
Integrative CRISPR Activation and Small Molecule Inhibitor Screening for lncRNA Mediating BRAF Inhibitor Resistance in Melanoma
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Targeted Arrayed sgRNA Design, Synthesis, and Plasmid Library Preparation
2.3. Lentivirus Generation
2.4. SAM Cas9 Expressing SKMEL-239 Stable Cell Line Generation
2.5. RNA Extraction, Library Preparation, and Sequencing
2.6. RNA-Seq Computational Analysis
2.7. Chromatin-Immunoprecipitation Sequencing (ChIP-Seq)
2.8. Cell Viability Assay
2.9. RNA Isolation and qRT-PCR
2.10. Western Blot Analysis
2.11. Small Molecule Inhibitor Cell Survival Assay (COMAS)
2.12. Statistics and Reproducibility
3. Results
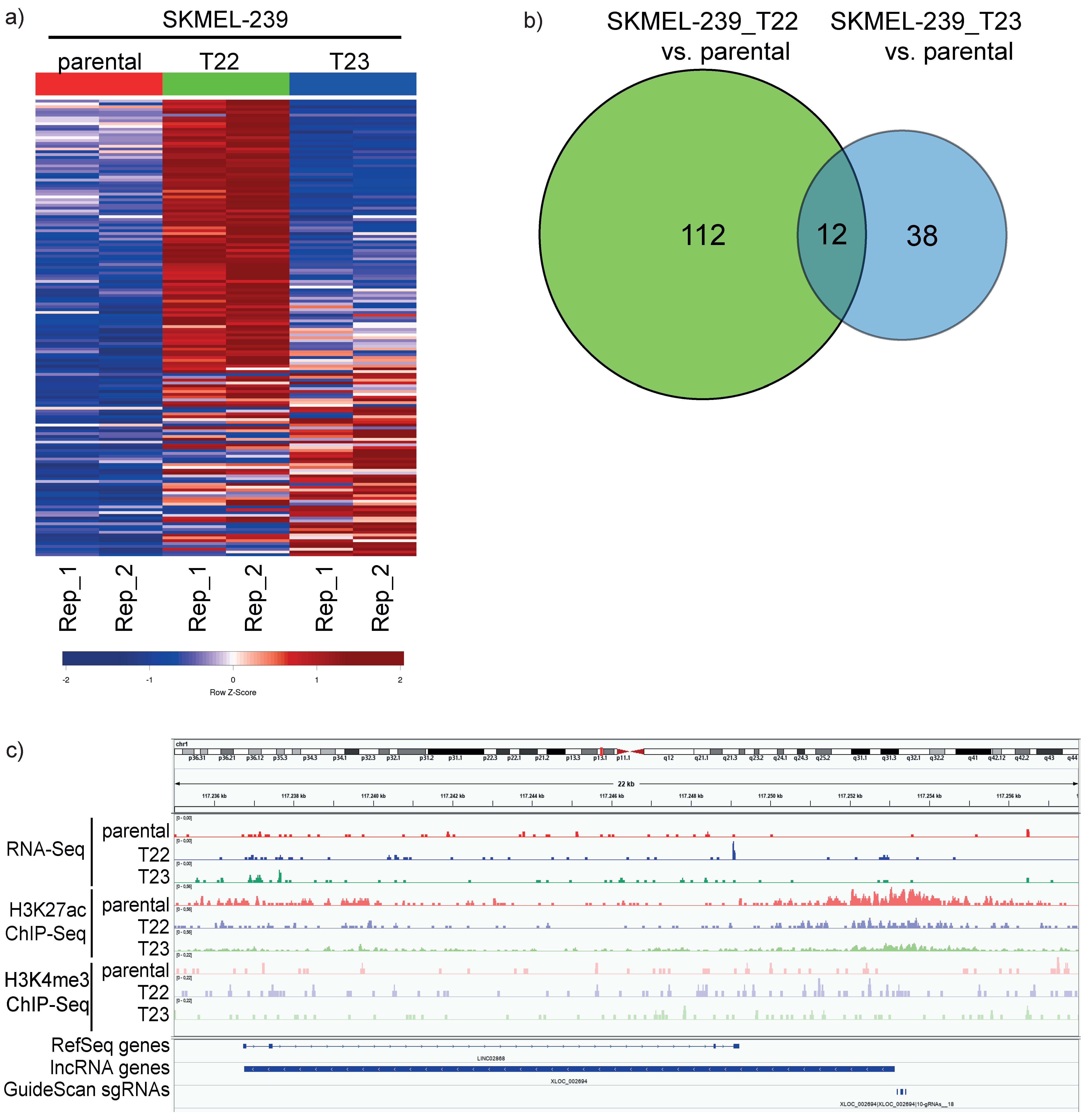
3.1. Integrated Analysis of RNA-Seq and CHIP-Seq for lncRNA Discovery in Vemurafenib-Resistant Melanoma Cell Lines
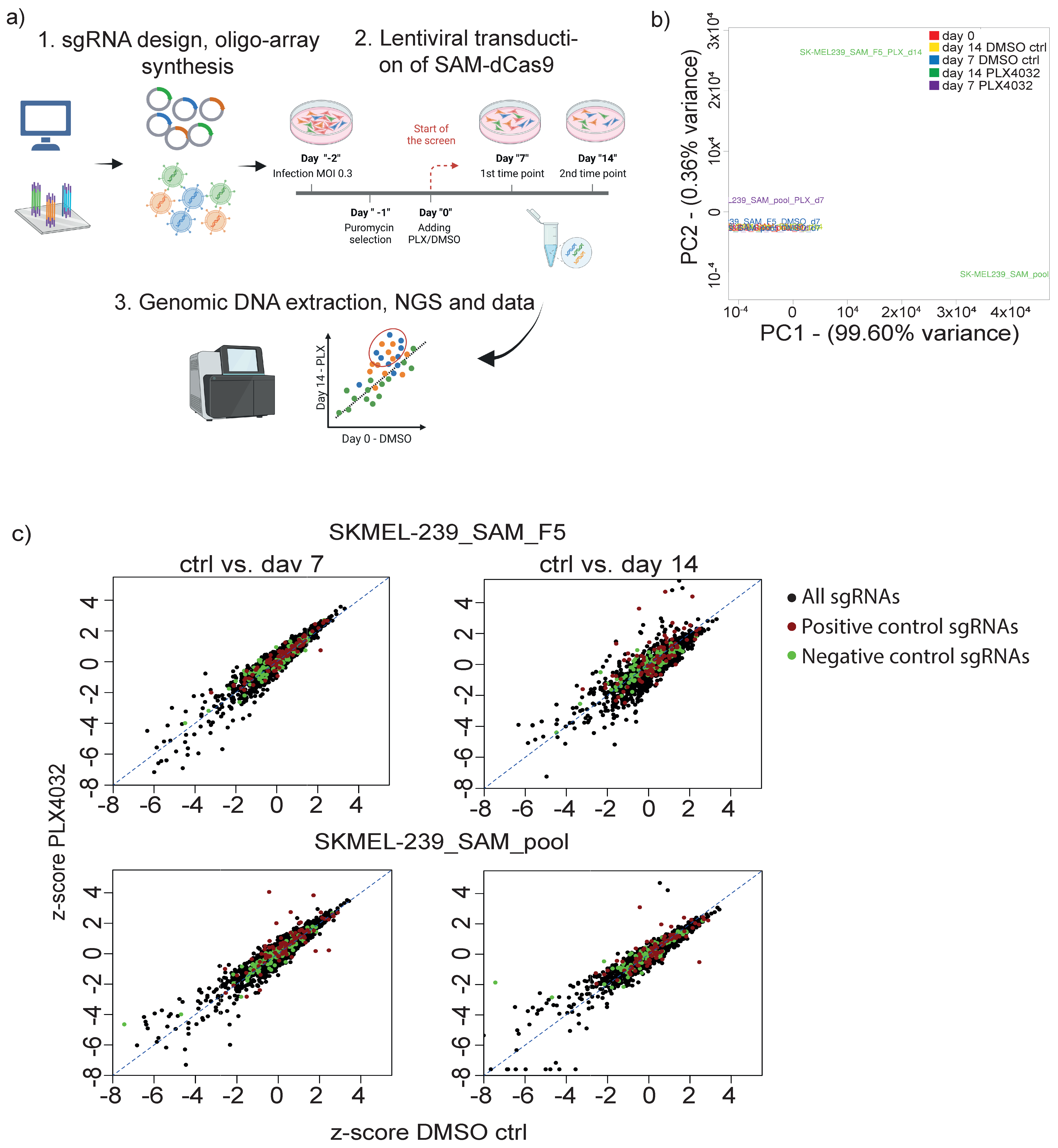
3.2. Targeted CRISPRa Screening Approach to Identify Putative lncRNAs Involved in Vemurafenib Resistance
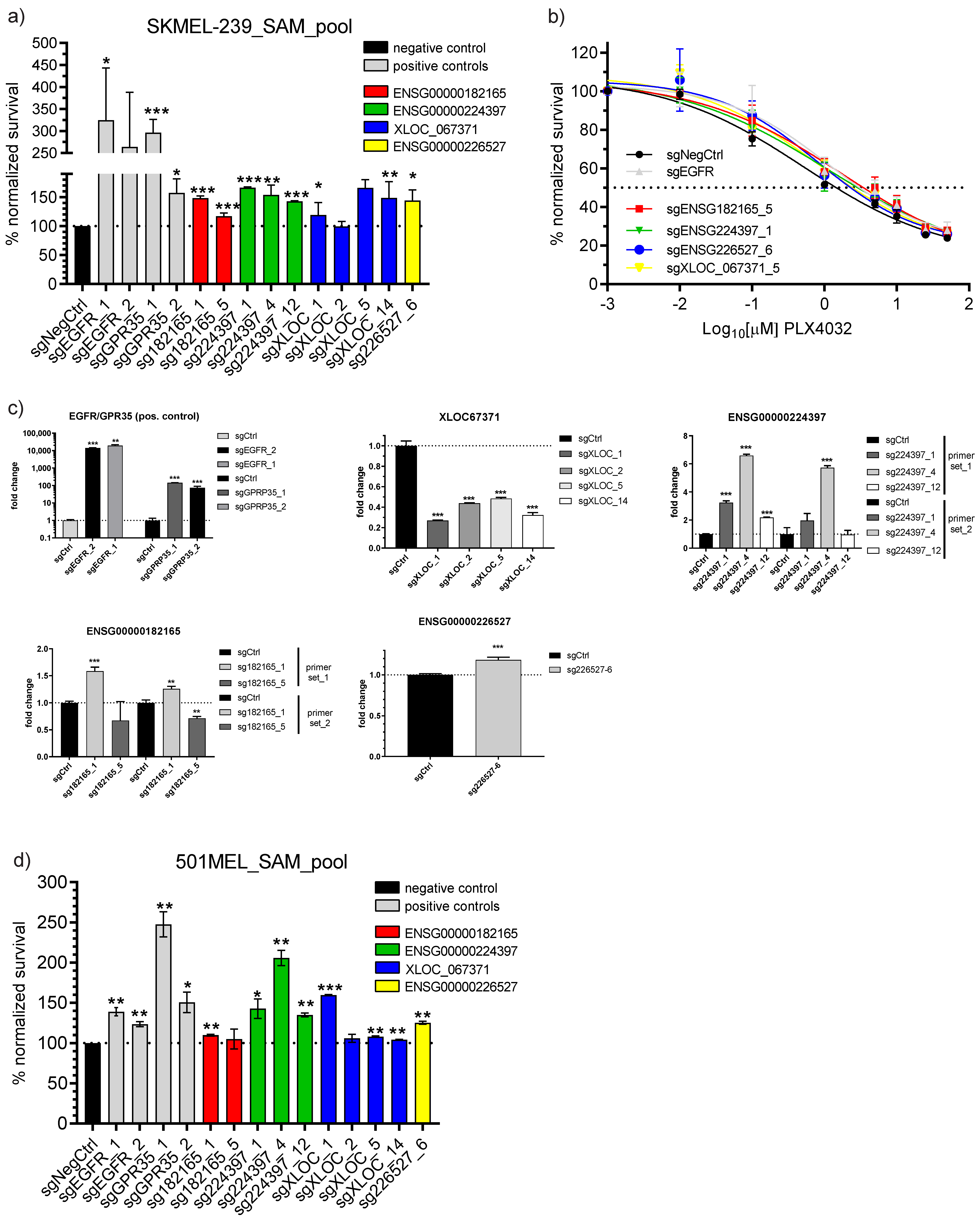
3.3. Single sgRNA Vemurafenib Resistance Cell Survival Validation
3.4. Broad Panel Inhibitor Screen to Identify Potential lncRNA Mode of Action and Rescue Therapeutic Windows
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Lashway, S.G.; Harris, R.B.; Farland, L.V.; O’Rourke, M.K.; Dennis, L.K. Age and Cohort Trends of Malignant Melanoma in the United States. Cancers 2021, 13, 3866. [Google Scholar] [CrossRef] [PubMed]
- Sacchetto, L.; Zanetti, R.; Comber, H.; Bouchardy, C.; Brewster, D.H.; Broganelli, P.; Chirlaque, M.D.; Coza, D.; Galceran, J.; Gavin, A.; et al. Trends in incidence of thick, thin and in situ melanoma in Europe. Eur. J. Cancer 2018, 92, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment. Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Rosen, N. Mutant BRAF melanomas—Dependence and resistance. Cancer Cell 2011, 19, 11–15. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.C.; Ni, J.J.; Cui, W.Y.; Wang, B.Y.; Zhuo, W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef]
- Goding, C.R. Targeting the lncRNA SAMMSON Reveals Metabolic Vulnerability in Melanoma. Cancer Cell 2016, 29, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.; Serafim, R.B.; Kawakami, A.; Goncalves Pereira, C.; Roszik, J.; Valente, V.; Vazquez, V.L.; Fisher, D.E.; Espreafico, E.M. The lncRNA RMEL3 protects immortalized cells from serum withdrawal-induced growth arrest and promotes melanoma cell proliferation and tumor growth. Pigment. Cell Melanoma Res. 2019, 32, 303–314. [Google Scholar] [CrossRef]
- Wu, C.F.; Tan, G.H.; Ma, C.C.; Li, L. The non-coding RNA llme23 drives the malignant property of human melanoma cells. J. Genet. Genom. 2013, 40, 179–188. [Google Scholar] [CrossRef]
- Sarkar, D.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Epigenetic regulation in human melanoma: Past and future. Epigenetics 2015, 10, 103–121. [Google Scholar] [CrossRef]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.Y.; Shang, J.; Xu, Y.D. LncRNA SNHG5 promotes cisplatin resistance in gastric cancer via inhibiting cell apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4185–4191. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zeng, T.; Lin, Z.; Yan, L.; Wang, F.; Tang, L.; Wang, L.; Tang, D.; Chen, P.; Yang, M. Long non-coding RNA SNHG5 regulates chemotherapy resistance through the miR-32/DNAJB9 axis in acute myeloid leukemia. Biomed. Pharmacother. 2020, 123, 109802. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qian, J.; Li, J.; Zhu, C. Knockdown of lncRNA-HOTAIR downregulates the drug-resistance of breast cancer cells to doxorubicin via the PI3K/AKT/mTOR signaling pathway. Exp. Ther. Med. 2019, 18, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Wang, Y.; Xu, Y.N.; Lu, Q.Y. Overexpression of LncRNA-HOTAIR promotes chemoresistance in acute leukemia cells. Int. J. Clin. Exp. Pathol. 2020, 13, 3044–3051. [Google Scholar]
- Bester, A.C.; Lee, J.D.; Chavez, A.; Lee, Y.R.; Nachmani, D.; Vora, S.; Victor, J.; Sauvageau, M.; Monteleone, E.; Rinn, J.L.; et al. An Integrated Genome-wide CRISPRa Approach to Functionalize lncRNAs in Drug Resistance. Cell 2018, 173, 649–664.e20. [Google Scholar] [CrossRef]
- Joung, J.; Engreitz, J.M.; Konermann, S.; Abudayyeh, O.O.; Verdine, V.K.; Aguet, F.; Gootenberg, J.S.; Sanjana, N.E.; Wright, J.B.; Fulco, C.P.; et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 2017, 548, 343–346. [Google Scholar] [CrossRef]
- Perez, A.R.; Pritykin, Y.; Vidigal, J.A.; Chhangawala, S.; Zamparo, L.; Leslie, C.S.; Ventura, A. GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol. 2017, 35, 347–349. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Chun-Wei Lee, S.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 369–384.e367. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kloetgen, A.; Thandapani, P.; Ntziachristos, P.; Ghebrechristos, Y.; Nomikou, S.; Lazaris, C.; Chen, X.; Hu, H.; Bakogianni, S.; Wang, J.; et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat. Genet. 2020, 52, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Huang, H.T.; Liang, Y.; Trimarchi, T.; Aifantis, I.; Tsirigos, A. lncRNA-screen: An interactive platform for computationally screening long non-coding RNAs in large genomics datasets. BMC Genom. 2017, 18, 434. [Google Scholar] [CrossRef]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes. Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef]
- Majidinia, M.; Yousefi, B. Long non-coding RNAs in cancer drug resistance development. DNA Repair 2016, 45, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Gao, L.; Ma, X.; Huang, J.J.; Chen, J.; Zeng, L.; Ashby, C.R., Jr.; Zou, C.; Chen, Z.S. Long non-coding RNAs regulate drug resistance in cancer. Mol. Cancer 2020, 19, 54. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, O.; Nizri, T.; Doniger, T.; Jacob, J.; Rechavi, G.; Ginsberg, D. The long non-coding RNA ERIC is regulated by E2F and modulates the cellular response to DNA damage. Mol. Cancer 2013, 12, 131. [Google Scholar] [CrossRef]
- Xue, X.; Yang, Y.A.; Zhang, A.; Fong, K.W.; Kim, J.; Song, B.; Li, S.; Zhao, J.C.; Yu, J. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene 2016, 35, 2746–2755. [Google Scholar] [CrossRef]
- Han, S.; Yan, Y.; Ren, Y.; Hu, Y.; Wang, Y.; Chen, L.; Zhi, Z.; Zheng, Y.; Shao, Y.; Liu, J. LncRNA SAMMSON Mediates Adaptive Resistance to RAF Inhibition in BRAF-Mutant Melanoma Cells. Cancer Res. 2021, 81, 2918–2929. [Google Scholar] [CrossRef]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef]
- Zhong, J.; Yan, W.; Wang, C.; Liu, W.; Lin, X.; Zou, Z.; Sun, W.; Chen, Y. BRAF Inhibitor Resistance in Melanoma: Mechanisms and Alternative Therapeutic Strategies. Curr. Treat. Options Oncol. 2022, 23, 1503–1521. [Google Scholar] [CrossRef]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shamloo, S.; Kloetgen, A.; Petroulia, S.; Hockemeyer, K.; Sievers, S.; Tsirigos, A.; Aifantis, I.; Imig, J. Integrative CRISPR Activation and Small Molecule Inhibitor Screening for lncRNA Mediating BRAF Inhibitor Resistance in Melanoma. Biomedicines 2023, 11, 2054. https://doi.org/10.3390/biomedicines11072054
Shamloo S, Kloetgen A, Petroulia S, Hockemeyer K, Sievers S, Tsirigos A, Aifantis I, Imig J. Integrative CRISPR Activation and Small Molecule Inhibitor Screening for lncRNA Mediating BRAF Inhibitor Resistance in Melanoma. Biomedicines. 2023; 11(7):2054. https://doi.org/10.3390/biomedicines11072054
Chicago/Turabian StyleShamloo, Sama, Andreas Kloetgen, Stavroula Petroulia, Kathryn Hockemeyer, Sonja Sievers, Aristotelis Tsirigos, Ioannis Aifantis, and Jochen Imig. 2023. "Integrative CRISPR Activation and Small Molecule Inhibitor Screening for lncRNA Mediating BRAF Inhibitor Resistance in Melanoma" Biomedicines 11, no. 7: 2054. https://doi.org/10.3390/biomedicines11072054
APA StyleShamloo, S., Kloetgen, A., Petroulia, S., Hockemeyer, K., Sievers, S., Tsirigos, A., Aifantis, I., & Imig, J. (2023). Integrative CRISPR Activation and Small Molecule Inhibitor Screening for lncRNA Mediating BRAF Inhibitor Resistance in Melanoma. Biomedicines, 11(7), 2054. https://doi.org/10.3390/biomedicines11072054