

The Spectrum of Molecular Pathways in Gliomas—An Up-to-Date Review

Abstract
:1. Introduction
2. Receptors and Different Modes of Their Activation
3. Signaling Pathways
4. Angiogenic and Apoptotic Pathways
5. Metabolic Pathways
6. Epigenetic Processes
7. Extracellular Vesicles
8. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Park, J.W. Metabolic rewiring in adult-type diffuse gliomas. Int. J. Mol. Sci. 2023, 24, 7348. [Google Scholar] [CrossRef] [PubMed]
- Afonso, M.; Brito, M.A. Therapeutic options in neuro-oncology. Int. J. Mol. Sci. 2022, 23, 5351. [Google Scholar] [CrossRef] [PubMed]
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor tyrosine kinase signaling and targeting in glioblastoma multiforme. Int. J. Mol. Sci. 2021, 22, 1831. [Google Scholar] [CrossRef]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor development and angiogenesis in adult brain tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Luo, L.; Zhang, X.; Wei, C.; Zhang, Z.; Han, L. CircRNA: An emerging star in the progression of glioma. Biomed. Pharmacother. 2022, 151, 113150. [Google Scholar] [CrossRef]
- Schwark, K.; Messinger, D.; Cummings, J.R.; Bradin, J.; Kawakibi, A.; Babila, C.M.; Lyons, S.; Ji, S.; Cartaxo, R.T.; Kong, S.; et al. Receptor tyrosine kinase (RTK) targeting in pediatric high-grade glioma and diffuse midline glioma: Preclinical models and precision medicine. Front. Oncol. 2022, 12, 922928. [Google Scholar] [CrossRef]
- Lu, Q.R.; Qian, L.; Zhou, X. Convergence of developmental origins and oncogenic pathways in malignant brain tumors. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e342. [Google Scholar] [CrossRef]
- Sie, M.; den Dunnen, W.F.A.; Lourens, H.J.; Meeuwsen-de Boer, T.G.J.; Scherpen, F.J.G.; Zomerman, W.W.; Kampen, K.; Hoving, E.W.; De Bont, E.S.J.M. Growth-factor-driven rescue to receptor tyrosine kinase (RTK) inhibitors through Akt and Erk phosphorylation in pediatric low grade astrocytoma and ependymoma. PLoS ONE 2015, 10, e0122555. [Google Scholar] [CrossRef]
- Kang, S.; Hong, J.; Lee, J.M.; Moon, H.E.; Jeon, B.; Choi, J.; Yoon, N.A.; Paek, S.H.; Roh, E.J.; Lee, C.J.; et al. Trifluoperazine, a well-known antipsychotic, inhibits glioblastoma invasion by binding to calmodulin and disinhibiting calcium release channel IP3R. Mol. Cancer Ther. 2017, 16, 217–227. [Google Scholar] [CrossRef]
- Bryant, J.A.; Finn, R.S.; Slamon, D.J.; Cloughesy, T.F.; Charles, A.C. EGF activates intracellular and intercellular calcium signaling by distinct pathways in tumor cells. Cancer Biol. Ther. 2004, 3, 1243–1249. [Google Scholar] [CrossRef]
- Sciaccaluga, M.; D’Alessandro, G.; Pagani, F.; Ferrara, G.; Lopez, N.; Warr, T.; Gorello, P.; Porzia, A.; Mainiero, F.; Santoro, A.; et al. Functional cross talk between CXCR4 and PDGFR on glioblastoma cells is essential for migration. PLoS ONE 2013, 8, e73426. [Google Scholar] [CrossRef]
- Cherry, A.E.; Stella, N. G protein-coupled receptors as oncogenic signals in glioma: Emerging therapeutic avenues. Neuroscience 2014, 278, 222–236. [Google Scholar] [CrossRef]
- Huang, J.; Hu, J.; Bian, X.; Chen, K.; Gong, W.; Dunlop, N.M.; Howard, O.Z.; Wang, J.M. Transactivation of the epidermal growth factor receptor by formylpeptide receptor exacerbates the malignant behaviour of human glioblastoma cells. Cancer Res. 2007, 67, 5906–5913. [Google Scholar] [CrossRef]
- Tilak, M.; Alural, B.; Wismer, S.E.; Brasher, M.I.; New, L.A.; Sheridan, S.D.; Perlis, R.H.; Coppolino, M.G.; Lalonde, J.; Jones, N. Adaptor protein ShcD/SHC4 interacts with Tie2 receptor to synergistically promote glioma cell invasion. Mol. Cancer Res. 2021, 19, 757–770. [Google Scholar] [CrossRef]
- Wills, M.K.; Tong, J.; Tremblay, S.L.; Moran, M.F.; Jones, N. The ShcD signaling adaptor facilitates ligand-independent phosphorylation of the EGF receptor. Mol. Biol. Cell 2014, 25, 739–752. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Huang, G.; Ho, B.; Yemma, M.; Morrison, C.D.; Lee, J.; Eliceiri, B.P.; Cance, W.G. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol. Cancer Ther. 2013, 12, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Li, X.; Yang, J.; Yuan, D.; Zhou, Y.; Zhang, Y.; Shi, G.; Zhang, R.; Liu, J.; Fu, P.; et al. PPFIBP1 induces glioma cell migration and invasion through FAK/Src/JNK signaling pathway. Cell Death Dis. 2021, 12, 827. [Google Scholar] [CrossRef]
- Peng, P.; Zhu, H.; Liu, D.; Chen, Z.; Zhang, X.; Guo, Z.; Dong, M.; Wan, L.; Zhang, P.; Liu, G.; et al. TGFBI secreted by tumor-associated macrophages promotes glioblastoma stem cell-driven tumor growth via integrin αvβ5-Src-Stat3 signaling. Theranostics 2022, 12, 4221–4236. [Google Scholar] [CrossRef]
- Nakada, M.; Nambu, E.; Furuyama, N.; Yoshida, Y.; Takino, T.; Hayashi, Y.; Sato, H.; Sai, Y.; Tsuji, T.; Miyamoto, K.-I.; et al. Integrin α3 is overexpressed in glioma stem-like cells and promotes invasion. Br. J. Cancer 2013, 108, 2516–2524. [Google Scholar] [CrossRef]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131, e142116. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Poleszak, K.; Pasierbinska, M.; Kaminska, B. Integrin signaling in glioma pathogenesis: From biology to therapy. Int. J. Mol. Sci. 2020, 21, 888. [Google Scholar] [CrossRef]
- Mair, D.B.; Ames, H.M.; Li, R. Mechanisms of invasion and motility of high-grade gliomas in the brain. Mol. Biol. Cell 2018, 29, 2509–2515. [Google Scholar] [CrossRef]
- Groblewska, M.; Litman-Zawadzka, A.; Mroczko, B. The role of selected chemokines and their receptors in the development of gliomas. Int. J. Mol. Sci. 2020, 21, 3704. [Google Scholar] [CrossRef]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef]
- Odemis, V.; Lipfert, J.; Kraft, R.; Hajek, P.; Abraham, G.; Hattermann, K.; Mentlein, R.; Engele, J. The presumed atypical chemokine receptor CXCR7 signals through G(i/o) proteins in primary rodent astrocytes and human glioma cells. Glia 2012, 60, 372–381. [Google Scholar] [CrossRef]
- Sen, B.; Xie, Z.; Case, N.; Thompson, W.R.; Uzer, G.; Styner, M.; Rubin, J. mTORC2 regulates mechanically induced cytoskeletal reorganization and lineage selection in marrow-derived mesenchymal stem cells. J. Bone Miner. Res. 2014, 29, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009, 100, 2235–2241. [Google Scholar] [CrossRef]
- Capper, D.; Reifenberger, G.; French, P.J.; Schweizer, L.; Weller, M.; Touat, M.; Niclou, S.P.; Euskirchen, P.; Haberler, C.; E Hegi, M.; et al. EANO guideline on rational molecular testing of gliomas, glioneuronal, and neuronal tumors in adults for targeted therapy selection. Neuro Oncol. 2023, 25, 813–826. [Google Scholar] [CrossRef]
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma targeted therapy: Insight into future of molecular approaches. Mol. Cancer 2022, 21, 39. [Google Scholar] [CrossRef]
- Mai, W.X.; Gosa, L.; Daniels, V.W.; Ta, L.; Tsang, J.E.; Higgins, B.; Gilmore, W.B.; Bayley, N.A.; Harati, M.D.; Lee, J.T.; et al. Cytoplasmic p53 couples oncogene-driven glucose metabolism to apoptosis and is a therapeutic target in glioblastoma. Nat. Med. 2017, 23, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-suppressor functions of the TP53 pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef]
- Di Fiore, R.; D’Anneo, A.; Tesoriere, G.; Vento, R. RB1 in cancer: Different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J. Cell. Physiol. 2013, 228, 1676–1687. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef]
- Wu, H.; Wei, M.; Li, Y.; Ma, Q.; Zhang, H. Research progress on the regulation mechanism of key signal pathways affecting the prognosis of glioma. Front. Mol. Neurosci. 2022, 15, 910543. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Zhong, Y.; Fu, J.; Cao, Y.; Fu, G.; Tian, X.; Wang, B. Activation of JAK/STAT signal pathway predicts poor prognosis of patients with gliomas. Med. Oncol. 2011, 28, 15–23. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, Q.; Lin, J. Runx1 promotes the development of glioma cells by regulating JAK-STAT signaling pathway. Arch. Med. Sci. 2019, 18, 761–776. [Google Scholar] [CrossRef]
- Zhu, X.; Yang, H.; Zhang, M.; Wu, X.; Jiang, L.; Liu, X.; Lv, K. YTHDC1-mediated VPS25 regulates cell cycle by targeting JAK-STAT signaling in human glioma cells. Cancer Cell Int. 2021, 21, 645. [Google Scholar] [CrossRef]
- Delen, E.; Doğanlar, O. The dose dependent effects of ruxolitinib on the invasion and tumorigenesis in gliomas cells via inhibition of interferon gamma-depended JAK/STAT signaling pathway. J. Korean Neurosurg. Soc. 2020, 63, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Assa-Kunik, E.; Torres, I.L.; Schejter, E.D.; Johnston, D.S.; Shilo, B.Z. Drosophila follicle cells are patterned by multiple levels of Notch signaling and antagonism between the Notch and JAK/STAT pathways. Development 2007, 134, 1161–1169. [Google Scholar] [CrossRef]
- Parmigiani, E.; Taylor, V.; Giachino, C. Oncogenic and tumor-suppressive functions of NOTCH signaling in glioma. Cells 2020, 9, 2304. [Google Scholar] [CrossRef]
- Kipper, F.C.; Kieran, M.W.; Thomas, A.; Panigrahy, D. Notch signaling in malignant gliomas: Supporting tumor growth and the vascular environment. Cancer Metastasis Rev. 2022, 41, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Los, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR survival pathways as therapeutic targets in glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Guillevin, R.; Vallée, J.N. Vasculogenesis and angiogenesis initiation under normoxic conditions through Wnt/β-catenin pathway in gliomas. Rev. Neurosci. 2018, 29, 71–91. [Google Scholar] [CrossRef]
- McCord, M.; Mukouyama, Y.S.; Gilbert, M.R.; Jackson, S. Targeting WNT signaling for multifaceted glioblastoma therapy. Front. Cell. Neurosci. 2017, 11, 318. [Google Scholar] [CrossRef]
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Iorio, P.D.; Caciagli, F.; Ciccarelli, R. The role of Wnt signal in glioblastoma development and progression: A possible new pharmacological target for the therapy of this tumor. Genes 2018, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Pedini, G.; Buccarelli, M.; Bianchi, F.; Pacini, L.; Cencelli, G.; D’Alessandris, Q.G.; Martini, M.; Giannetti, S.; Sasso, F.; Melocchi, V.; et al. FMRP modulates the Wnt signaling pathway in glioblastoma. Cell Death Dis. 2022, 13, 719. [Google Scholar] [CrossRef]
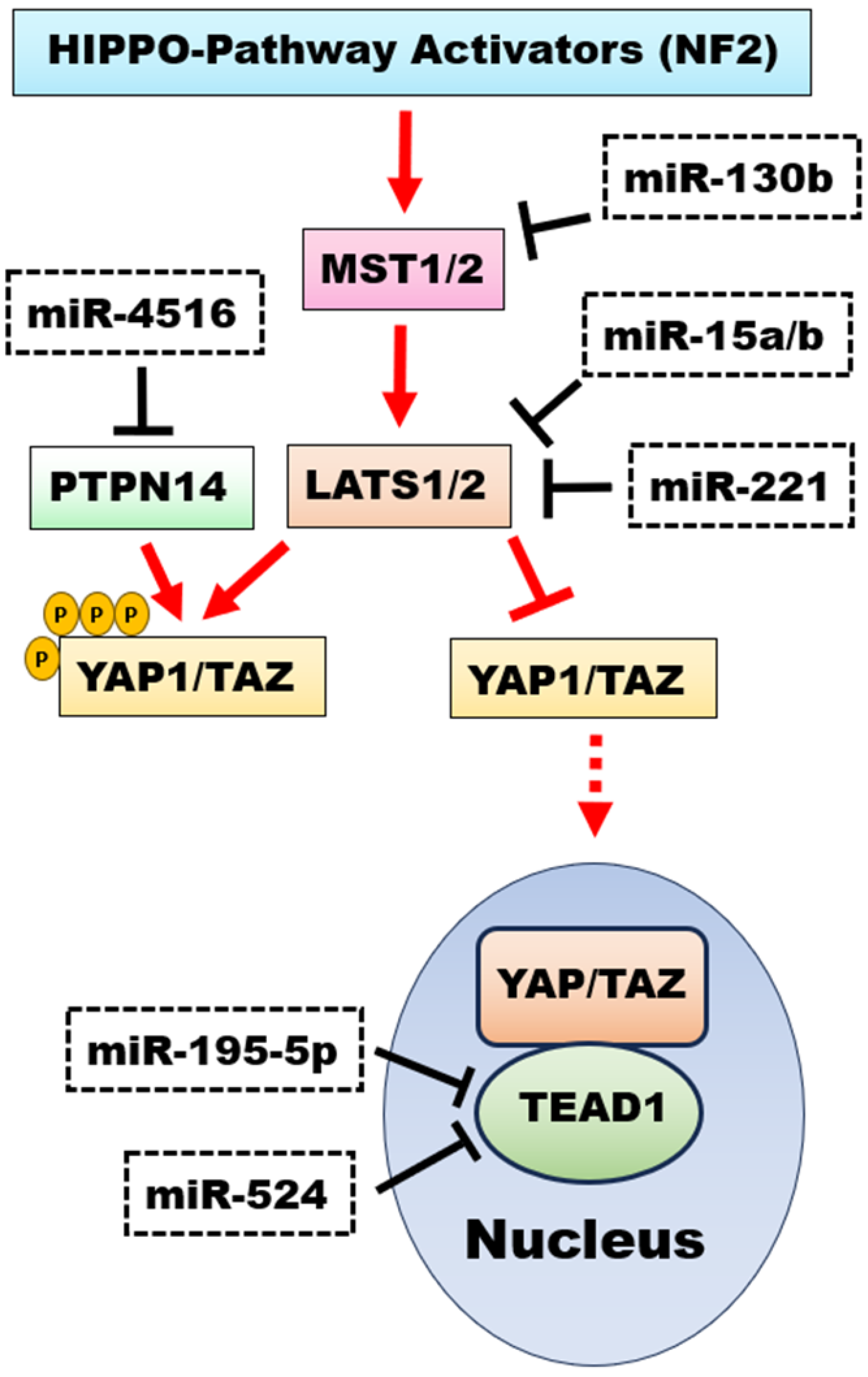
- Masliantsev, K.; Karayan-Tapon, L.; Guichet, P.O. Hippo signaling pathway in gliomas. Cells 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, T.; Meng, W.; Li, M.; Hong, T.; Zhang, N. Recent advances of the Hippo/YAP signaling pathway in brain development and glioma. Cell. Mol. Neurobiol. 2020, 40, 495–510. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, X.; Hu, J.; Zhou, W.; Jiang, Y.; Li, G.; Lu, D. Promoter hypermethylation-mediated down-regulation of LATS1 and LATS2 in human astrocytoma. Neurosci. Res. 2006, 56, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, R.; Lu, S.; Zhang, W.; Wang, D.; Ge, Y.; Jiang, F.; Qin, X.; Liu, Y. S100 calcium binding protein A16 promotes cell proliferation by triggering LATS1 ubiquitin degradation mediated by CUL4A ligase to inhibit Hippo pathway in glioma development. Int. J. Biol. Sci. 2023, 19, 2034–2052. [Google Scholar] [CrossRef] [PubMed]
- Bulnes, S.; Bengoetxea, H.; Ortuzar, N.; Argandona, E.G.; Garcia-Blanco, A.; Rico-Barrio, I.; Lafuente, J.V. Angiogenic signaling pathways altered in gliomas: Selection mechanisms for more aggressive neoplastic subpopulations with invasive phenotype. J. Signal Transduct. 2012, 2012, 597915. [Google Scholar] [CrossRef]
- Keller, S.; Schmidt, M.H.H. EGFR and EGFRvIII promote angiogenesis and cell invasion in glioblastoma: Combination therapies for an effective treatment. Int. J. Mol. Sci. 2017, 18, 1295. [Google Scholar] [CrossRef]
- Scholz, A.; Harter, P.N.; Cremer, S.; Yalcin, B.H.; Gurnik, S.; Yamaji, M.; Di Tacchio, M.; Sommer, K.; Baumgarten, P.; Bähr, O.; et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 2016, 8, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Guo, B.; Ye, J.; Liu, H.; Deng, W.; Lin, C.; Zhong, X.; Wang, L. Vasorin stimulates malignant progression and angiogenesis in glioma. Cancer Sci. 2019, 110, 2558–2572. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, C.; Zheng, Q.; Ma, Z.; Qi, M.; Di, G.; Ling, S.; Xu, H.; Qi, B.; Yao, C.; et al. RhoJ facilitates angiogenesis in glioblastoma via JNK/VEGFR2 mediated activation of PAK and ERK signaling pathways. Int. J. Biol. Sci. 2022, 18, 942–955. [Google Scholar] [CrossRef]
- Chen, L.; Xie, X.; Wang, T.; Xu, L.; Zhai, Z.; Wu, H.; Deng, L.; Lu, Q.; Chen, Z.; Yang, X.; et al. ARL13B promotes angiogenesis and glioma growth by activating VEGFA-VEGFR2 signaling. Neuro Oncol. 2023, 25, 871–885. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, Y.; Li, Y.; Lian, W.; Zheng, K.; Zhang, Y.; Zhang, Y.; Lin, C.; Liu, C.; Sun, F.; et al. Prrx1 promotes stemness and angiogenesis via activating TGF-β/Smad pathway and upregulating proangiogenic factors in glioma. Cell Death Dis. 2021, 12, 615. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, H.; Sun, M.; Deng, X.; Wu, X.; Ma, Y.; Li, M.; Shuoa, S.M.; You, Q.; Miao, L. CXCL5/CXCR2 axis in tumor microenvironment as potential diagnostic biomarker and therapeutic target. Cancer Commun. 2020, 40, 69–80. [Google Scholar] [CrossRef]
- Acker, G.; Zollfrank, J.; Jelgersma, C.; Nieminen-Kelhä, M.; Kremenetskaia, I.; Mueller, S.; Ghori, A.; Vajkoczy, P.; Brandenburg, S. The CXCR2/CXCL2 signaling pathway—An alternative therapeutic approach in high-grade glioma. Eur. J. Cancer 2020, 126, 106–115. [Google Scholar] [CrossRef]
- Fischer, I.; Gagner, J.P.; Law, M.; Newcomb, E.W.; Zagzag, D. Angiogenesis in gliomas: Biology and molecular pathophysiology. Brain Pathol. 2005, 15, 297–310. [Google Scholar] [CrossRef]
- Ghoochani, A.; Hatipoglu-Majernik, G.; Sehm, T.; Wach, S.; Buchfelder, M.; Taubert, H.; Eyupoglu, I.Y.; Savaskan, N. Cabazitaxel operates anti-metastatic and cytotoxic via apoptosis induction and stalls brain tumor angiogenesis. Oncotarget 2016, 7, 38306–38318. [Google Scholar] [CrossRef] [PubMed]
- Zhen, H.N.; Zhang, X.; Hu, P.Z.; Yang, T.T.; Fei, Z.; Zhang, J.N.; Fu, L.A.; He, X.S.; Ma, F.C.; Wang, X.L. Survivin expression and its relation with proliferation, apoptosis, and angiogenesis in brain gliomas. Cancer 2005, 104, 2775–2783. [Google Scholar] [CrossRef]
- Trejo-Solís, C.; Serrano-Garcia, N.; Escamilla-Ramirez, A.; Castillo-Rodríguez, R.A.; Jimenez-Farfan, D.; Palencia, G.; Calvillo, M.; Alvarez-Lemus, M.A.; Flores-Nájera, A.; Cruz-Salgado, A.; et al. Autophagic and apoptotic pathways as targets for chemotherapy in glioblastoma. Int. J. Mol. Sci. 2018, 19, 3773. [Google Scholar] [CrossRef] [PubMed]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [CrossRef] [PubMed]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef] [PubMed]
- Krakstad, C.; Chekenya, M. Survival signaling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Stegh, A.H.; Kim, H.; Bachoo, R.M.; Forloney, K.L.; Zhang, J.; Schulze, H.; Park, K.; Hannon, G.J.; Yuan, J.; Louis, D.N.; et al. Bcl2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma. Genes Dev. 2007, 21, 98–111. [Google Scholar] [CrossRef]
- Ruano, Y.; Mollejo, M.; Camacho, F.I.; Rodríguez de Lope, A.; Fiano, C.; Ribalta, T.; Martínez, P.; Hernández-Moneo, J.L.; Meléndez, B. Identification of survival-related genes of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma multiforme. Cancer 2008, 112, 1575–1584. [Google Scholar] [CrossRef]
- Saggioro, F.P.; Neder, L.; Stavale, J.N.; Paixao-Becker, A.N.; Malheiros, S.M.; Soares, F.A.; Pittella, J.E.H.; Matias, C.C.M.; Colli, B.O.; Carlotti, C.G.; et al. Fas, FasL, and cleaved caspases 8 and 3 in glioblastomas: A tissue microarray-based study. Pathol. Res. Pract. 2014, 210, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chowdhury, T.; Yu, H.J.; Kahng, J.Y.; Lee, C.E.; Choi, S.A.; Kim, K.M.; Kang, H.; Lee, J.H.; Lee, S.T.; et al. The telomere maintenance mechanism spectrum and its dynamics in gliomas. Genome Med. 2022, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S.; Kuwahara, K.; Yamada, S.; Abe, M.; Hirose, Y. Correlation between IDH, ATRX, and TERT promoter mutations in glioma. Brain Tumor Pathol. 2020, 37, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, X.; Yan, X.; Sun, M.; Fan, Y.; Huang, Y. Significance of TERT and ATRX mutations in glioma. Oncol. Lett. 2019, 17, 95–102. [Google Scholar] [CrossRef]
- Fan, H.C.; Chen, C.M.; Chi, C.S.; Tsai, J.D.; Chiang, K.L.; Chang, Y.K.; Lin, S.Z.; Harn, H.J. Targeting telomerase and ATRX/DAXX inducing tumor senescence and apoptosis in the malignant glioma. Int. J. Mol. Sci. 2019, 20, 200. [Google Scholar] [CrossRef] [PubMed]
- Kurt, I.C.; Sur, I.; Kaya, E.; Cingoz, A.; Kazancioglu, S.; Kahya, Z.; Toparlak, O.D.; Senbabaoglu, F.; Kaya, Z.; Ozyerli, E.; et al. KDM2B, an H3K36-specific demethylase, regulates apoptotic response of GBM cells to TRAIL. Cell Death Dis. 2017, 8, e2897. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Dungen, K.V.; Bressler, K.R.; Fredriksen, M.; Khandige-Sharma, D.; Balasingam, N.; Thakor, N. Eukaryotic initiation factor 5B (eIF5B) provides a critical cell survival switch to glioblastoma cells via regulation of apoptosis. Cell Death Dis. 2019, 10, 57. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro Oncol. 2013, 15, 172–188. [Google Scholar] [CrossRef]
- Kefas, B.; Comeau, L.; Erdle, N.; Montgomery, E.; Amos, S.; Purow, B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro Oncol. 2010, 12, 1102–1112. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Dunphy, M.P.; Zhang, H.; Pitter, K.L.; Zanzonico, P.; Campos, C.; Carlin, S.D.; La Rocca, G.; Lyashchenko, S.; Ploessl, K.; et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci. Transl. Med. 2015, 7, 274ra17. [Google Scholar] [CrossRef]
- Martins, F.; Gonçalves, L.G.; Pojo, M.; Serpa, J. Take advantage of glutamine anaplerosis, the kernel of the metabolic rewiring in malignant gliomas. Biomolecules 2020, 10, 1370. [Google Scholar] [CrossRef] [PubMed]
- Obara-Michlewska, M.; Szeliga, M. Targeting glutamine addiction in gliomas. Cancers 2020, 12, 310. [Google Scholar] [CrossRef]
- Mukherjee, P.; Augur, Z.M.; Li, M.; Hill, C.; Greenwood, B.; Domin, M.A.; Kondakci, G.; Narain, N.R.; Kiebish, M.A.; Bronson, R.T.; et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun. Biol. 2019, 2, 200. [Google Scholar] [CrossRef]
- Chou, F.J.; Liu, Y.; Lang, F.; Yang, C. D-2-Hydroxyglutarate in glioma biology. Cells 2021, 10, 2345. [Google Scholar] [CrossRef]
- Ohba, S.; Mukherjee, J.; Johannessen, T.C.; Mancini, A.; Chow, T.T.; Wood, M.; Jones, L.; Mazor, T.; Marshall, R.E.; Viswanath, P.; et al. Mutant IDH1 expression drives TERT promoter reactivation as part of the cellular transformation process. Cancer Res. 2016, 76, 6680–6689. [Google Scholar] [CrossRef] [PubMed]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- De Souza, C.F.; Sabedot, T.S.; Malta, T.M.; Stetson, L.; Morozova, O.; Sokolov, A.; Laird, P.W.; Wiznerowicz, M.; Iavarone, A.; Snyder, J.; et al. A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell Rep. 2018, 23, 637–651. [Google Scholar] [CrossRef]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, Z.; Chai, R.C.; Liu, Y.Q.; Li, G.Z.; Jiang, H.Y.; Jiang, T. Prognostic power of a lipid metabolism gene panel for diffuse gliomas. J. Cell. Mol. Med. 2019, 23, 7741–7748. [Google Scholar] [CrossRef]
- Ahmad, F.; Patrick, S.; Sheikh, T.; Sharma, V.; Pathak, P.; Malgulwar, P.B.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Telomerase reverse transcriptase (TERT)–enhancer of zeste homolog 2 (EZH2) network regulates lipid metabolism and DNA damage responses in glioblastoma. J. Neurochem. 2017, 143, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef]
- ENZYME—Enzyme Nomenclature Database. Available online: https://enzyme.expasy.org/ (accessed on 1 August 2023).
- Ozair, A.; Bhat, V.; Alisch, R.S.; Khosla, A.A.; Kotecha, R.R.; Odia, Y.; McDermott, M.W.; Ahluwalia, M.S. DNA methylation and histone modification in low-grade gliomas: Current understanding and potential clinical targets. Cancers 2023, 15, 1342. [Google Scholar] [CrossRef]
- Della Monica, R.; Cuomo, M.; Buonaiuto, M.; Costabile, D.; Franca, R.A.; Del Basso De Caro, M.; Catapano, G.; Chiariotti, L.; Visconti, R. MGMT and whole-genome DNA methylation impacts on diagnosis, prognosis and therapy of glioblastoma multiforme. Int. J. Mol. Sci. 2022, 23, 7148. [Google Scholar] [CrossRef]
- Brain Classifier Version 12.5. Available online: https://www.molecularneuropathology.org/mnp/classifiers/11 (accessed on 1 August 2023).
- Wenger, A.; Caren, H. Methylation profiling in diffuse gliomas: Diagnostic value and considerations. Cancers 2022, 14, 5679. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.J.; Wojtas, B. Global DNA methylation patterns in human gliomas and their interplay with other epigenetic modifications. Int. J. Mol. Sci. 2019, 20, 3478. [Google Scholar] [CrossRef]
- Li, F.; Chen, S.; Yu, J.; Gao, Z.; Sun, Z.; Yi, Y.; Long, T.; Zhang, C.; Li, Y.; Pan, Y.; et al. Interplay of m6 A and histone modifications contributes to temozolomide resistance in glioblastoma. Clin. Transl. Med. 2021, 11, e553. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wu, T.P.; Gimple, R.C.; Li, Z.; Prager, B.C.; Wu, Q.; Yu, Y.; Wang, P.; Wang, Y.; Gorkin, D.U.; et al. N6-methyladenine DNA modification in glioblastoma. Cell 2018, 175, 1228–1243. [Google Scholar] [CrossRef]
- Wei, C.; Wang, B.; Peng, D.; Zhang, X.; Li, Z.; Luo, L.; He, Y.; Liang, H.; Du, X.; Li, S.; et al. Pan-cancer analysis shows that ALKBH5 is a potential prognostic and immunotherapeutic biomarker for multiple cancer types including gliomas. Front. Immunol. 2022, 13, 849592. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Jia, F.; Hou, Y.; Sang, Y.; Alvarez, A.A.; Zhang, W.; Gao, W.Q.; Hu, B.; Cheng, S.Y.; Ge, J.; et al. Histone acetyltransferase KAT6A upregulates PI3K/AKT signaling through TRIM24 binding. Cancer Res. 2017, 77, 6190–6201. [Google Scholar] [CrossRef]
- McCornack, C.; Woodiwiss, T.; Hardi, A.; Yano, H.; Kim, A.H. The function of histone methylation and acetylation regulators in GBM pathophysiology. Front. Oncol. 2023, 13, 1144184. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, P.; Tang, F.; Lian, H.; Chen, X.; Zhang, Y.; He, X.; Liu, W.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016, 379, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wu, Y.; Wang, M.; Sun, Z.; Zou, J.; Zhang, Y.; Cui, H. HDAC9 promotes glioblastoma growth via TAZ-mediated EGFR pathway activation. Oncotarget 2015, 6, 7644–7656. [Google Scholar] [CrossRef]
- Aquilina, K.; Chakrapani, A.; Carr, L.; Kurian, M.A.; Hargrave, D. Convection-enhanced delivery in children: Techniques and applications. Adv. Tech. Stand. Neurosurg. 2022, 45, 199–228. [Google Scholar] [CrossRef]
- Mo, F.; Pellerino, A.; Soffietti, R.; Ruda, R. Blood-brain barrier in brain tumors: Biology and clinical relevance. Int. J. Mol. Sci. 2021, 22, 12654. [Google Scholar] [CrossRef] [PubMed]
- Fults, D.W. Stemming the growth of pediatric gliomas through histone modification. Neuro Oncol. 2021, 23, 341–342. [Google Scholar] [CrossRef]
- Kommidi, H.; Tosi, U.; Maachani, U.B.; Guo, H.; Marnell, C.S.; Law, B.; Souweidane, M.M.; Ting, R. 18F-radiolabeled panobinostat allows for positron emission tomography guided delivery of a histone deacetylase inhibitor. ACS Med. Chem. Lett. 2018, 9, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.; Jain, P.; Kilburn, L.; Bonner, E.R.; Gupta, N.; Crawford, J.R.; Banerjee, A.; Packer, R.J.; Villanueva-Meyer, J.; Luks, T.; et al. Upfront biology-guided therapy in diffuse intrinsic pontine glioma: Therapeutic, molecular, and biomarker outcomes from PNOC003. Clin. Cancer Res. 2022, 28, 3965–3978. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. (Eds.) WHO Classification of Tumours of the Central Nervous System, Revised 4th ed.; International Agency for Research on Cancer, IARC: Lyon, France, 2016; pp. 12–14. [Google Scholar]
- Varlet, P.; Baker, S.J.; Ellison, D.W.; Jabado, N.; Jones, C.; Jones, D.T.W.; Leske, H.; Orr, B.A.; Solomon, D.A.; Suva, M.L.; et al. Diffuse midline glioma, H3K27-altered. In Central Nervous System Tumours, 5th ed.; WHO Classification of Tumours Series; WHO Classification of Tumours Editorial Board; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6, pp. 69–73. Available online: https://publications.iarc.fr/601 (accessed on 1 August 2023).
- Nafe, R.; Porto, L.; Samp, P.F.; You, S.J.; Hattingen, E. Adult-type and pediatric-type diffuse gliomas: What the neuroradiologist should know. Clin. Neuroradiol. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Qiu, L.; Han, J. Histone H3G34 mutation in brain and bone tumors. Adv. Exp. Med. Biol. 2021, 1283, 63–71. [Google Scholar] [CrossRef]
- Dai, L.; Liang, W.; Shi, Z.; Li, X.; Zhou, S.; Hu, W.; Yang, Z.; Wang, X. Systematic characterization and biological functions of non-coding RNAs in glioblastoma. Cell Prolif. 2023, 56, e13375. [Google Scholar] [CrossRef]
- Ghaemi, S.; Fekrirad, Z.; Zamani, N.; Rahmani, R.; Arefian, E. Non-coding RNAs enhance the apoptosis efficacy of therapeutic agents used for the treatment of glioblastoma multiform. J. Drug Target. 2022, 30, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Salami, R.; Salami, M.; Mafi, A.; Vakili, O.; Asemi, Z. Circular RNAs and glioblastoma multiforme: Focus on molecular mechanisms. Cell Commun. Signal. 2022, 20, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, X.; Geng, X.; Shi, L.; Li, Q.; Liu, F.; Fang, C.; Wang, H. Advances in circular RNAs and their role in glioma (Review). Int. J. Oncol. 2020, 57, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Peng, G. Long non-coding RNA FAM66C regulates glioma growth via the miRNA/LATS1 signaling pathway. Biol. Chem. 2021, 403, 679–689. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, Y.; Mijiti, M.; Wang, Z.; Wu, P.F.; Jiafu, D. Upregulation of miR-130b enhances stem cell-like phenotype in glioblastoma by inactivating the Hippo signaling pathway. Biochem. Biophys. Res. Commun. 2015, 465, 194–199. [Google Scholar] [CrossRef]
- Aguennouz, M.; Polito, F.; Visalli, M.; Vita, G.; Raffa, G.; Oteri, R.; Ghazi, B.; Scalia, G.; Angileri, F.F.; Barresi, V.; et al. MicroRNA-10 and -221 modulate differential expression of Hippo signaling pathway in human astroglial tumors. Cancer Treat. Res. Commun. 2020, 24, 100203. [Google Scholar] [CrossRef]
- Cui, T.; Bell, E.H.; McElroy, J.; Becker, A.P.; Gulati, P.M.; Geurts, M.; Mladkova, N.; Gray, A.; Liu, K.; Yang, L.; et al. MiR-4516 predicts poor prognosis and functions as a novel oncogene via targeting PTPN14 in human glioblastoma. Oncogene 2019, 38, 2923–2936. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.D.; Fu, Z.; Zhou, Y.; Huang, X.; Jiang, X. Long non-coding RNA LINC00473/miR-195-5p promotes glioma progression via YAP1-TEAD1-Hippo signaling. Int. J. Oncol. 2020, 56, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wang, Q.; Wang, Y.; Huang, K.; Yang, C.; Li, Y.; Yi, K.; Kang, C. EGFR/c-myc axis regulates TGFβ/Hippo/Notch pathway via epigenetic silencing miR-524 in gliomas. Cancer Lett. 2017, 406, 12–21. [Google Scholar] [CrossRef]
- Ebrahimpour, A.; Sarfi, M.; Rezatabar, S.; Tehrani, S.S. Novel insights into the interaction between long non-coding RNAs and microRNAs in glioma. Mol. Cell. Biochem. 2021, 476, 2317–2335. [Google Scholar] [CrossRef]
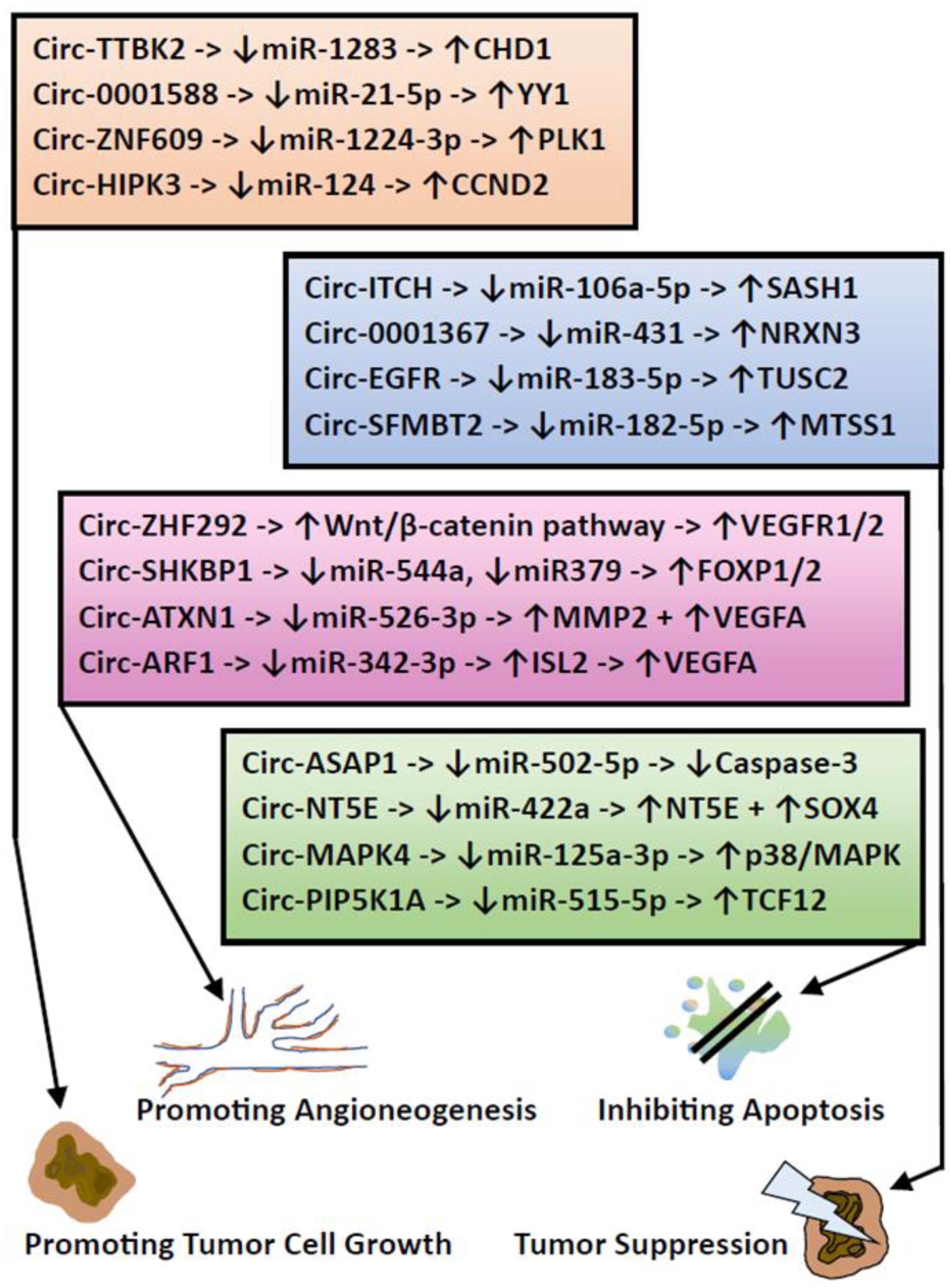
- Abi, A.; Ghaedi, K.; Khosravi, A.; Hayat, S.M.G. Circular RNAs and glioma: Small molecule with big actions. Curr. Mol. Med. 2021, 21, 25–44. [Google Scholar] [CrossRef]
- Han, C.; Wang, S.; Wang, H.; Zhang, J. Knockdown of circ-TTBK2 inhibits glioma progression by regulating miR-1283 and CHD1. Cancer Manag. Res. 2020, 12, 10055–10065. [Google Scholar] [CrossRef]
- Wang, Q.; Zheng, D.; Li, Y.; Zhang, Y.; Sui, R.; Chen, Y.; Liang, H.; Shi, J.; Pan, R.; Xu, X.; et al. Circular RNA circ_0001588 sponges miR-211-5p to facilitate the progression of glioblastoma via up-regulating YY1 expression. J. Gene Med. 2021, 23, e3371. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Li, H.; Lu, F.; Zhang, S.; Tang, J. Circular RNA ZNF609 promotes the malignant progression of glioma by regulating miR-1224-3p/PLK1 signaling. J. Cancer 2021, 12, 3354–3366. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, S.; Sun, H.; Bai, Y.; Song, Z.; Liu, X. Circular RNA CircHIPK3 elevates CCND2 expression and promotes cell proliferation and invasion through miR-124 in glioma. Front. Genet. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Yang, P.; Qiu, Z.; Jiang, Y.; Dong, L.; Yang, W.; Gu, C.; Li, G.; Zhu, Y. Silencing of cZNF292 circular RNA suppresses human glioma tube formation via the Wnt/β-catenin signaling pathway. Oncotarget 2016, 7, 63449–63455. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhao, L.; Liu, Y.; Liu, X.; Zheng, J.; Yu, H.; Cai, H.; Ma, J.; Liu, L.; Wang, P.; et al. Circ-SHKBP1 regulates the angiogenesis of U87 glioma-exposed endothelial cells through miR-544a/FOXP1 and miR-379/FOXP2 pathways. Mol. Ther. Nucleic Acids 2018, 10, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shen, S.; Zhu, L.; Su, R.; Zheng, J.; Ruan, X.; Shao, L.; Wang, D.; Yang, C.; Liu, Y. SRSF10 inhibits biogenesis of circ-ATXN1 to regulate glioma angiogenesis via miR-526b-3p/MMP2 pathway. J. Exp. Clin. Cancer Res. 2020, 39, 121. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhou, J.; Zhao, J.; Zhang, H.; Li, L.; Li, H.; Chen, L.; Hu, J.; Zheng, W.; Jing, Z. The U2AF2 /circRNA ARF1/miR-342-3p/ISL2 feedback loop regulates angiogenesis in glioma stem cells. J. Exp. Clin. Cancer Res. 2020, 39, 182. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lu, C.; Zhou, P.; Zhao, L.; Lyu, X.; Yin, J.; Shi, Z.; You, Y. EIF4A3-induced circular RNA ASAP1 promotes tumorigenesis and temozolomide resistance of glioblastoma via NRAS/MEK1/ERK1-2 signaling. Neuro Oncol. 2021, 23, 611–624. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, S.; Chen, X.; Li, N.; Li, J.; Jia, R.; Pan, Y.; Liang, H. CircNT5E Acts as a sponge of miR-422a to promote glioblastoma tumorigenesis. Cancer Res. 2018, 78, 4812–4825. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Huang, Z.; He, M.; Liao, J.; Zhang, Q.; Wang, S.; Xie, L.; Ouyang, L.; Koeffler, H.P.; Yin, D.; et al. Circular RNA MAPK4 (circ-MAPK4) inhibits cell apoptosis via MAPK signaling pathway by sponging miR-125a-3p in gliomas. Mol. Cancer 2020, 19, 17. [Google Scholar] [CrossRef]
- Zheng, K.; Xie, H.; Wu, W.; Wen, X.; Zeng, Z.; Shi, Y. CircRNA PIP5K1A promotes the progression of glioma through upregulation of the TCF12/PI3K/AKT pathway by sponging miR-515-5p. Cancer Cell Int. 2021, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, M.; Cui, S.T.; Zheng, Y.; Liu, Z.; Luo, L.S. CircRNA Circ-ITCH inhibits the proliferation and invasion of glioma cells through targeting the miR-106a-5p/SASH1 axis. Cell Transplant. 2021, 30, 963689720983785. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, P.; Dong, X.; Li, H.; Li, S.; Cheng, S.; Yuan, J.; Yang, X.; Qian, Z.; Dong, J. Circ_0001367 inhibits glioma proliferation, migration and invasion by sponging miR-431 and thus regulating NRXN3. Cell Death Dis. 2021, 12, 536. [Google Scholar] [CrossRef]
- Guo, Q.; Guo, J.; Liu, W.; Hu, S.; Hu, X.; Wang, Q.; Jiang, X. Circ-EGFR functions as an inhibitory factor in the malignant progression of glioma by regulating the miR-183-5p/TUSC2 axis. Cell. Mol. Neurobiol. 2022, 42, 2245–2256. [Google Scholar] [CrossRef]
- Zhang, S.; Qin, W.; Yang, S.; Guan, N.; Sui, X.; Guo, W. Circular RNA SFMBT2 inhibits the proliferation and metastasis of glioma cells through Mir-182-5p/Mtss1 pathway. Technol. Cancer Res. Treat. 2020, 19, 1533033820945799. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Graner, M.W. Roles of extracellular vesicles in high-grade gliomas: Tiny particles with outsized influence. Annu. Rev. Genomics. Hum. Genet. 2019, 20, 331–357. [Google Scholar] [CrossRef]
- Barthel, L.; Hadamitzky, M.; Dammann, P.; Schedlowski, M.; Sure, U.; Thakur, B.K.; Hetze, S. Glioma: Molecular signature and crossroads with tumor microenvironment. Cancer Metastasis Rev. 2022, 41, 53–75. [Google Scholar] [CrossRef]
- Guo, X.; Sui, R.; Piao, H. Tumor-derived small extracellular vesicles: Potential roles and mechanism in glioma. J. Nanobiotechnology 2022, 20, 383. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, Y.; Yao, B.; Sun, P.; Hao, Y.; Piao, H.; Zhao, X. Role of exosomes in the progression, diagnosis, and treatment of gliomas. Med. Sci. Monit. 2020, 26, e924023. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Cifola, I.; Caratelli, S.; Sconocchia, G.; D’Agnano, I.; Cenciarelli, C. Glioma extracellular vesicles for precision medicine: Prognostic and theragnostic application. Discov. Oncol. 2022, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Dong, H.; Cao, H.; Ji, X.; Luan, S.; Liu, J. Exosomes in pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Med. Sci. Monit. 2019, 25, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Nieland, L.; Morsett, L.M.; Broekman, M.L.D.; Breakefield, X.O.; Abels, E.R. Extracellular vesicle-mediated bilateral communication between glioblastoma and astrocytes. Trends Neurosci. 2021, 44, 215–226. [Google Scholar] [CrossRef]
- Lang, H.L.; Hu, G.W.; Chen, Y.; Liu, Y.; Tu, W.; Lu, Y.M.; Wu, L.; Xu, G.H. Glioma cells promote angiogenesis through the release of exosomes containing long non-coding RNA POU3F3. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 959–972. [Google Scholar]
- Sun, X.; Ma, X.; Wang, J.; Zhao, Y.; Wang, Y.; Bihl, J.C.; Chen, Y.; Jiang, C. Glioma stem cells-derived exosomes promote the angiogenic ability of endothelial cells through miR-21/VEGF signal. Oncotarget 2017, 8, 36137–36148. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Ge, X.; Shi, Z.; Yu, C.; Lu, C.; Wei, Y.; Zeng, A.; Wang, X.; Yan, W.; Zhang, J.; et al. Extracellular vesicles derived from hypoxic glioma stem-like cells confer temozolomide resistance on glioblastoma by delivering miR-30b-3p. Theranostics 2021, 11, 1763–1779. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhai, Y.; Hao, Y.; Wang, Q.; Han, F.; Zheng, W.; Hong, J.; Cui, L.; Jin, W.; Ma, S.; et al. Specific anti-glioma targeted-delivery strategy of engineered small extracellular vesicles dual-functionalised by Angiopep-2 and TAT peptides. J. Extracell. Vesicles 2022, 11, e12255. [Google Scholar] [CrossRef]
- Hong, S.; You, J.Y.; Paek, K.; Park, J.; Kang, S.J.; Han, E.H.; Choi, N.; Chung, S.; Rhee, W.J.; Kim, J.A. Inhibition of tumor progression and M2 microglial polarization by extracellular vesicle-mediated microRNA-124 in a 3D microfluidic glioblastoma microenvironment. Theranostics 2021, 11, 9687–9704. [Google Scholar] [CrossRef]
- Chen, X.; Chen, J.; Chen, Y.; You, H.; Lin, Y.; Wu, Z.; Kang, D.; Ding, C. Exosomal circular RNAs in glioma: Coexistence of opportunities and challenges for application. Chin. Med. J. Engl. 2022, 135, 1528–1530. [Google Scholar] [CrossRef]
- Gao, X.; Xia, X.; Li, F.; Zhang, M.; Zhou, H.; Wu, X.; Zhong, J.; Zhao, Z.; Zhao, K.; Liu, D.; et al. Circular RNA-encoded oncogenic E-cadherin variant promotes glioblastoma tumorigenicity through activation of EGFR-STAT3 signaling. Nat. Cell Biol. 2021, 23, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.H.; Yang, L.; Chen, J.X.; Li, Q.R.; Zhu, L.R.; Xu, Q.F.; Huang, G.H.; Zhang, Z.X.; Xiang, Y.; Du, L.; et al. Bortezomib inhibits growth and sensitizes glioma to temozolomide (TMZ) via down-regulating the FOXM1-Survivin axis. Cancer Commun. 2019, 39, 81. [Google Scholar] [CrossRef]
- Richard, Q.; Laurenge, A.; Mallat, M.; Sanson, M.; Castro-Vega, L.J. New insights into the immune TME of adult-type diffuse gliomas. Curr. Opin. Neurol. 2022, 35, 794–802. [Google Scholar] [CrossRef]
- Lang, F.; Liu, Y.; Chou, F.J.; Yang, C. Genotoxic therapy and resistance mechanism in gliomas. Pharmacol. Ther. 2021, 228, 107922. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Stagni, V.; Barila, D. Targeting the DNA damage response to overcome cancer drug resistance in glioblastoma. Int. J. Mol. Sci. 2020, 21, 4910. [Google Scholar] [CrossRef] [PubMed]
- Eich, M.; Roos, W.P.; Nikolova, T.; Kaina, B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol. Cancer Ther. 2013, 12, 2529–2540. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. Introduction to CNS tumours. In Central Nervous System Tumours, 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6, pp. 7–14. Available online: https://publications.iarc.fr/601 (accessed on 1 August 2023).
- Louis, D.N.; Aldape, K.D.; Capper, D.; Giannini, C.; Horbinski, C.M.; Ng, H.K.; Perry, A.; Reifenberger, G.; Sarkar, C.; Soffietti, R.; et al. Glioblastoma, IDH-wildtype. In Central Nervous System Tumours, 5th ed.; WHO Classification of Tumours Series; WHO Classification of Tumours Editorial Board; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6, pp. 39–55. Available online: https://publications.iarc.fr/601 (accessed on 1 August 2023).
- Brat, D.J.; Cahill, D.P.; Cimino, P.J.; Huse, J.T.; Kleinschmidt-DeMasters, B.K.; Komori, T.; Reuss, D.E.; Suva, M.L.; von Deimling, A.; Weller, M. Astrocytoma, IDH-mutant. In Central Nervous System Tumours, 5th ed.; WHO Classification of Tumours Series; WHO Classification of Tumours Editorial Board; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6, pp. 19–27. Available online: https://publications.iarc.fr/601 (accessed on 1 August 2023).
- Ryall, S.; Zapotocky, M.; Fukuoka, K.; Nobre, L.; Guerreiro-Stucklin, A.; Bennet, J.; Siddaway, R.; Li, C.; Pajovic, S.; Arnoldo, A.; et al. Integrated molecular and clinical analysis of 1000 pediatric low-grade gliomas. Cancer Cell 2020, 37, 569–583.e5. [Google Scholar] [CrossRef] [PubMed]
- Jacques, T.S.; Capper, D.; Giannini, C.; Orr, B.A.; Tabori, U. Diffuse low-grade glioma, MAPK pathway-altered. In Central Nervous System Tumours, 5th ed.; WHO Classification of Tumours Series; WHO Classification of Tumours Editorial Board; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6, pp. 65–68. Available online: https://publications.iarc.fr/601 (accessed on 1 August 2023).
- Korshunov, A.; Capper, D.; Jones, D.T.W.; Leske, H.; Orr, B.A.; Rodriguez, F.J.; Solomon, D.A.; Sturm, D.; Warren, K.E.; Weller, M. Diffuse hemispheric glioma, H3G34-mutant. In Central Nervous System Tumours, 5th ed.; WHO Classification of Tumours Series; WHO Classification of Tumours Editorial Board; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6, pp. 74–76. Available online: https://publications.iarc.fr/601 (accessed on 1 August 2023).
- Capper, D.; Jones, D.T.W.; Rodriguez, F.J.; Varlet, P. High-grade astrocytoma with piloid features. In Central Nervous System Tumours, 5th ed.; WHO Classification of Tumours Series; WHO Classification of Tumours Editorial Board; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6, pp. 90–93. Available online: https://publications.iarc.fr/601 (accessed on 1 August 2023).
- Brągiel-Pieczonka, A.; Lipka, G.; Stapinska-Syniec, A.; Czyzewski, M.; Zybura-Broda, K.; Sobstyl, M.; Rylski, M.; Grabiec, M. The profiles of TET-mediated DNA hydroxymethylation in human gliomas. Front. Oncol. 2022, 12, 621460. [Google Scholar] [CrossRef]
- Crake, R.L.I.; Burgess, E.R.; Wiggins, G.A.R.; Magon, N.J.; Das, A.B.; Vissers, M.C.M.; Morrin, H.R.; Royds, J.A.; Slatter, T.L.; Robinson, B.A.; et al. Ascorbate content of clinical glioma tissues is related to tumour grade and to global levels of 5-hydroxymethyl cytosine. Sci. Rep. 2022, 12, 14845. [Google Scholar] [CrossRef]
- Zhuo, S.; Chen, Z.; Yang, Y.; Zhang, J.; Tang, J.; Yang, K. Clinical and biological significances of a ferroptosis-related gene signature in glioma. Front. Oncol. 2020, 10, 590861. [Google Scholar] [CrossRef] [PubMed]
- Wan, R.J.; Peng, W.; Xia, Q.X.; Zhou, H.H.; Mao, X.Y. Ferroptosis-related gene signature predicts prognosis and immunotherapy in glioma. CNS Neurosci. Ther. 2021, 27, 973–986. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Ge, F.H.; Li, W.X.; Xu, Z.; Wang, X.L.; Shen, J.L.; Xu, A.B.; Hao, R.R. MicroRNA-147a targets SLC40A1 to induce ferroptosis in human glioblastoma. Anal. Cell. Pathol. 2022, 2022, 2843990. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, J.; Li, R.; Liu, Y.; Zhou, L.; Wang, C.; Lv, C.; Gao, L.; Cui, D. CircLRFN5 inhibits the progression of glioblastoma via PRRX2/GCH1 mediated ferroptosis. J. Exp. Clin. Cancer Res. 2022, 41, 307. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Gazzah, A.; Lassen, U.; Stein, A.; Wen, P.Y.; Dietrich, S.; de Jonge, M.J.A.; Blay, J.-Y.; et al. Dabrafenib plus trametinib in BRAFV600E-mutated rare cancers: The phase 2 ROAR trial. Nat. Med. 2023, 29, 1103–1112. [Google Scholar] [CrossRef]
- Bouffet, E.; Geoerger, B.; Moertel, C.; Whitlock, J.A.; Aerts, I.; Hargrave, D.; Osterloh, L.; Tan, E.; Choi, J.; Russo, M.; et al. Efficacy and safety of trametinib monotherapy or in combination with dabrafenib in pediatric BRAF V600-mutant Low-Grade Glioma. J. Clin. Oncol. 2023, 41, 664–674. [Google Scholar] [CrossRef]
- Narita, Y.; Muragaki, Y.; Kagawa, N.; Asai, K.; Nagane, M.; Matsuda, M.; Ueki, K.; Kuroda, J.; Date, I.; Kobayashi, H.; et al. Safety and efficacy of depatuxizumab mafodotin in Japanese patients with malignant glioma: A nonrandomized, phase 1/2 trial. Cancer Sci. 2021, 112, 5020–5033. [Google Scholar] [CrossRef]
- Martínez-García, M.; Velasco, G.; Pineda, E.; Gil-Gil, M.; Alameda, F.; Capellades, J.; Martín-Soberón, M.C.; López-Valero, I.; Ambel, E.T.; Foro, P.; et al. Safety and efficacy of crizotinib in combination with temozolomide and radiotherapy in patients with newly diagnosed glioblastoma: Phase Ib GEINO 1402 Trial. Cancers 2022, 14, 2393. [Google Scholar] [CrossRef]
- El-Khouly, F.E.; Veldhuijzen van Zanten, S.E.M.; Jansen, M.H.A.; Bakker, D.P.; Sanchez Aliaga, E.; Hendrikse, N.H.; Vandertop, W.P.; van Vuurden, D.G.; Kaspers, G.J.L. A phase I/II study of bevacizumab, irinotecan and erlotinib in children with progressive diffuse intrinsic pontine glioma. J. Neurooncol. 2021, 153, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Yuan, Y.; Wu, J.; Mendoza, T.; Vera, E.; Omuro, A.; Lieberman, F.; Robins, H.I.; Gerstner, E.R.; Wu, J.; et al. A phase II study of dose-dense temozolomide and lapatinib for recurrent low-grade and anaplastic supratentorial, infratentorial, and spinal cord ependymoma. Neuro Oncol. 2021, 23, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, S.; Mason, W.; MacNeil, M.; Harlos, C.; Tsang, R.; Sederias, J.; Luchman, H.A.; Weiss, S.; Rossiter, J.P.; Tu, D.; et al. A phase I study of vistusertib (dual mTORC1/2 inhibitor) in patients with previously treated glioblastoma multiforme: A CCTG study. Investig. New Drugs 2020, 38, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Li, S.; Lin, Y.; Li, Y.; Mao, Y.; Zhang, J.; Lei, T.; Wang, H.; Su, Y.; Yang, Y.; et al. A phase I dose-escalation study of SYHA1813, a VEGFR and CSF1R inhibitor, in patients with recurrent high-grade gliomas or advanced solid tumors. Investig. New Drugs 2023, 41, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Trippett, T.; Toledano, H.; Campbell Hewson, Q.; Verschuur, A.; Langevin, A.M.; Aerts, I.; Howell, L.; Gallego, S.; Rossig, C.; Smith, A.; et al. Cobimetinib in pediatric and young adult patients with relapsed or refractory solid tumors (iMATRIX-cobi): A multicenter, phase I/II study. Target. Oncol. 2022, 17, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Su, J.M.; Kilburn, L.B.; Mansur, D.B.; Krailo, M.; Buxton, A.; Adekunle, A.; Gajjar, A.; Adamson, P.C.; Weigel, B.; Fox, E.; et al. Phase I/II trial of vorinostat and radiation and maintenance vorinostat in children with diffuse intrinsic pontine glioma: A children’s oncology group report. Neuro Oncol. 2022, 24, 655–664. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Tumor | Diagnostic Morphological and Molecular Criteria |
|---|---|
| Glioblastoma, IDH-wildtype, and WHO grade 4 | Diffusely infiltrating astrocytic glioma with IDH-wildtype and no H3-histone alteration; at least one of the following five criteria: (1) microvascular proliferation, (2) necrosis, (3) TERT promotor mutation, (4) EGFR amplification, (5) chromosome +7/−10 copy number alterations. |
| Astrocytoma, IDH-mutant, and WHO grade 4 | Diffusely infiltrating astrocytic glioma with IDH1- or IDH2-mutation and without 1p/19q-codeletion; at least one of the following three criteria: (1) microvascular proliferation, (2) necrosis, (3) homozygous deletion of cyclin-dependent kinase 2A or 2B (CDKN2A/B). |
| Diffuse low-grade glioma, and MAPK pathway altered | Diffuse glioma without histological features of a high-grade glioma and without microvascular proliferation or necrosis; genetic alterations in the MAPK-pathway; IDH-wildtype and no H3-histone alterations; no homozygous deletion of CDKN2A/B. |
| Diffuse hemispheric glioma and H3G34-mutant | Infiltrative glioma with hemispheric location; missense mutation of the histone H3-3A with replacement of glycine (G) by arginine or valine at position 34 of histone H3.3 (H3G34-mutation); the DNA-methylation profile can support the diagnosis. |
| High-grade astrocytoma with piloid features | Astrocytic glioma with a specific DNA-methylation profile of a high-grade astrocytoma with piloid features; in addition, several gene mutations may occur, such as MAPK pathway mutations or homozygous deletion of CDKN2A/B, but the diagnosis is based solely on confirmation of the specific DNA methylation profile. |
| First Author, Year, and Phase of Study | Therapeutic Agent | Type of the Agent |
|---|---|---|
| Subbiah, 2023, Phase II [178] | Dabrafenib and Trametinib | BRAF kinase-inhibitor and MEK-inhibitor |
| The overall response rate was 54% for BRAFV600E mutant low-grade gliomas and 33% for BRAFV600E mutant high-grade gliomas. Adverse reactions occurred in the majority of patients, most commonly (>20%) fever and nausea. | ||
| Bouffet, 2022, Phase I/II [179] | Dabrafenib and Trametinib | BRAF kinase inhibitor and MEK-inhibitor |
| Adequate efficacy and tolerability in BRAF V600 mutated low-grade gliomas. Significantly fewer adverse reactions were observed in patients receiving combination therapy compared to patients receiving monotherapy (22% vs. 54%). | ||
| Narita, 2021, Phase I/II [180] | Depatux-M | EGFR-inhibitor |
| EGFR-modified high-grade gliomas treated with Depatux-M in combination with TMZ; mild efficacy and tolerable safety profiles were reported. | ||
| Martinez-Garcia, 2022, Phase I [181] | Crizotinib | MET and ALK-inhibitor |
| Combination of crizotinib with TMZ and radiotherapy. The combination therapy was well tolerated; median overall survival time in glioblastoma patients was 22.6 months, which was longer than expected. | ||
| El-Khouly, 2021, Phase I/II [182] | Bevacizumab and Irinotecan and Erlotinib | VEGF-inhibitor, Topoisomerase I-inhibitor, and EGFR-inhibitor |
| Children with diffuse midline glioma treated with a triple combination reported to be well tolerated; median overall survival was 13.8 months with long-term survivors up to 33 months. | ||
| Gilbert, 2021, Phase II [183] | Lapatinib | Inhibitor of MGMT and ErbB1/2 |
| Adult patients with low- and high-grade ependymoma treated with a combination of lapatinib and TMZ. Combination therapy was well tolerated; objective response with several complete or partial remissions and reduced symptom burden was reported. | ||
| Lapointe, 2020, Phase I [184] | Vistusertib | mTOR1/2-inhibitor |
| Combination of vistusertib with TMZ in GBM patients after first relapse, with favourable safety profile at doses tested. | ||
| Kang, 2023, Phase I [185] | SYHA1813 | VEGF inhibitor |
| In high-grade gliomas, a significant decrease in soluble VEGFR2, an increase in VEGFA, and preliminary anti-tumor activity have been observed; adverse reactions were moderate but acceptable. | ||
| Trippett, 2022, Phase I/II [186] | Cobimetinib | MEK-inhibitor |
| Several tumors with MAPK changes, a clinical response was observed, especially in patients with low-grade gliomas; the therapy was well tolerated. | ||
| Su, 2022, Phase I/II [187] | Vorinostat | Histone-deacetylase- inhibitor |
| Children with diffuse midline glioma were treated with vorinostat and radiation; the treatment was well tolerated but did not improve the outcome. | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nafe, R.; Hattingen, E. The Spectrum of Molecular Pathways in Gliomas—An Up-to-Date Review. Biomedicines 2023, 11, 2281. https://doi.org/10.3390/biomedicines11082281
Nafe R, Hattingen E. The Spectrum of Molecular Pathways in Gliomas—An Up-to-Date Review. Biomedicines. 2023; 11(8):2281. https://doi.org/10.3390/biomedicines11082281
Chicago/Turabian StyleNafe, Reinhold, and Elke Hattingen. 2023. "The Spectrum of Molecular Pathways in Gliomas—An Up-to-Date Review" Biomedicines 11, no. 8: 2281. https://doi.org/10.3390/biomedicines11082281
APA StyleNafe, R., & Hattingen, E. (2023). The Spectrum of Molecular Pathways in Gliomas—An Up-to-Date Review. Biomedicines, 11(8), 2281. https://doi.org/10.3390/biomedicines11082281