Schacht, J.; Jaffe, C.L.; Adir, N. Structural basis for selective targeting of leishmanial ribosomes: Aminoglycoside derivatives as promising therapeutics. Nucleic Acids Res. 2015, 43, 8601–8613. [Google Scholar] [CrossRef]
- Blaha, G.; Gürel, G.; Schroeder, S.J.; Moore, P.B.; Steitz, T.A. Mutations outside the anisomycin-binding site can make ribosomes drug-resistant. J. Mol. Biol. 2008, 379, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Cheng, Q.; Yao, F.; Wang, X.; Kong, L.; Cao, B.; Xu, M.; Lin, S.; Deng, Z.; Chooi, Y.-H. Biosynthesis of the pyrrolidine protein synthesis inhibitor anisomycin involves novel gene ensemble and cryptic biosynthetic steps. Proc. Natl. Acad. Sci. USA 2017, 114, 4135–4140. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.L.; Moore, P.B.; Steitz, T.A. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 2003, 330, 1061–1075. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Corbeil, A.; do Monte-Neto, R.L.; Fernandez-Prada, C. Of Drugs and Trypanosomatids: New Tools and Knowledge to Reduce Bottlenecks in Drug Discovery. Genes 2020, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- De Muylder, G.; Ang, K.K.; Chen, S.; Arkin, M.R.; Engel, J.C.; McKerrow, J.H. A screen against Leishmania intracellular amastigotes: Comparison to a promastigote screen and identification of a host cell-specific hit. PLoS Negl. Trop. Dis. 2011, 5, e1253. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
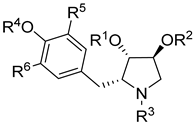
| Analog | R1 | R2 | R3 | R4 | R5 | R6 | IC50 (μM) | LC50 (μM) | SI | ||
| L. tarentolae | E. coli | RR | |||||||||
| Paromomycin | 3.62 ± 0.09 | 0.051 ± 0.005 | 21 ± 1.1 | 35.2 | 0.2 | ||||||
| 1 | Ac | H | H | Me | H | H | 0.55 ± 0.03 | >500 | 0.1 ± 0.02 | 0.22 ± 0.015 | 2.5 |
| 2 | H | H | H | Me | H | H | >100 | >1000 | >1000 | >>600 | - |
| 8 | Ac | H | Me | Me | H | H | >100 | >1000 | 25.5 ±3.6 | >>600 | - |
| 9 | Ac | Ac | H | Me | H | H | >100 | >1000 | 25.7 ± 8.5 | >>600 | - |
| 10 | Me | Me | H | Me | H | H | >1000 | >1000 | >1000 | >>600 | - |
| 11 | Me | H | H | Me | H | H | 24 ± 2.5 | >1000 | 70.9 ± 8.3 | >>600 | 0.3 |
| 16 | Ac | H | H | Me | I | H | >1000 | >1000 | 73.7 ± 1.7 | >>600 | - |
| 17 | Ac | H | H | H | H | H | 53.7 ± 6.1 | >200 | 6.2 ± 0.45 | 35±7.2 | 8.6 |
| 19 | Ac | H | H | H | -CH2N(CH3)2 | >1000 | >200 | 70.7 ± 7.3 | >>600 | - | |
| 20 | Ac | H | H | H | -CH2N(CH2CH2)2O | >1000 | >1000 | >1000 | >>600 | - | |
| 22a | Ac | H | H | i-Pr | H | H | 1.13 ± 0.12 | >200 | 0.9 ± 0.3 | 0.3 ± 0.12 | 1.2 |
| 22b | Ac | H | H | n-octyl | H | H | 33.5 ± 8.2 | >200 | 9.5 ± 3.5 | 5.2 ± 1.5 | 3.5 |
| 22c | Ac | H | H | Bn | H | H | 7.96 ± 1.02 | >200 | 0.9 ± 0.2 | 1.9 ± 0.3 | 8.8 |
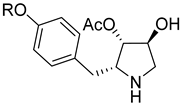 | ||||||
|---|---|---|---|---|---|---|
| # | R | IC50 (μM) | ||||
| Ldi WT 1 | LmF WT 2 | Ldi Sb-Res 3 | Ldi MF-Res 4 | Ldi AmB-Res 5 | ||
| Miltefosine | 7.16 ± 0.21 | 8.47 ± 1.18 | 8.21 ± 0.53 | >200 | 30.34 ± 4.28 | |
| Antimony | 75.80 ± 6.13 | 25.02 ± 1.13 | >2000 | 85.33 ± 7.10 | 90.25 ± 8.30 | |
| Anisomycin (1, nM) | 123.9 ± 5.34 | 109.56 ± 9.16 | 294.9 ± 12.45 | 467.5 ± 30.11 | 168.6 ± 10.90 | |
| 22a (nM) | i-Pr | 121.80 ± 5.62 | 180.17 ± 4.69 | 128.80 ± 3.16 | 132.70 ± 7.80 | 118.90 ± 14.07 |
| 22b | n-Octyl | 1.11 ± 0.03 | 1.75 ± 0.12 | 1.75 ± 0.08 | 1.83 ± 0.06 | 1.78 ± 0.58 |
| 22d | -CH2CO2H | 14.9 ± 2.18 | 16.55 ± 2.23 | 15.98 ± 1.03 | 18.17 ± 2.33 | 19.23 ± 1.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, A.M.T.; Shalev-Benami, M.; Rosa-Teijeiro, C.; Ibarra-Meneses, A.V.; Yonath, A.; Bashan, A.; Jaffe, C.L.; Olivier, M.; Fernandez-Prada, C.; Lubell, W.D. Systematic Exploration of Functional Group Relevance for Anti-Leishmanial Activity of Anisomycin. Biomedicines 2023, 11, 2541. https://doi.org/10.3390/biomedicines11092541
Nguyen AMT, Shalev-Benami M, Rosa-Teijeiro C, Ibarra-Meneses AV, Yonath A, Bashan A, Jaffe CL, Olivier M, Fernandez-Prada C, Lubell WD. Systematic Exploration of Functional Group Relevance for Anti-Leishmanial Activity of Anisomycin. Biomedicines. 2023; 11(9):2541. https://doi.org/10.3390/biomedicines11092541
Chicago/Turabian StyleNguyen, Anh Minh Thao, Moran Shalev-Benami, Chloé Rosa-Teijeiro, Ana Victoria Ibarra-Meneses, Ada Yonath, Anat Bashan, Charles L. Jaffe, Martin Olivier, Christopher Fernandez-Prada, and William D. Lubell. 2023. "Systematic Exploration of Functional Group Relevance for Anti-Leishmanial Activity of Anisomycin" Biomedicines 11, no. 9: 2541. https://doi.org/10.3390/biomedicines11092541
APA StyleNguyen, A. M. T., Shalev-Benami, M., Rosa-Teijeiro, C., Ibarra-Meneses, A. V., Yonath, A., Bashan, A., Jaffe, C. L., Olivier, M., Fernandez-Prada, C., & Lubell, W. D. (2023). Systematic Exploration of Functional Group Relevance for Anti-Leishmanial Activity of Anisomycin. Biomedicines, 11(9), 2541. https://doi.org/10.3390/biomedicines11092541