
Combination Therapy with Trastuzumab and Niraparib: Quantifying Early Proliferative Alterations in HER2+ Breast Cancer Models
 ,
,  , , and
, , and
Abstract
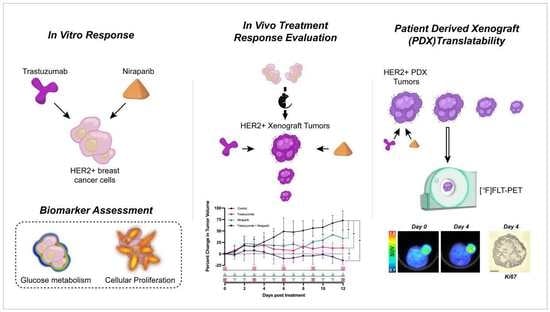
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Treatments, and In Vitro Assays
2.2. Animal Models
2.3. FLT–PET Imaging and Analysis
2.4. Histological Assessment
2.5. Statistics and Code
3. Results
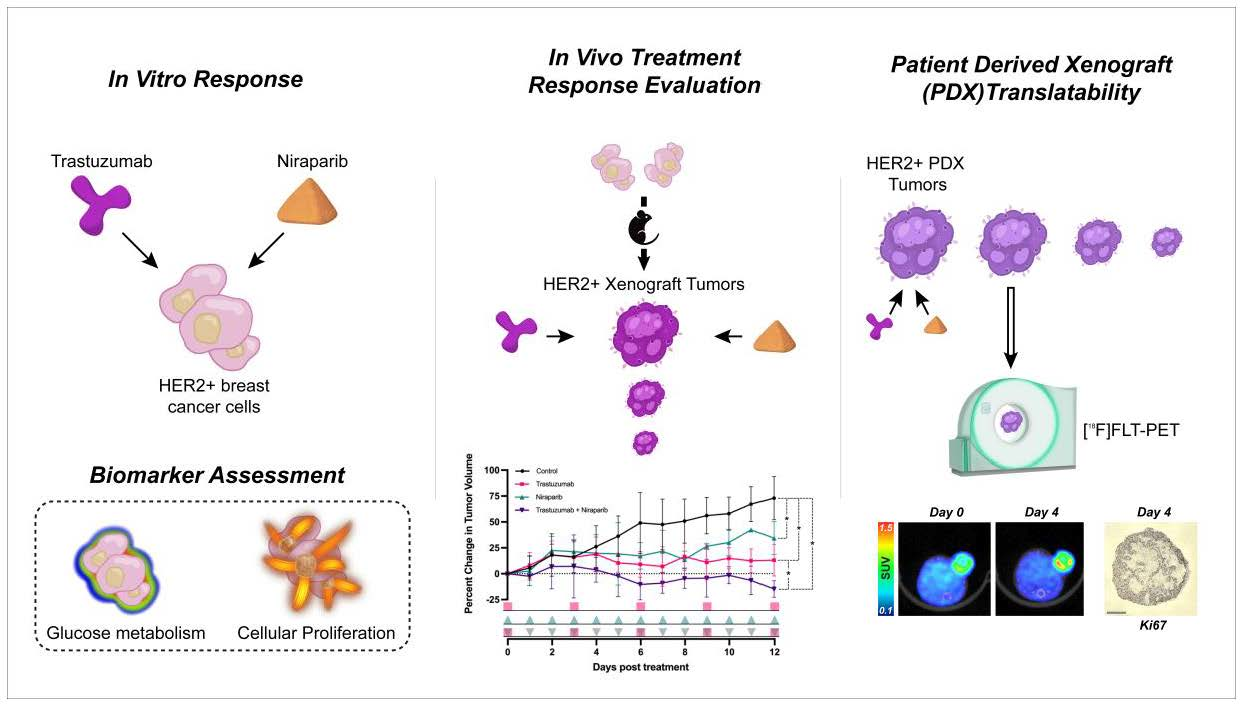
3.1. Trastuzumab and Niraparib Inhibited Growth in HER2+ Breast Cancer Cell Lines
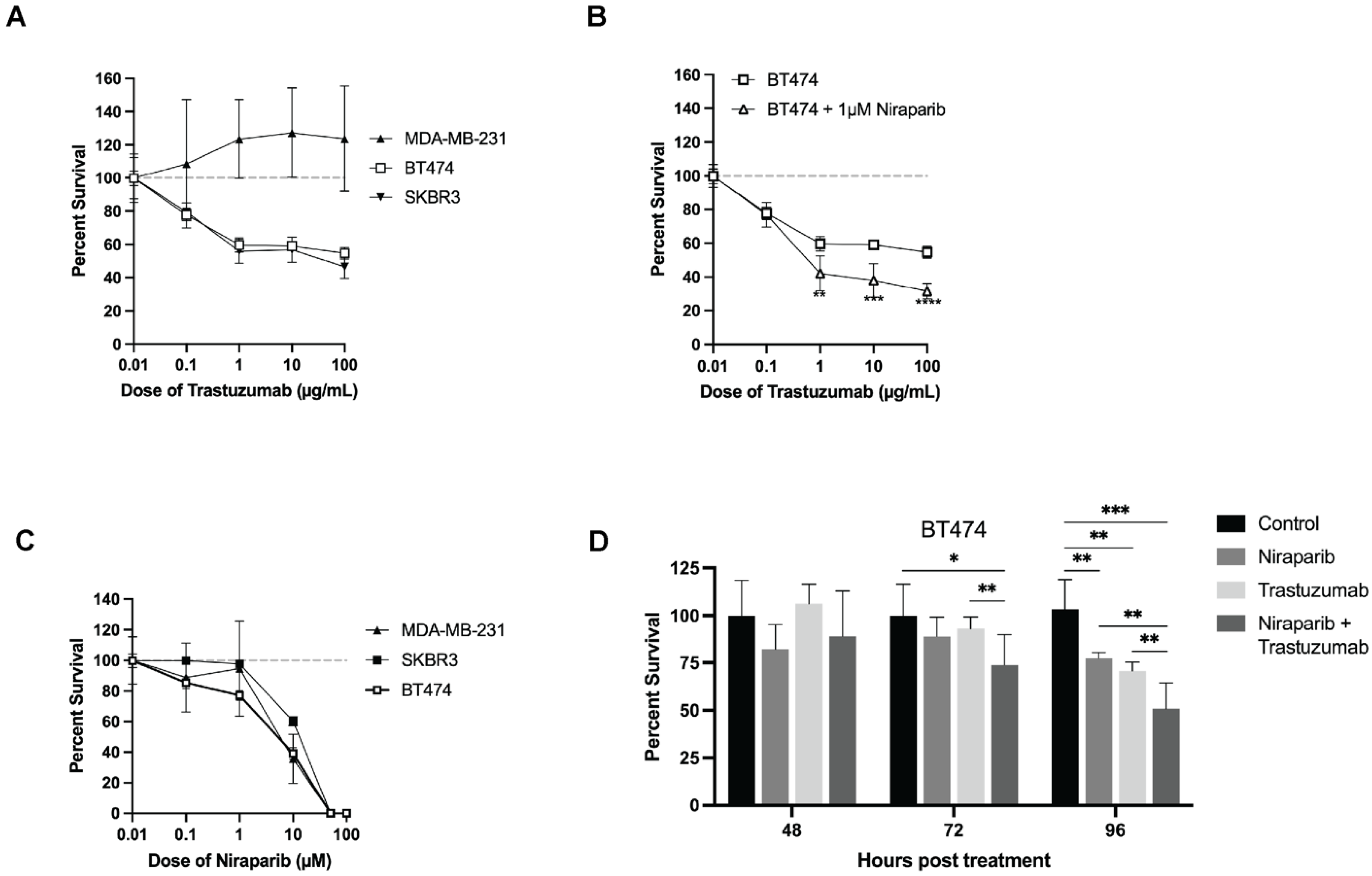
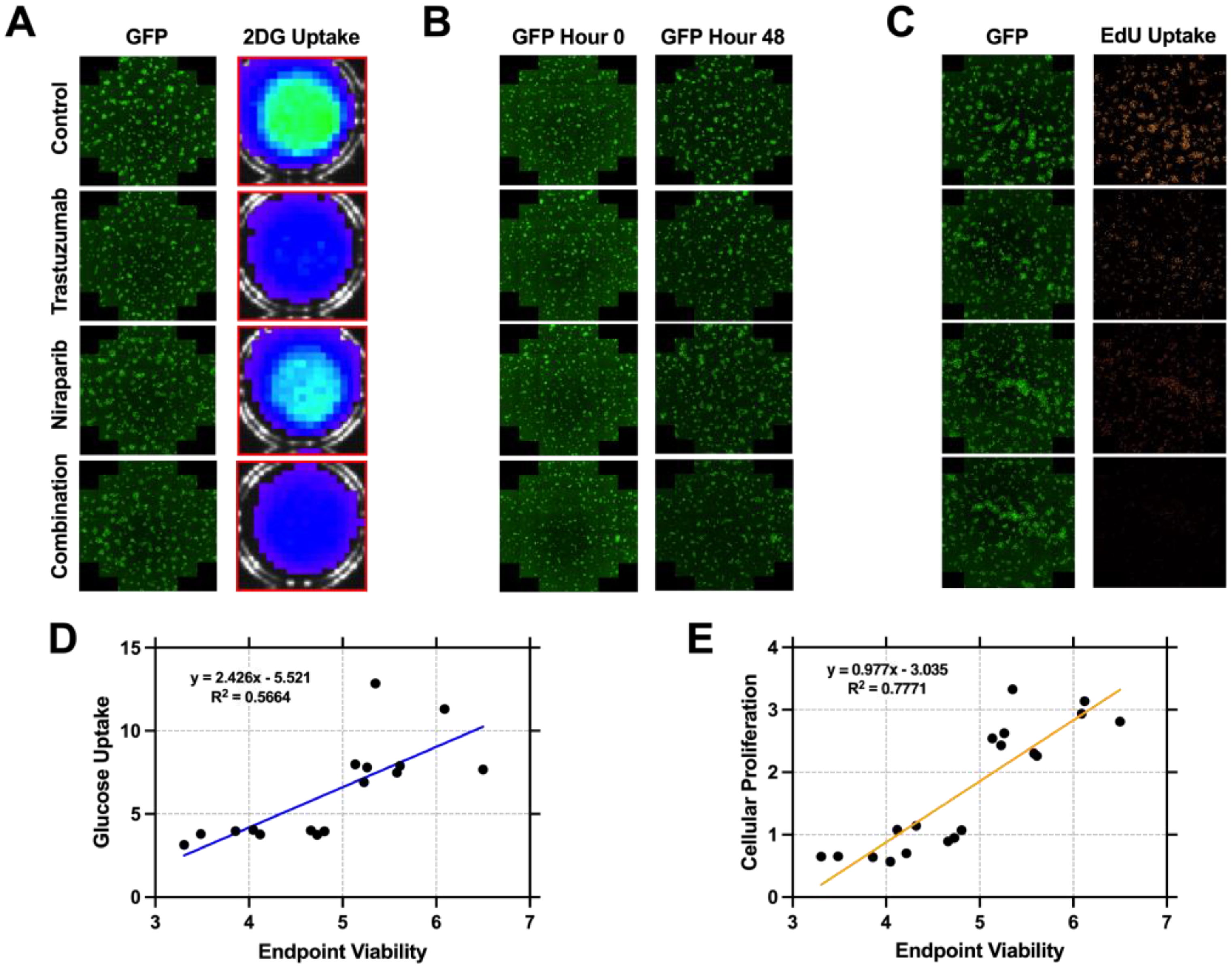
3.2. Cellular Proliferation Serves as a Metric of Early Response Evaluation In Vitro
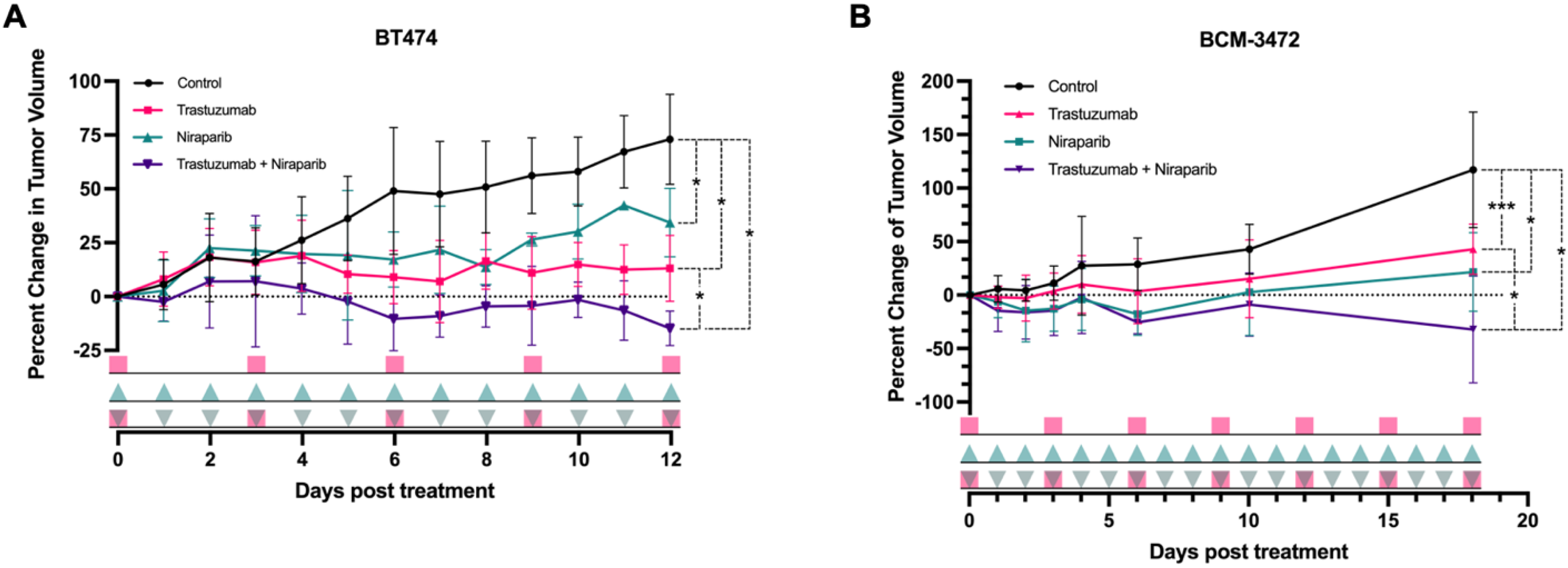
3.3. Trastuzumab and Niraparib Displayed Anti–Tumoral Effects in HER2+ Xenograft and PDX HER2+ Models
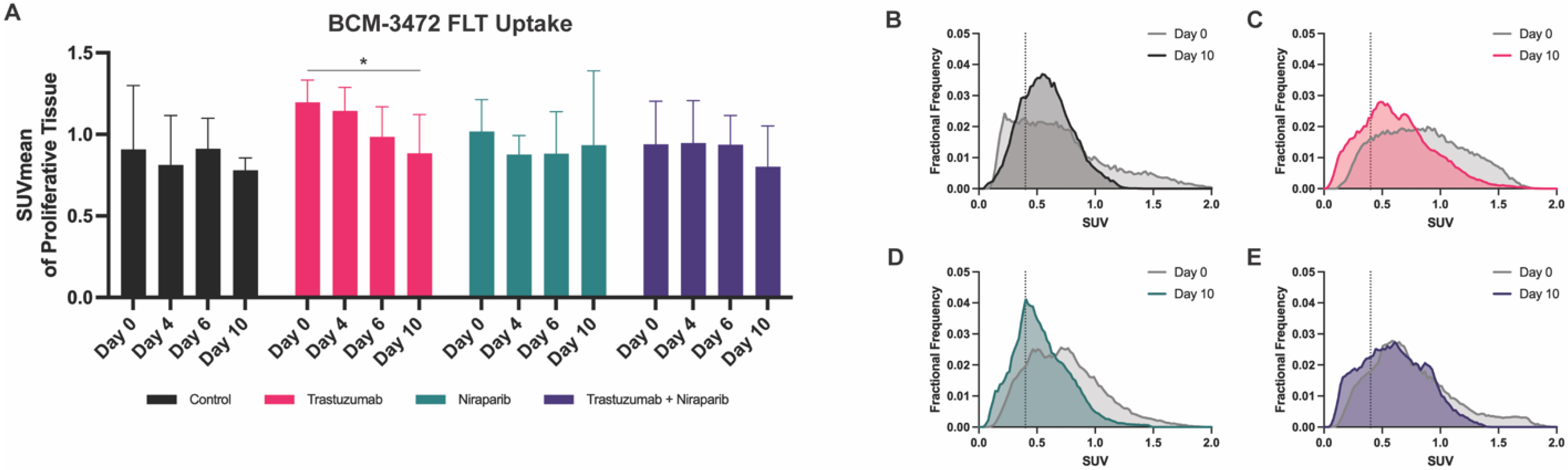
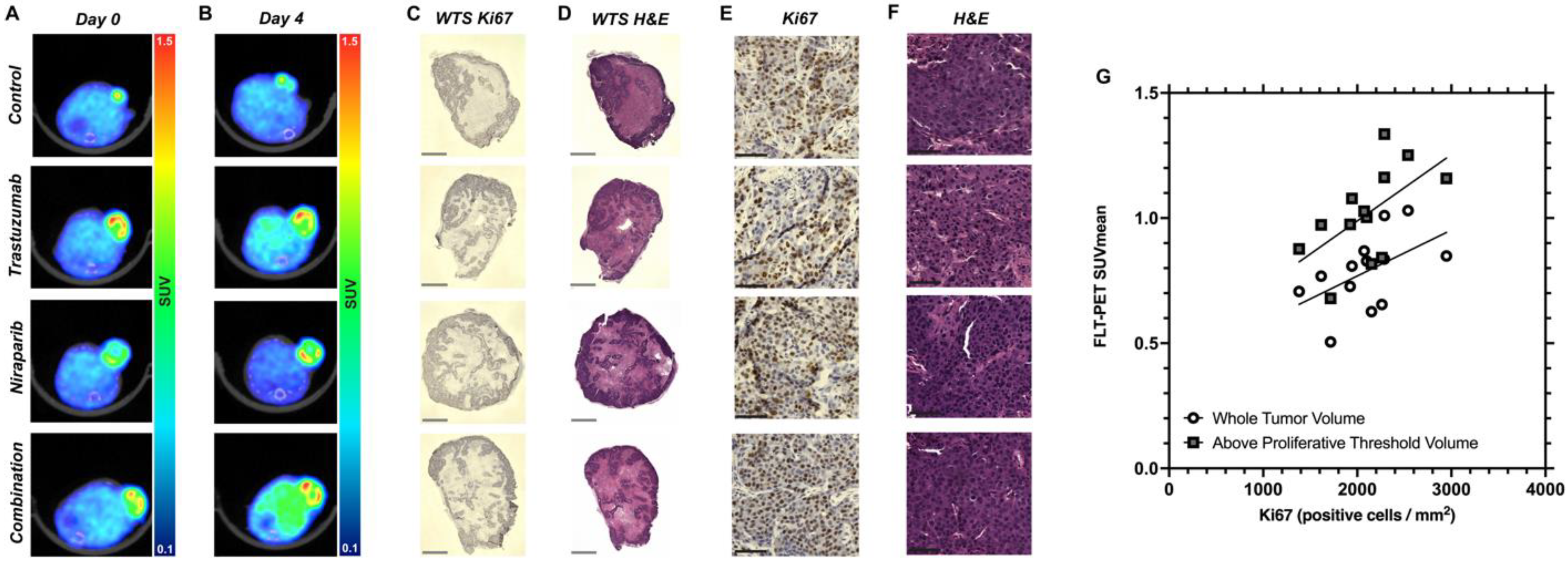
3.4. Decreasing Uptake of FLT–PET Observed in BCM–3472 PDX Model Prior to Significant Changes in Tumor Volume
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baselga, J.; Albanell, J. Mechanism of action of anti–HER2 monoclonal antibodies. Ann. Oncol. 2001, 12, S35–S41. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, P.R.; Mayer, I.A.; Mernaugh, R. Resistance to Trastuzumab in Breast Cancer. Clin. Cancer Res. 2009, 15, 7479–7491. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Nami, B.; Maadi, H.; Wang, Z. Mechanisms Underlying the Action and Synergism of Trastuzumab and Pertuzumab in Targeting HER2–Positive Breast Cancer. Cancers 2018, 10, 342. [Google Scholar] [CrossRef]
- Wilken, J.A.; Maihle, N.J. Primary trastuzumab resistance: New tricks for an old drug. Ann. N. Y. Acad. Sci. 2010, 1210, 53–65. [Google Scholar] [CrossRef]
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940. [Google Scholar] [CrossRef] [PubMed]
- Wielgos, M.E.; Zhang, Z.; Rajbhandari, R.; Cooper, T.S.; Zeng, L.; Forero, A.; Esteva, F.J.; Osborne, C.K.; Schiff, R.; LoBuglio, A.F.; et al. Trastuzumab–Resistant HER2+ Breast Cancer Cells Retain Sensitivity to Poly (ADP–Ribose) Polymerase (PARP) Inhibition. Mol. Cancer Ther. 2018, 17, 921–930. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui–Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP–Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Yap, T.A.; de Bono, J.S. Poly(ADP–ribose) polymerase inhibitors in cancer treatment: A clinical perspective. Eur. J. Cancer 2010, 46, 9–20. [Google Scholar] [CrossRef]
- Nowsheen, S.; Cooper, T.; Bonner, J.A.; LoBuglio, A.F.; Yang, E.S. HER2 Overexpression Renders Human Breast Cancers Sensitive to PARP Inhibition Independently of Any Defect in Homologous Recombination DNA Repair. Cancer Res. 2012, 72, 4796–4806. [Google Scholar] [CrossRef] [PubMed]
- Veuger, S.J.; Hunter, J.E.; Durkacz, B.W. Ionizing radiation–induced NF–κB activation requires PARP–1 function to confer radioresistance. Oncogene 2009, 28, 832–842. [Google Scholar] [CrossRef]
- Kitamura, T.; Sekimata, M.; Kikuchi, S.-I.; Homma, Y. Involvement of poly(ADP–ribose) polymerase 1 in ERBB2 expression in rheumatoid synovial cells. Am. J. Physiol. Physiol. 2005, 289, C82–C88. [Google Scholar] [CrossRef]
- Li, J.; Holzschu, D.L.; Sugiyama, T. PCNA is efficiently loaded on the DNA recombination intermediate to modulate polymerase δ, η, and ζ activities. Proc. Natl. Acad. Sci. USA 2013, 110, 7672–7677. [Google Scholar] [CrossRef]
- García–Parra, J.; Dalmases, A.; Morancho, B.; Arpí, O.; Menendez, S.; Sabbaghi, M.; Zazo, S.; Chamizo, C.; Madoz, J.; Eroles, P.; et al. Poly (ADP–ribose) polymerase inhibition enhances trastuzumab antitumour activity in HER2 overexpressing breast cancer. Eur. J. Cancer 2014, 50, 2725–2734. [Google Scholar] [CrossRef] [PubMed]
- Stringer–Reasor, E.M.; Li, Y.; Witherspoon, F.; Specht, J.M.; Mesias, J.D.S.A.; Nanda, R.; Dees, E.C.; Wolff, A.C.; Krop, I.E.; Lin, N.U.; et al. Trial in progress: A phase 1b/2 study of the PARP inhibitor niraparib in combination with trastuzumab in patients with metastatic HER2+ breast cancer (TBCRC 050). J. Clin. Oncol. 2021, 39, TPS1098. [Google Scholar] [CrossRef]
- Whisenant, J.G.; McIntyre, J.O.; Peterson, T.E.; Kang, H.; Sánchez, V.; Manning, H.C.; Arteaga, C.L.; Yankeelov, T.E. Utility of 18FLT–PET to Assess Treatment Response in Trastuzumab–resistant and Sensitive HER2–overexpressingHuman Breast Cancer Xenografts. Mol. Imaging Biol. 2015, 17, 119–128. [Google Scholar] [CrossRef][Green Version]
- Sorace, A.G.; Elkassem, A.A.; Galgano, S.J.; Lapi, S.E.; Larimer, B.M.; Partridge, S.C.; Quarles, C.C.; Reeves, K.; Napier, T.S.; Song, P.N.; et al. Imaging for Response Assessment in Cancer Clinical Trials. Semin. Nucl. Med. 2020, 50, 488–504. [Google Scholar] [CrossRef]
- Shah, C.; Miller, T.W.; Wyatt, S.K.; McKinley, E.T.; Olivares, M.G.; Sanchez, V.; Nolting, D.D.; Buck, J.R.; Zhao, P.; Ansari, M.S.; et al. Imaging Biomarkers Predict Response to Anti–HER2 (ErbB2) Therapy in Preclinical Models of Breast Cancer. Clin. Cancer Res. 2009, 15, 4712–4721. [Google Scholar] [CrossRef]
- Wesolowski, R.; Stover, D.G.; Lustberg, M.B.; Shoben, A.; Zhao, M.; Mrozek, E.; Layman, R.M.; Macrae, E.; Duan, W.; Zhang, J.; et al. Phase I Study of Veliparib on an Intermittent and Continuous Schedule in Combination with Carboplatin in Metastatic Breast Cancer: A Safety and [18F]–Fluorothymidine Positron Emission Tomography Biomarker Study. Oncologist 2020, 25, e1158–e1169. [Google Scholar] [CrossRef]
- Fujii, E.; Kato, A.; Suzuki, M. Patient–derived xenograft (PDX) models: Characteristics and points to consider for the process of establishment. J. Toxicol. Pathol. 2020, 33, 153–160. [Google Scholar] [CrossRef]
- Chen, J.; Jin, Y.; Li, S.; Qiao, C.; Peng, X.; Li, Y.; Gu, Y.; Wang, W.; You, Y.; Yin, J.; et al. Patient–Derived Xenografts Are a Reliable Preclinical Model for the Personalized Treatment of Epithelial Ovarian Cancer. Front. Oncol. 2021, 11, 744256. [Google Scholar] [CrossRef] [PubMed]
- Marchand, P.; Ouadi, A.; Pellicioli, M.; Schuler, J.; Laquerriere, P.; Boisson, F.; Brasse, D. Automated and efficient radiosynthesis of [18F]FLT using a low amount of precursor. Nucl. Med. Biol. 2016, 43, 520–527. [Google Scholar] [CrossRef]
- Oh, S.J.; Mosdzianowski, C.; Chi, D.Y.; Kim, J.Y.; Kang, S.H.; Ryu, J.S.; Yeo, J.S.; Moon, D.H. Fully automated synthesis system of 3′–deoxy–3′–[18F]fluorothymidine. Nucl. Med. Biol. 2004, 31, 803–809. [Google Scholar] [CrossRef]
- Machulla, H.-J.; Blocher, A.; Kuntzsch, M.; Piert, M.; Wei, R.; Grierson, J. Simplified Labeling Approach for Synthesizing 3′–Deoxy–3′–[18F]fluorothymidine ([18F]FLT). J. Radioanal. Nucl. Chem. 2000, 243, 843–846. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Schmidt, U.; Weigert, M.; Broaddus, C.; Myers, G. Cell Detection with Star–convex Polygons. In Medical Image Computing and Computer Assisted Intervention, Proceedings of the MICCAI 2018: 21st International Conference, Granada, Spain, 16–20 September 2018; Springer International Publishing: Berlin, Germany, 2018; Volume 11071, pp. 265–273. [Google Scholar] [CrossRef]
- Zazo, S.; González–Alonso, P.; Martín–Aparicio, E.; Chamizo, C.; Cristóbal, I.; Arpí, O.; Rovira, A.; Albanell, J.; Eroles, P.; Lluch, A.; et al. Generation, characterization, and maintenance of trastuzumab–resistant HER2+ breast cancer cell lines. Am. J. Cancer Res. 2016, 6, 2661–2678. [Google Scholar] [PubMed]
- Barthel, H.; Cleij, M.C.; Collingridge, D.R.; Hutchinson, O.C.; Osman, S.; He, Q.; Luthra, S.K.; Brady, F.; Price, P.M.; Aboagye, E.O. 3′–deoxy–3′–[18F]fluorothymidine as a new marker for monitoring tumor response to antiproliferative therapy in vivo with positron emission tomography. Cancer Res. 2003, 63, 3791–3798. [Google Scholar]
- Leyton, J.; Latigo, J.R.; Perumal, M.; Dhaliwal, H.; He, Q.; Aboagye, E.O. Early Detection of Tumor Response to Chemotherapy by 3′–Deoxy–3′–[18F]Fluorothymidine Positron Emission Tomography: The Effect of Cisplatin on a Fibrosarcoma Tumor Model In vivo. Cancer Res. 2005, 65, 4202–4210. [Google Scholar] [CrossRef]
- Apisarnthanarax, S.; Alauddin, M.M.; Mourtada, F.; Ariga, H.; Raju, U.; Mawlawi, O.; Han, D.; Bornmann, W.G.; Ajani, J.A.; Milas, L.; et al. Early Detection of Chemoradioresponse in Esophageal Carcinoma by 3′–Deoxy–3′–3H–Fluorothymidine Using Preclinical Tumor Models. Clin. Cancer Res. 2006, 12, 4590–4597. [Google Scholar] [CrossRef] [PubMed]
- Molthoff, C.F.M.; Klabbers, B.M.; Berkhof, J.; Felten, J.T.; van Gelder, M.; Windhorst, A.D.; Slotman, B.J.; Lammertsma, A.A. Monitoring Response to Radiotherapy in Human Squamous Cell Cancer Bearing Nude Mice: Comparison of 2′–deoxy–2′–[18F]fluoro–d–glucose (FDG) and 3′–[18F]fluoro–3′–deoxythymidine (FLT). Mol. Imaging Biol. 2007, 9, 340–347. [Google Scholar] [CrossRef]
- Waldherr, C.; Mellinghoff, I.K.; Tran, C.; Halpern, B.S.; Rozengurt, N.; Safaei, A.; Weber, W.A.; Stout, D.; Satyamurthy, N.; Barrio, J.; et al. Monitoring antiproliferative responses to kinase inhibitor therapy in mice with 3’–deoxy–3’–18F–fluorothymidine PET. J. Nucl. Med. 2005, 46, 114–120. [Google Scholar]
- Wei, L.H.; Su, H.; Hildebrandt, I.J.; Phelps, M.E.; Czernin, J.; Weber, W.A. Changes in Tumor Metabolism as Readout for Mammalian Target of Rapamycin Kinase Inhibition by Rapamycin in Glioblastoma. Clin. Cancer Res. 2008, 14, 3416–3426. [Google Scholar] [CrossRef]
- Ullrich, R.T.; Zander, T.; Neumaier, B.; Koker, M.; Shimamura, T.; Waerzeggers, Y.; Borgman, C.L.; Tawadros, S.; Li, H.; Sos, M.L.; et al. Early detection of erlotinib treatment response in NSCLC by 3′–deoxy–3′–[F]–fluoro–L–thymidine ([F]FLT) positron emission tomography (PET). PLoS ONE 2008, 3, e3908. [Google Scholar] [CrossRef]
- Brepoels, L.; Stroobants, S.; Verhoef, G.; De Groot, T.; Mortelmans, L.; De Wolf–Peeters, C. 18F–FDG and 18F–FLT Uptake Early after Cyclophosphamide and mTOR Inhibition in an Experimental Lymphoma Model. J. Nucl. Med. 2009, 50, 1102–1109. [Google Scholar] [CrossRef][Green Version]
- Weber, W.A. Monitoring Tumor Response to Therapy with 18F–FLT PET. J. Nucl. Med. 2010, 51, 841–844. [Google Scholar] [CrossRef][Green Version]
- Syed, A.K.; Whisenant, J.G.; Barnes, S.L.; Sorace, A.G.; Yankeelov, T.E. Multiparametric Analysis of Longitudinal Quantitative MRI Data to Identify Distinct Tumor Habitats in Preclinical Models of Breast Cancer. Cancers 2020, 12, 1682. [Google Scholar] [CrossRef]


 pink squares), followed by the niraparib cohort (
pink squares), followed by the niraparib cohort ( green triangles), and last, the combination cohort (
green triangles), and last, the combination cohort ( pink squares with green triangles).
pink squares), followed by the niraparib cohort ( green triangles), and last, the combination cohort ( pink squares with green triangles).
pink squares with green triangles).
pink squares), followed by the niraparib cohort ( green triangles), and last, the combination cohort ( pink squares with green triangles).


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansur, A.; Song, P.N.; Lu, Y.; Burns, A.C.; Sligh, L.; Yang, E.S.; Sorace, A.G. Combination Therapy with Trastuzumab and Niraparib: Quantifying Early Proliferative Alterations in HER2+ Breast Cancer Models. Biomedicines 2023, 11, 2090. https://doi.org/10.3390/biomedicines11082090
Mansur A, Song PN, Lu Y, Burns AC, Sligh L, Yang ES, Sorace AG. Combination Therapy with Trastuzumab and Niraparib: Quantifying Early Proliferative Alterations in HER2+ Breast Cancer Models. Biomedicines. 2023; 11(8):2090. https://doi.org/10.3390/biomedicines11082090
Chicago/Turabian StyleMansur, Ameer, Patrick N. Song, Yun Lu, Andrew C. Burns, Luke Sligh, Eddy S. Yang, and Anna G. Sorace. 2023. "Combination Therapy with Trastuzumab and Niraparib: Quantifying Early Proliferative Alterations in HER2+ Breast Cancer Models" Biomedicines 11, no. 8: 2090. https://doi.org/10.3390/biomedicines11082090
APA StyleMansur, A., Song, P. N., Lu, Y., Burns, A. C., Sligh, L., Yang, E. S., & Sorace, A. G. (2023). Combination Therapy with Trastuzumab and Niraparib: Quantifying Early Proliferative Alterations in HER2+ Breast Cancer Models. Biomedicines, 11(8), 2090. https://doi.org/10.3390/biomedicines11082090