
Involvement of the Opioid Peptide Family in Cancer Progression




Abstract
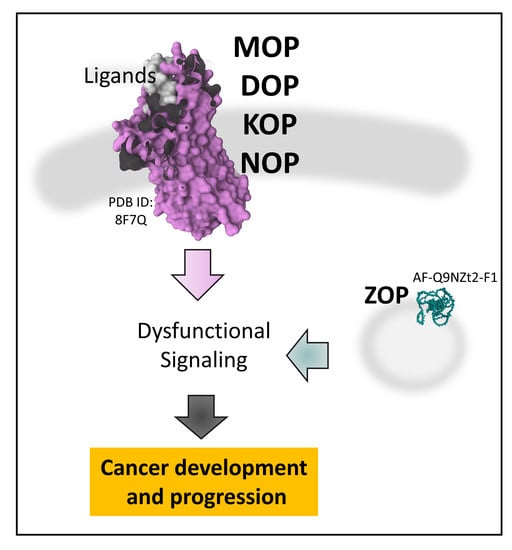
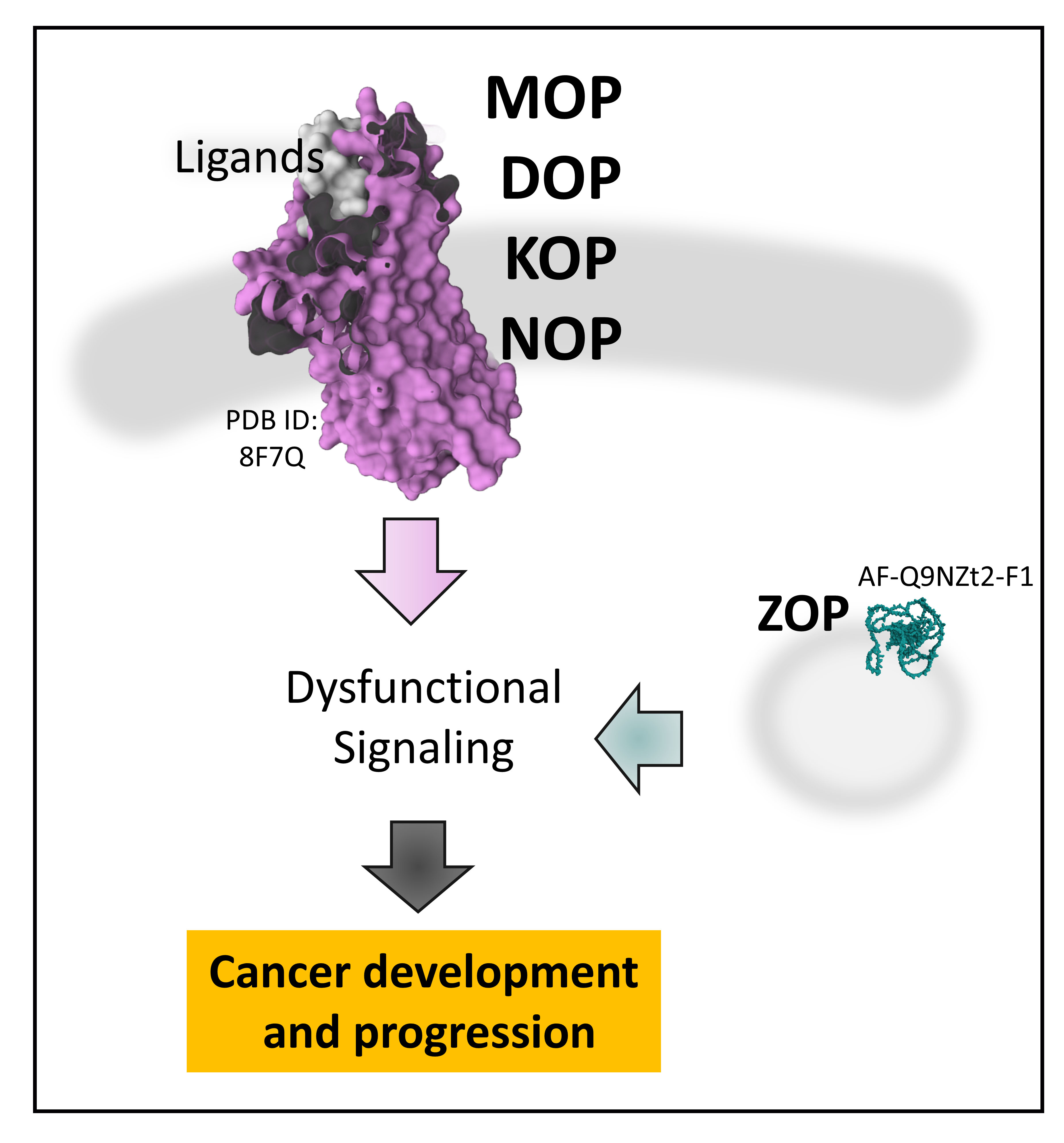
1. Introduction
2. The Endogenous Opioid System: Peptides and Receptors
2.1. Endomorphins
2.2. β-Endorphins
2.3. Enkephalins
2.4. Dynorphins
2.5. Nociceptins
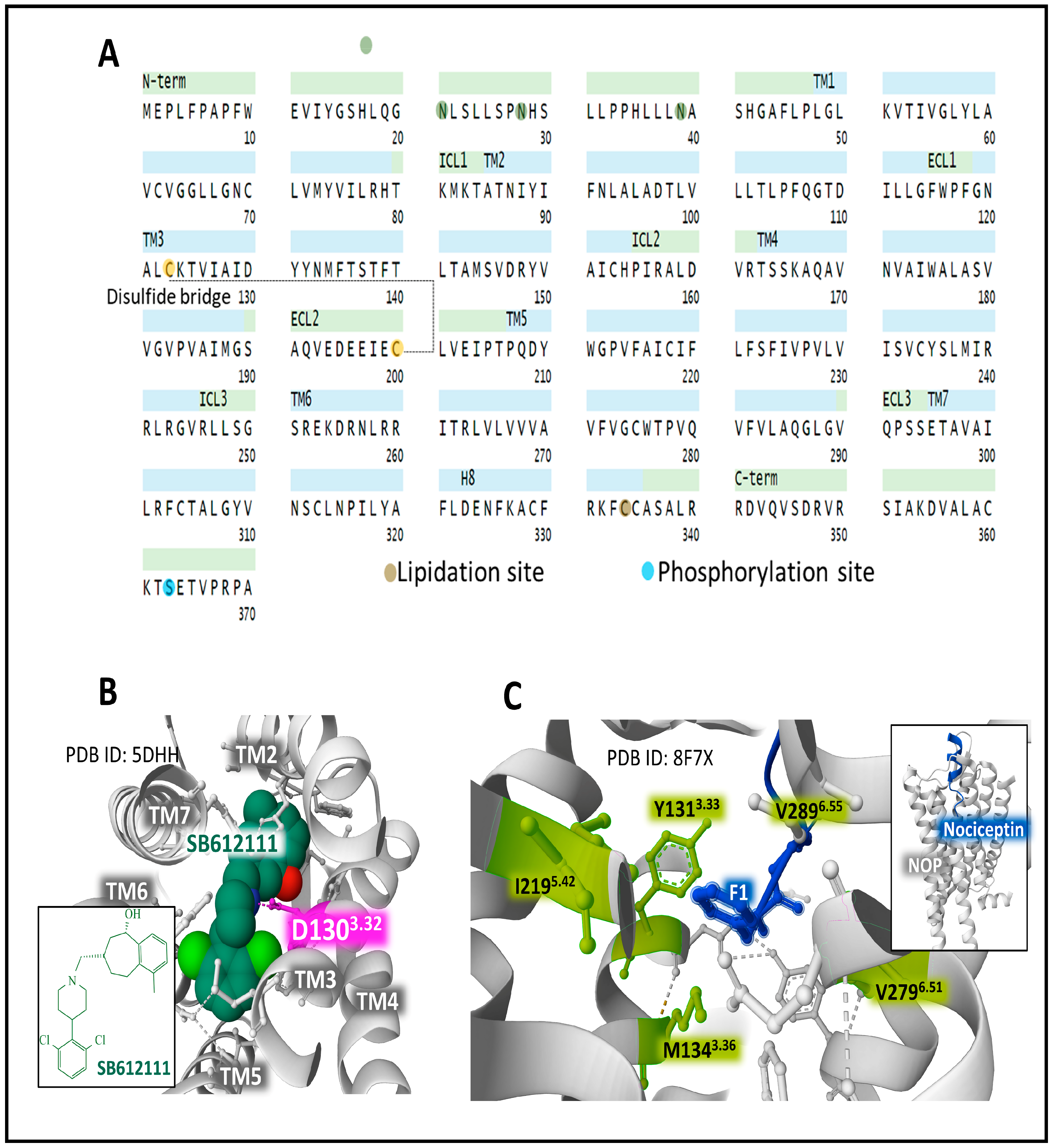
2.6. The Structure and Dynamics of the Opioid Receptors (OP)
2.6.1. The μ Receptor (MOP)
2.6.2. The δ Receptor (DOP.)
2.6.3. The κ Receptor (KOP)
2.6.4. The Nociceptin/Orphanin FQ Receptor (NOP.)
2.7. The Distribution of OR
2.8. Signaling Pathways of OR
3. Involvement of Opioid Peptides in Cancer
3.1. Bone Cancer
Endorphin
3.2. Brain Tumors
3.2.1. Enkephalin
3.2.2. Endorphin
3.2.3. Dynorphin
3.3. Breast Cancer
3.3.1. Enkephalin
3.3.2. Endorphin
3.3.3. Dynorphin
3.4. Cervical Cancer
3.4.1. Enkephalin
3.4.2. Endorphin
3.5. Colorectal Cancer
3.5.1. Enkephalin
3.5.2. Endorphin
3.5.3. Dynorphin
3.6. Cutaneous Squamous Cell Carcinoma
Enkephalin
3.7. Gastric Cancer
3.7.1. Enkephalin
3.7.2. Endorphin
3.8. Head and Neck Cancer
3.8.1. Enkephalin
3.8.2. Endorphin
3.8.3. Dynorphin
3.9. Larynx Cancer
3.9.1. Enkephalin
3.9.2. Endorphin
3.10. Leukemia
3.10.1. Enkephalin
3.10.2. Endorphin
3.11. Liver Cancer
3.11.1. Enkephalin
3.11.2. Endorphin
3.12. Lung Cancer
3.12.1. Enkephalin
3.12.2. Endorphin
3.12.3. Dynorphin
3.13. Melanoma
3.13.1. Enkephalin
3.13.2. Endorphin
3.14. Myeloma
Dynorphin
3.15. Neuroblastoma
3.15.1. Enkephalin
3.15.2. Endorphin
3.15.3. Dynorphin
3.16. Ovarian Cancer
3.16.1. Enkephalin
3.16.2. Endorphin
3.17. Pancreatic Cancer
3.17.1. Enkephalin
3.17.2. Dynorphin
3.18. Pheochromocytoma
3.18.1. Enkephalin
3.18.2. Endorphin
3.18.3. Dynorphin
3.19. Pituitary Cancer
3.19.1. Enkephalin
3.19.2. Endorphin
3.20. Prostate Cancer
3.20.1. Enkephalin
3.20.2. Endorphin
3.20.3. Dynorphin
3.21. Renal Cancer
Enkephalin
3.22. Retinoblastoma
3.22.1. Enkephalin
3.22.2. Endorphin
3.23. Testicular Cancer
Dynorphin
3.24. Thymic Cancer
3.24.1. Enkephalin
3.24.2. Endorphin
3.24.3. Dynorphin
3.25. Thyroid Cancer
3.25.1. Enkephalin
3.25.2. Endorphin
4. Antitumor Therapeutic Strategies
5. Future Research
5.1. Basic Research
5.2. MET/LEU Research
5.3. Beta-END Research
5.4. DYN Research
5.5. Signaling Pathways
5.6. Tumor Cell Migration
5.7. Angiogenesis
5.8. Polymorphisms
5.9. Substances Regulating the Opioid System
5.10. Endopeptidases
5.11. The Opioid System as a Cancer-Predictive Factor
5.12. Structural Dynamics of OR
5.13. Other Research Lines
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coveñas, R.; Muñoz, M. Involvement of the Substance P/Neurokinin-1 Receptor System in Cancer. Cancers 2022, 14, 3539. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.; Pei, G.; Kim, S.T.; German, A.; Robinson, P. Substance P Antagonism as a Novel Therapeutic Option to Enhance Efficacy of Cisplatin in Triple Negative Breast Cancer and Protect PC12 Cells against Cisplatin-Induced Oxidative Stress and Apoptosis. Cancers 2021, 13, 3871. [Google Scholar] [CrossRef] [PubMed]
- García-Aranda, M.; Téllez, T.; McKenna, L.; Redondo, M. Neurokinin-1 Receptor (NK-1R) Antagonists as a New Strategy to Overcome Cancer Resistance. Cancers 2022, 14, 2255. [Google Scholar] [CrossRef]
- Beirith, I.; Renz, B.W.; Mudusetti, S.; Ring, N.S.; Kolorz, J.; Koch, D.; Bazhin, A.V.; Berger, M.; Wang, J.; Angele, M.K.; et al. Identification of the Neurokinin-1 Receptor as Targetable Stratification Factor for Drug Repurposing in Pancreatic Cancer. Cancers 2021, 13, 2703. [Google Scholar] [CrossRef]
- Sánchez, M.L.; Coveñas, R. The Galaninergic System: A Target for Cancer Treatment. Cancers 2022, 14, 3755. [Google Scholar] [CrossRef]
- Sánchez, M.L.; Rodríguez, F.D.; Coveñas, R. Peptidergic Systems and Cancer: Focus on Tachykinin and Calcitonin/Calcitonin Gene-Related Peptide Families. Cancers 2023, 15, 1694. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.L.; Coveñas, R. The Neurotensinergic System: A Target for Cancer Treatment. Curr. Med. Chem. 2022, 29, 3231–3260. [Google Scholar] [CrossRef]
- Robinson, P.; Coveñas, R.; Muñoz, M. Combination Therapy of Chemotherapy or Radiotherapy and the Neurokinin-1 Receptor Antagonist Aprepitant: A New Antitumor Strategy? Curr. Med. Chem. 2023, 29, 1798–1812. [Google Scholar] [CrossRef]
- Muñoz, M.F.; Argüelles, S.; Rosso, M.; Medina, R.; Coveñas, R.; Ayala, A.; Muñoz, M. The Neurokinin-1 Receptor Is Essential for the Viability of Human Glioma Cells: A Possible Target for Treating Glioblastoma. BioMed Res. Int. 2022, 2022, 6291504. [Google Scholar] [CrossRef]
- Bostwick, D.G.; Null, W.E.; Holmes, D.; Weber, E.; Barchas, J.D.; Bensch, K. Expression of Opioid Peptides in Tumors. N. Engl. J. Med. 1987, 317, 1439–1443. [Google Scholar] [CrossRef]
- Roth, K.A.; Bensch, K.G.; Hoffman, A.R. Characterization of Opioid Peptides in Human Thyroid Medullary Carcinoma. Cancer 1987, 59, 1594–1598. [Google Scholar] [CrossRef]
- Cronin-Fenton, D. Opioids, and Breast Cancer Recurrence. Curr. Opin. Support. Palliat. Care 2019, 13, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; McLaughlin, P.J.; Goodman, S.R.; Rhodes, R.E. Opioid Receptors and Endogenous Opioids in Diverse Human and Animal Cancers23. JNCI J. Natl. Cancer Inst. 1987, 79, 1059–1065. [Google Scholar] [CrossRef]
- Zagon, I.S.; Smith, J.P.; McLaughlin, P.J. Human Pancreatic Cancer Cell Proliferation in Tissue Culture Is Tonically Inhibited by Opioid Growth Factor. Int. J. Oncol. 1999, 14, 577–584. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.J.; Levin, R.J.; Zagon, I.S. Regulation of Human Head and Neck Squamous Cell Carcinoma Growth in Tissue Culture by Opioid Growth Factor. Int. J. Oncol. 1999, 14, 991–998. [Google Scholar] [CrossRef]
- Donahue, R.N.; McLaughlin, P.J.; Zagon, I.S. Cell Proliferation of Human Ovarian Cancer Is Regulated by the Opioid Growth Factor-Opioid Growth Factor Receptor Axis. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2009, 296, R1716–R1725. [Google Scholar] [CrossRef]
- Zagon, I.S.; Donahue, R.N.; McLaughlin, P.J. Opioid Growth Factor-Opioid Growth Factor Receptor Axis Is a Physiological Determinant of Cell Proliferation in Diverse Human Cancers. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2009, 297, R1154–R1161. [Google Scholar] [CrossRef] [PubMed]
- Schoos, A.; Gabriel, C.; Knab, V.M.; Fux, D.A. Activation of HIF-1 Alpha by Gamma-Opioid Receptors Induces COX-2 Expression in Breast Cancer Cells and Leads to Paracrine Activation of Vascular Endothelial Cells. J. Pharmacol. Exp. Ther. 2019, 370, 480–489. [Google Scholar] [CrossRef]
- UniProt Database. Available online: https://www.uniprot.org/ (accessed on 10 June 2023).
- Parker, K.E.; Pedersen, C.E.; Gomez, A.M.; Spangler, S.M.; Walicki, M.C.; Feng, S.Y.; Stewart, S.L.; Otis, J.M.; Al-Hasani, R.; McCall, J.G.; et al. A Paranigral VTA Nociceptin Circuit That Constrains Motivation for Reward. Cell 2019, 178, 653–671.e19. [Google Scholar] [CrossRef] [PubMed]
- Guide to Pharmacology IUPAHR. Available online: https://www.guidetopharmacology.org/ (accessed on 15 May 2023).
- Zagon, I.S.; Verderame, M.F.; Zimmer, W.E.; Mclaughlin, P.J. Molecular Characterization and Distribution of the Opioid Growth Factor Receptor (OGFr) in Mouse. Mol. Brain Res. 2000, 84, 106. [Google Scholar] [CrossRef]
- Borsodi, A.; Bruchas, M.; Caló, G.; Chavkin, C.; Christie, M.J.; Civelli, O.; Connor, M.; Cox, B.M.; Devi, L.A.; Evans, C.; et al. Opioid Receptors in GtoPdb v.2021.3. IUPHARBPS Guide Pharmacol. CITE 2021, 2021. [Google Scholar] [CrossRef]
- Bodnar, R.J. Endogenous Opiates and Behavior: 2021. Peptides 2023, 164, 171004. [Google Scholar] [CrossRef]
- Petrocelli, G.; Pampanella, L.; Abruzzo, P.M.; Ventura, C.; Canaider, S.; Facchin, F. Endogenous Opioids and Their Role in Stem Cell Biology and Tissue Rescue. Int. J. Mol. Sci. 2022, 23, 3819. [Google Scholar] [CrossRef]
- Zadina, J.E.; Hackler, L.; Ge, L.-J.; Kastin, A.J. A Potent and Selective Endogenous Agonist for the Μ-Opiate Receptor. Nature 1997, 386, 499–502. [Google Scholar] [CrossRef]
- Terskiy, A.; Wannemacher, K.M.; Yadav, P.N.; Tsai, M.; Tian, B.; Howells, R.D. Search of the Human Proteome for Endomorphin-1 and Endomorphin-2 Precursor Proteins. Life Sci. 2007, 81, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Kaiya, H. Chapter 21—Mexneurin. In Handbook of Hormones; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 173–174. [Google Scholar]
- PepDraw. Available online: https://pepdraw.com/ (accessed on 17 May 2023).
- Tanaka, S. Comparative Aspects of Intracellular Proteolytic Processing of Peptide Hormone Precursors: Studies of Proopiomelanocortin Processing. Zoolog. Sci. 2003, 20, 1183–1198. [Google Scholar] [CrossRef] [PubMed]
- Gene NCBI Database. Available online: https://www.ncbi.nlm.nih.gov/gene/ (accessed on 12 June 2023).
- Pilozzi, A.; Carro, C.; Huang, X. Roles of β-Endorphin in Stress, Behavior, Neuroinflammation, and Brain Energy Metabolism. Int. J. Mol. Sci. 2020, 22, 338. [Google Scholar] [CrossRef] [PubMed]
- Netto, C.A. Role of Brain Β-Endorphin in Memory Modulation Revisited. Neuroscience 2022, 497, 30–38. [Google Scholar] [CrossRef]
- Pert, C.B.; Snyder, S.H. Opiate Receptor: Demonstration in Nervous Tissue. Science 1973, 179, 1011–1014. [Google Scholar] [CrossRef]
- Hughes, J.; Smith, T.W.; Kosterlitz, H.W.; Fothergill, L.A.; Morgan, B.A.; Morris, H.R. Identification of Two Related Pentapeptides from the Brain with Potent Opiate Agonist Activity. Nature 1975, 258, 577–580. [Google Scholar] [CrossRef]
- Ndong, D.B.; Blais, V.; Holleran, B.; Proteau-Gagné, A.; Cantin-Savoie, I.; Robert, W.; Nadon, J.-F.; Beauchemin, S.; Leduc, R.; Pineyro, G.; et al. Exploration of the Fifth Position of Leu-Enkephalin and Its Role in Binding and Activating Delta (DOP) and Mu (MOP) Opioid Receptors. Pept. Sci. 2018, 111, e24070. [Google Scholar] [CrossRef]
- Zagon, I.S.; McLaughlin, P.J. Opioid Growth Factor and the Treatment of Human Pancreatic Cancer: A Review. World J. Gastroenterol. WJG 2014, 20, 2218–2223. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; Verderame, M.F.; Mclaughlin, P.J. The Biology of the Opioid Growth Factor Receptor (OGFr). Brain Res. Rev. 2002, 38, 351. [Google Scholar] [CrossRef]
- Schwarzer, C. 30 Years of Dynorphins—New Insights on Their Functions in Neuropsychiatric Diseases. Pharmacol. Ther. 2009, 123, 353–370. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, X.; Fan, J.; Sun, H.; Yao, Q.; Shi, J.; Qu, H.; Du, S.; Cheng, Y.; Ma, S.; et al. Dynorphin A (1–8) Inhibits Oxidative Stress and Apoptosis in MCAO Rats, Affording Neuroprotection through NMDA Receptor and κ-Opioid Receptor Channels. Neuropept. Edinb. 2021, 89, 102182. [Google Scholar] [CrossRef]
- Meunier, J.C.; Mollereau, C.; Toll, L.; Suaudeau, C.; Moisand, C.; Alvinerie, P.; Butour, J.L.; Guillemot, J.C.; Ferrara, P.; Monsarrat, B. Isolation and Structure of the Endogenous Agonist of Opioid Receptor-like ORL1 Receptor. Nature 1995, 377, 532–535. [Google Scholar] [CrossRef]
- Toll, L.; Cippitelli, A.; Ozawa, A. The NOP Receptor System in Neurological and Psychiatric Disorders: Discrepancies, Peculiarities and Clinical Progress in Developing Targeted Therapies. CNS Drugs 2021, 35, 591–607. [Google Scholar] [CrossRef]
- Tariq, S.; Nurulain, S.M.; Tekes, K.; Adeghate, E. Deciphering Intracellular Localization and Physiological Role of Nociceptin and Nocistatin. Peptides 2013, 43, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, O.N.A.; Awwad, H.O.; Zhang, Y.; Standifer, K.M. Therapeutic Potential of Nociceptin/Orphanin FQ Peptide (NOP) Receptor Modulators for Treatment of Traumatic Brain Injury, Traumatic Stress, and Their Co-Morbidities. Pharmacol. Ther. 2022, 231, 107982. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, D.; Yan, Y.; Li, Q.; Xing, W.; Liu, Y.; Chen, Y.; Wang, D.; Yuan, Y.; Xie, J.; et al. The Nociceptin Receptor Promotes Autophagy through NF-KB Signaling and Is Transcriptionally Regulated by E2F1 in HCC. Cell Death Discov. 2022, 8, 165. [Google Scholar] [CrossRef]
- García-Nafría, J.; Tate, C.G. Structure Determination of GPCRs: Cryo-EM Compared with X-ray Crystallography. Biochem. Soc. Trans. 2021, 49, 2345–2355. [Google Scholar] [CrossRef]
- Cheng, J.-X.; Cheng, T.; Li, W.-H.; Liu, G.-X.; Zhu, W.-L.; Tang, Y. Computational Insights into the Subtype Selectivity and “Message-Address-Efficacy” Mechanisms of Opioid Receptors through JDTic Binding and Unbinding. Acta Pharmacol. Sin. 2018, 39, 482. [Google Scholar] [CrossRef] [PubMed]
- Degrandmaison, J.; Rochon-Haché, S.; Parent, J.-L.; Gendron, L. Knock-In Mouse Models to Investigate the Functions of Opioid Receptors in Vivo. Front. Cell. Neurosci. 2022, 16, 807549. [Google Scholar] [CrossRef] [PubMed]
- GPCR Database. Available online: https://gpcrdb.org/ (accessed on 17 May 2023).
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR Sequence, Structure and Function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef] [PubMed]
- RCSB PDB. Available online: https://www.rcsb.org/ (accessed on 30 May 2023).
- Zhuang, Y.; Wang, Y.; He, B.; He, X.; Zhou, X.E.; Guo, S.; Rao, Q.; Yang, J.; Liu, J.; Zhou, Q.; et al. Molecular Recognition of Morphine and Fentanyl by the Human μ-Opioid Receptor. Cell 2022, 185, 4361–4375.e19. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- KingDraw-Free Chemical Structure Editor. Available online: http://www.kingdraw.cn/en/index.html (accessed on 17 May 2023).
- Pasternak, G.W.; Pan, Y.-X. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Liu, N.-J.; Gintzler, A.R. Relevance of Mu-Opioid Receptor Splice Variants and Plasticity of Their Signaling Sequelae to Opioid Analgesic Tolerance. Cell. Mol. Neurobiol. 2021, 41, 855–862. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal Structure of the Μ-Opioid Receptor Bound to a Morphinan Antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural Insights into µ-opioid Receptor Activation. Nature 2015, 524, 315. [Google Scholar] [CrossRef] [PubMed]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the Μ-Opioid Receptor–Gi Protein Complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Tan, K.; Floyd, C.; Bong, D.; Pino, M.J.; Wu, C. Probing Biased Activation of Mu-Opioid Receptor by the Biased Agonist PZM21 Using All Atom Molecular Dynamics Simulation. Life Sci. 2021, 269, 119026. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hetzer, F.; Huang, W.; Qu, Q.; Meyerowitz, J.; Kaindl, J.; Hübner, H.; Skiniotis, G.; Kobilka, B.K.; Gmeiner, P. Structure-Based Evolution of G Protein-Biased μ-Opioid Receptor Agonists. Angew. Chem. Int. Ed. 2022, 61, e202200269. [Google Scholar] [CrossRef]
- Azzam, A.A.H.; McDonald, J.; Lambert, D.G. Hot Topics in Opioid Pharmacology: Mixed and Biased Opioids. Br. J. Anaesth. BJA 2019, 122, e136–e145. [Google Scholar] [CrossRef]
- Granier, S.; Manglik, A.; Kruse, A.C.; Tong, S.K.; Foon, S.T.; Weis, W.I.; Kobilka, B.K. Structure of the δ-Opioid Receptor Bound to Naltrindole. Nature 2012, 485, 400–404. [Google Scholar] [CrossRef]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.-P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular Control of δ-Opioid Receptor Signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Poorten, O.V.D.; White, K.L.; et al. Elucidating the Active δ-Opioid Receptor Crystal Structure with Peptide and Small-Molecule Agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef]
- Wang, Y.; Zhuang, Y.; DiBerto, J.F.; Zhou, X.E.; Schmitz, G.P.; Yuan, Q.; Jain, M.K.; Liu, W.; Melcher, K.; Jiang, Y.; et al. Structures of the Entire Human Opioid Receptor Family. Cell 2023, 186, 413–427.e17. [Google Scholar] [CrossRef]
- Sofuoglu, M.; Portoghese, P.S.; Takemori, A.E. 7-Benzylidenenaltrexone (BNTX): A Selective Δ1 Opioid Receptor Antagonist in the Mouse Spinal Cord. Life Sci. 1993, 52, 769. [Google Scholar] [CrossRef]
- Zhou, W.; Li, Y.; Meng, X.; Liu, A.; Mao, Y.; Zhu, X.; Meng, Q.; Jin, Y.; Zhang, Z.; Tao, W. Switching of Delta Opioid Receptor Subtypes in Central Amygdala Microcircuits Is Associated with Anxiety States in Pain. J. Biol. Chem. 2021, 296, 100277. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Elladki, R.; Jiang, J.; Wei, G.-W. Machine-Learning Analysis of Opioid Use Disorder Informed by MOR, DOR, KOR, NOR and ZOR-Based Interactome Networks. Comput. Biol. Med. 2023, 157, 106745. [Google Scholar] [CrossRef] [PubMed]
- Huixian, W.U.; Wacker, D.; Mascarella, S.W.; Westkaemper, R.B.; Mosier, P.D.; Roth, B.L.; Cherezov, V.; Stevens, R.C.; Mileni, M.; Katritch, V.; et al. Structure of the Human κ-Opioid Receptor in Complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Che, T.; English, J.; Krumm, B.E.; Kim, K.; Pardon, E.; Olsen, R.H.J.; Wang, S.; Zhang, S.; Diberto, J.F.; Sciaky, N.; et al. Nanobody-Enabled Monitoring of Kappa Opioid Receptor States. Nat. Commun. 2020, 11, 1145. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.; White, K.L.; Doncescu, N.; Didenko, T.; Roth, B.L.; Czaplicki, G.; Stevens, R.C.; Wüthrich, K.; Milon, A. NMR Structure and Dynamics of the Agonist Dynorphin Peptide Bound to the Human Kappa Opioid Receptor. Proc. Natl. Acad. Sci. USA 2015, 112, 11852. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, H.; Zheng, Q.; Chen, Q.; Li, X.; Guo, J. κ-Opioid Receptors Improve Vascular Endothelial Dysfunction in Salt-Sensitive Hypertension via PI3K/Akt/ENOS Signaling Pathway. Oxid. Med. Cell. Longev. 2023, 2023, 5352959-13. [Google Scholar] [CrossRef]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67.e15. [Google Scholar] [CrossRef]
- Han, J.; Zhang, J.; Nazarova, A.L.; Bernhard, S.M.; Krumm, B.E.; Zhao, L.; Lam, J.H.; Rangari, V.A.; Majumdar, S.; Nichols, D.E.; et al. Ligand and G-Protein Selectivity in the κ-Opioid Receptor. Nature 2023, 617, 417. [Google Scholar] [CrossRef]
- Ji, J.; Lin, W.; Vrudhula, A.; Xi, J.; Yeliseev, A.; Grothusen, J.; Bu, W.; Liu, R. Molecular Interaction Between Butorphanol and κ-Opioid Receptor. Obstet. Anesthesia Dig. 2020, 131, 935–942. [Google Scholar] [CrossRef]
- Uprety, R.; Che, T.; Zaidi, S.A.; Grinnell, S.G.; Varga, B.R.; Faouzi, A.; Slocum, S.T.; Allaoa, A.; Varadi, A.; Nelson, M.; et al. Controlling Opioid Receptor Functional Selectivity by Targeting Distinct Subpockets of the Orthosteric Site. eLife 2021, 10, e56519. [Google Scholar] [CrossRef]
- Puls, K.; Wolber, G. Solving an Old Puzzle: Elucidation and Evaluation of the Binding Mode of Salvinorin A at the Kappa Opioid Receptor. Molecules 2023, 28, 718. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.L.; Thompson, A.A.; Trapella, C.; Guerrini, R.; Malfacini, D.; Patel, N.; Han, G.W.; Cherezov, V.; Caló, G.; Katritch, V.; et al. The Importance of Ligand-Receptor Conformational Pairs in Stabilization: Spotlight on the N/OFQ G Protein-Coupled Receptor. Structure 2015, 23, 2291–2299. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, S.; Albanese, V.; Illuminati, D.; Marzola, E.; Fabbri, M.; Ferrari, F.; Holanda, V.A.D.; Sturaro, C.; Malfacini, D.; Ruzza, C.; et al. Novel Mixed NOP/Opioid Receptor Peptide Agonists. J. Med. Chem. 2021, 64, 6656. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, N.; Ding, H.; Ko, M. Therapeutic Potentials of NOP and MOP Receptor Coactivation for the Treatment of Pain and Opioid Abuse. J. Neurosci. Res. 2022, 100, 191. [Google Scholar] [CrossRef] [PubMed]
- Kamakolanu, U.G.; Meyer, M.E.; Yasuda, D.; Polgar, W.E.; Marti, M.; Mercatelli, D.; Pisanò, C.A.; Brugnoli, A.; Morari, M.; Zaveri, N.T. Discovery and Structure–Activity Relationships of Nociceptin Receptor Partial Agonists That Afford Symptom Ablation in Parkinson’s Disease Models. J. Med. Chem. 2020, 63, 2688–2704. [Google Scholar] [CrossRef] [PubMed]
- Daibani, A.E.; Paggi, J.M.; Kim, K.; Laloudakis, Y.D.; Popov, P.; Bernhard, S.M.; Krumm, B.E.; Olsen, R.H.J.; Diberto, J.; Carroll, F.I.; et al. Molecular Mechanism of Biased Signaling at the Kappa Opioid Receptor. Nat. Commun. 2023, 14, 1338. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Papasergi-Scott, M.; He, F.; Seven, A.B.; Meyerowitz, J.G.; Panova, O.; Peroto, M.C.; Che, T.; Skiniotis, G. Structure Determination of Inactive-State GPCRs with a Universal Nanobody. Nat. Struct. Mol. Biol. 2022, 29, 1188. [Google Scholar] [CrossRef]
- Vo, Q.N.; Mahinthichaichan, P.; Shen, J.; Ellis, C.R. How μ-Opioid Receptor Recognizes Fentanyl. Nat. Commun. 2021, 12, 984. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Marcotte, I.; Separovic, F.; Auger, M.; Gagné, S.M. A Multidimensional 1H NMR Investigation of the Conformation of Methionine-Enkephalin in Fast-Tumbling Bicelles. Biophys. J. 2004, 86, 1587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Geng, J.; Shan, F.; Shan, Y.; Griffin, N.; Wu, B.; Wang, X. Methionine Enkephalin Suppresses Lung Cancer Metastasis by Regulating the Polarization of Tumor-Associated Macrophages and the Distribution of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment and Inhibiting Epithelial-Mesenchymal Transition. Int. Immunopharmacol. 2023, 118, 110064. [Google Scholar] [CrossRef] [PubMed]
- Budka, J.; Kowalski, S.; Chylinska, M.; Dzierzbicka, K.; Inkielewicz-Stepniak, I. Opioid Growth Factor and Its Derivatives as Potential Non-Toxic Multifunctional Anticancer and Analgesic Compounds. Curr. Med. Chem. 2021, 28, 673–686. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.J.; Sassani, J.W.; Diaz, D.; Zagon, I.S. Elevated Opioid Growth Factor Alters the Limbus in Type 1 Diabetic Rats. J. Diabetes Clin. Res. 2023, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.J.; Sassani, J.W.; Zagon, I.S. Dysregulation of the OGF-OGFr Pathway and Associated Diabetic Complications. J. Diabetes Clin. Res. 2021, 3, 64–67. [Google Scholar] [CrossRef]
- Bodnar, R.J. Endogenous Opiates and Behavior: 2020. Peptides 2022, 151, 170752. [Google Scholar] [CrossRef]
- Quirion, B.; Beaulieu, C.; Cote, L.; Parent, J.-L.; Gendron, L. Distribution of Delta and Mu Opioid Receptor mRNA in Rodent Dorsal Root Ganglia Neurons. Eur. J. Neurosci. 2022, 56, 4031–4044. [Google Scholar] [CrossRef]
- Tollefson, S.; Stoughton, C.; Himes, M.L.; McKinney, K.E.; Mason, S.; Ciccocioppo, R.; Narendran, R. Imaging Nociceptin Opioid Peptide Receptors in Alcohol Use Disorder with [11C]NOP-1A and Positron Emission Tomography: Findings from a Second Cohort. Biol. Psychiatry 2023. [Google Scholar] [CrossRef]
- Zamfir, M.; Sharif, B.; Locke, S.; Ehrlich, A.T.; Ochandarena, N.E.; Scherrer, G.; Ribeiro-Da-Silva, A.; Kieffer, B.L.; Séguéla, P. Distinct and Sex-Specific Expression of Mu Opioid Receptors in Anterior Cingulate and Somatosensory S1 Cortical Areas. Pain 2022, 164, 703. [Google Scholar] [CrossRef]
- Kareem, Z.Y.; McLaughlin, P.J.; Kumari, R. Opioid Growth Factor Receptor: Anatomical Distribution and Receptor Colocalization in Neurons of the Adult Mouse Brain. Neuropept. Edinb. 2023, 99, 102325. [Google Scholar] [CrossRef]
- Olabarrieta, E.; Totorikaguena, L.; Matorras, R.; Agirregoitia, E.; Agirregoitia, N. Delta and Kappa Opioid Receptors in Human Endometrium during the Menstrual Cycle: Expression and Localization. Eur. J. Obstet. Amp Gynecol. Reprod. Biol. 2023, 283, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.K.; Smith, C.M.; Rahmatullah, M.; Nissapatorn, V.; Wilairatana, P.; Spetea, M.; Gueven, N.; Dietis, N. Opioid Analgesia and Opioid-Induced Adverse Effects: A Review. Pharmaceuticals 2021, 14, 1091. [Google Scholar] [CrossRef] [PubMed]
- Lešnik, S.; Bertalan, É.; Bren, U.; Bondar, A.-N. Opioid Receptors and Protonation-Coupled Binding of Opioid Drugs. Int. J. Mol. Sci. 2021, 22, 13353. [Google Scholar] [CrossRef]
- Gopalakrishnan, L.; Chatterjee, O.; Ravishankar, N.; Suresh, S.; Raju, R.; Mahadevan, A.; Prasad, T.S.K. Opioid Receptors Signaling Network. J. Cell Commun. Signal. 2022, 16, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Miess, E.; Gondin, A.B.; Yousuf, A.; Steinborn, R.; Mösslein, N.; Yang, Y.; Göldner, M.; Ruland, J.G.; Bünemann, M.; Krasel, C.; et al. Multisite Phosphorylation Is Required for Sustained Interaction with GRKs and Arrestins during Rapid μ-Opioid Receptor Desensitization. Sci. Signal. 2018, 11, eaas9609. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Vargas, N.N.; Gong, J.; Wisdom, M.J.; Jensen, D.D.; Latorre, R.; Hegron, A.; Teng, S.; DiCello, J.J.; Rajasekhar, P.; Veldhuis, N.A.; et al. Endosomal Signaling of Delta Opioid Receptors Is an Endogenous Mechanism and Therapeutic Target for Relief from Inflammatory Pain. Proc. Natl. Acad. Sci. USA 2020, 117, 15281–15292. [Google Scholar] [CrossRef]
- Shiraki, A.; Shimizu, S. The Molecular Associations in Clathrin-Coated Pit Regulate β-Arrestin-Mediated MAPK Signaling Downstream of μ-Opioid Receptor. Biochem. Biophys. Res. Commun. 2023, 640, 64–72. [Google Scholar] [CrossRef]
- Radoux-Mergault, A.; Oberhauser, L.; Aureli, S.; Gervasio, F.L.; Stoeber, M. Subcellular Location Defines GPCR Signal Transduction. Sci. Adv. 2023, 9, eadf6059. [Google Scholar] [CrossRef]
- Miyoshi, K.; Shimizu, S.; Shiraki, A.; Egi, M. Ubiquitination of the μ-Opioid Receptor Regulates Receptor Internalization without Affecting Gi/o-Mediated Intracellular Signaling or Receptor Phosphorylation. Biochem. Biophys. Res. Commun. 2023, 643, 96–104. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.; Shan, F. Interaction of Opioid Growth Factor (OGF) and Opioid Antagonist and Their Significance in Cancer Therapy. Int. Immunopharmacol. 2019, 75, 105785. [Google Scholar] [CrossRef]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased Opioid Ligands. Molecules 2020, 25, 4257. [Google Scholar] [CrossRef] [PubMed]
- Groom, S.; Blum, N.K.; Conibear, A.E.; Disney, A.A.; Hill, R.R.; Husbands, S.M.; Li, Y.; Toll, L.; Kliewer, A.; Schulz, S.; et al. A Novel G Protein-biased Agonist at the μ Opioid Receptor Induces Substantial Receptor Desensitisation through G Protein-coupled Receptor Kinase. Br. J. Pharmacol. 2020, 180, 943. [Google Scholar] [CrossRef] [PubMed]
- Meqbil, Y.; Rijn, R. van Opportunities and Challenges for In Silico Drug Discovery at Delta Opioid Receptors. Pharmaceuticals 2022, 15, 873. [Google Scholar] [CrossRef]
- Deo, O.; Alvi, S.; Pham, V.; Christopoulos, A.; Thal, D.M.; Jörg, M.; Capuano, B.; Valant, C.; Scammells, P.J. The Design, Synthesis, and Evaluation of Novel 9-Arylxanthenedione-Based Allosteric Modulators for the δ-Opioid Receptor. J. Med. Chem. 2022, 65, 12367. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Miyano, K.; Hirayama, S.; Karasawa, Y.; Ohshima, K.; Uezono, E.; Komatsu, A.; Nonaka, M.; Fujii, H.; Yamaguchi, K.; et al. Evaluation of the Intracellular Signaling Activities of κ-Opioid Receptor Agonists, Nalfurafine Analogs; Focusing on the Selectivity of G-Protein- and β-Arrestin-Mediated Pathways. Molecules 2022, 27, 7065. [Google Scholar] [CrossRef]
- Brust, T.F. Biased Ligands at the Kappa Opioid Receptor: Fine-Tuning Receptor Pharmacology. In The Kappa Opioid Receptor; Springer: Cham, Switzerland, 2020; pp. 115–135. [Google Scholar]
- Kelly, E.; Conibear, A.; Henderson, G. Biased Agonism: Lessons from Studies of Opioid Receptor Agonists. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 491–515. [Google Scholar] [CrossRef]
- Puig, S.; Barker, K.E.; Szott, S.R.; Kann, P.T.; Morris, J.S.; Gutstein, H.B. Spinal Opioid Tolerance Depends upon Platelet-Derived Growth Factor Receptor-β Signaling, Not μ-Opioid Receptor Internalization. Mol. Pharmacol. 2020, 98, 487–496. [Google Scholar] [CrossRef]
- Gamble, M.C.; Williams, B.R.; Singh, N.; Posa, L.; Freyberg, Z.; Logan, R.W.; Puig, S. Mu-Opioid Receptor and Receptor Tyrosine Kinase Crosstalk: Implications in Mechanisms of Opioid Tolerance, Reduced Analgesia to Neuropathic Pain, Dependence, and Reward. Front. Syst. Neurosci. 2022, 16, 1059089. [Google Scholar] [CrossRef]
- Gaborit, M.; Massotte, D. Therapeutic Potential of Opioid Receptor Heteromers in Chronic Pain and Associated Comorbidities. Br. J. Pharmacol. 2023, 180, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Kunselman, J.M.; Gupta, A.; Gomes, I.; Devi, L.A.; Puthenveedu, M.A. Compartment-Specific Opioid Receptor Signaling Is Selectively Modulated by Different Dynorphin Peptides. eLife 2021, 10, e60270. [Google Scholar] [CrossRef]
- Lec, P.M.; Lenis, A.T.; Golla, V.; Brisbane, W.; Shuch, B.; Garraway, I.P.; Reiter, R.E.; Chamie, K. The Role of Opioids and Their Receptors in Urological Malignancy: A Review. J. Urol. 2020, 204, 1150–1159. [Google Scholar] [CrossRef]
- Carli, M.; Donnini, S.; Pellegrini, C.; Coppi, E.; Bocci, G. Opioid Receptors beyond Pain Control: The Role in Cancer Pathology and the Debated Importance of Their Pharmacological Modulation. Pharmacol. Res. 2020, 159, 104938. [Google Scholar] [CrossRef]
- Lewis, J.W.; Shavit, Y.; Terman, G.W.; Nelson, L.R.; Gale, R.P.; Liebeskind, J.C. Apparent Involvement of Opioid Peptides in Stress-Induced Enhancement of Tumor Growth. Peptides 1983, 4, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Cabot, P.J.; Carter, L.; Schäfer, M.; Stein, C. Methionine-Enkephalin-and Dynorphin A-Release from Immune Cells and Control of Inflammatory Pain. Pain 2001, 93, 207–212. [Google Scholar] [CrossRef]
- Samuelsson, H.; Ekman, R.; Hedner, T. CSF Neuropeptides in Cancer Pain: Effects of Spinal Opioid Therapy. Acta Anaesthesiol. Scand. 1993, 37, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Viet, C.T.; Dang, D.; Ye, Y.; Ono, K.; Campbell, R.R.; Schmidt, B.L. Demethylating Drugs as Novel Analgesics for Cancer Pain. Clin. Cancer Res. 2014, 20, 4882–4893. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.L.; Fell, G.L.; Kemény, L.V.; Fung, C.Y.; Held, K.D.; Biggs, P.J.; Rivera, P.D.; Bilbo, S.D.; Igras, V.; Willers, H.; et al. β-Endorphin Mediates Radiation Therapy Fatigue. Sci. Adv. 2022, 8, eabn6025. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.; Shao, L.; Yang, Y.; Wang, R.; Yang, C.; Zhang, B. Correlations of Cancer Pain Degree with Levels of β-EP, CGRP, and PGE2 and the Effects of Oxycontin on Them. J BUON 2018, 23, 1552–1557. [Google Scholar]
- Yamano, S.; Viet, C.T.; Dang, D.; Dai, J.; Hanatani, S.; Takayama, T.; Kasai, H.; Imamura, K.; Campbell, R.; Ye, Y.; et al. Ex Vivo Nonviral Gene Delivery of μ-Opioid Receptor to Attenuate Cancer-Induced Pain. Pain 2017, 158, 240–251. [Google Scholar] [CrossRef]
- Wu, T.-T.; Wang, Z.-G.; Ou, W.-L.; Wang, J.; Yao, G.-Q.; Yang, B.; Rao, Z.-G.; Gao, J.-F.; Zhang, B.-C. Intravenous Flurbiprofen Axetil Enhances Analgesic Effect of Opioids in Patients with Refractory Cancer Pain by Increasing Plasma β-Endorphin. Asian Pac. J. Cancer Prev. 2015, 15, 10855–10860. [Google Scholar] [CrossRef]
- Apryani, E.; Ali, U.; Wang, Z.-Y.; Wu, H.-Y.; Mao, X.-F.; Ahmad, K.A.; Li, X.-Y.; Wang, Y.-X. The Spinal Microglial IL-10/β-Endorphin Pathway Accounts for Cinobufagin-Induced Mechanical Antiallodynia in Bone Cancer Pain Following Activation of A7-Nicotinic Acetylcholine Receptors. J. Neuroinflamm. 2020, 17, 75. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Hu, W.; He, H.; Gong, Z.; Wang, J.; Yu, X.; Ai, T.; Zhan, L. A Study on the Mechanism of Cinobufagin in the Treatment of Paw Cancer Pain by Modulating Local β -Endorphin Expression In Vivo. Evid. Based Complement. Alternat. Med. 2013, 2013, 851256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xia, C.; Zhang, X.; Li, W.; Miao, X.; Zhou, Q. Wrist–Ankle Acupuncture Attenuates Cancer-Induced Bone Pain by Regulating Descending Pain-Modulating System in a Rat Model. Chin. Med. 2020, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Du, J.-Y.; Liang, Y.; Fang, J.-F.; Jiang, Y.-L.; Shao, X.-M.; He, X.-F.; Fang, J.-Q. Effect of Systemic Injection of Heterogenous and Homogenous Opioids on Peripheral Cellular Immune Response in Rats with Bone Cancer Pain: A Comparative Study. Exp. Ther. Med. 2016, 12, 2568–2576. [Google Scholar] [CrossRef]
- Gustin, T.; Bachelot, T.; Verna, J.M.; Molin, L.F.; Brunet, J.F.; Berger, F.R.; Benabid, A.L. Immunodetection of Endogenous Opioid Peptides in Human Brain Tumors and Associated Cyst Fluids. Cancer Res. 1993, 53, 4715–4719. [Google Scholar]
- Lu, W.; Xie, H.; Tie, X.; Wang, R.; Wu, A.; Shan, F. NFAT-1 Hyper-Activation by Methionine Enkephalin (MENK) Significantly Induces Cell Apoptosis of Rats C6 Glioma in Vivo and in Vitro. Int. Immunopharmacol. 2018, 56, 1–8. [Google Scholar] [CrossRef]
- Molin, L.; Verna, J.-M.; Nissou, M.-F.; Benabid, A.L. Met-Enkephalin Receptors in Human Gliomas. NeuroReport 1994, 5, 2474–2476. [Google Scholar] [CrossRef]
- Mar, E.-C.; Suh, H.H.; Hong, J.-S. Regulation of Proenkephalin Expression in C6 Rat Glioma Cells. Mol. Cell. Neurosci. 1992, 3, 518–528. [Google Scholar] [CrossRef]
- Yin, J.; Lee, J.A.; Howells, R.D. Stimulation of C-Fos and c-Jun Gene Expression and Down-Regulation of Proenkephalin Gene Expression in C6 Glioma Cells by Endothelin-1. Mol. Brain Res. 1992, 14, 213–220. [Google Scholar] [CrossRef]
- Lee, Y.S.; Wurster, R.D. Differential Effects of Methionine Enkephalin on the Growth of Brain Tumor Cells. J. Neurooncol. 1994, 19, 11–15. [Google Scholar] [CrossRef]
- Westphal, M.; Li, C.H. Beta-Endorphin: Characterization of Binding Sites Specific for the Human Hormone in Human Glioblastoma SF126 Cells. Proc. Natl. Acad. Sci. USA 1984, 81, 2921–2923. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, S.; Rao, A.P.K.; Sampathkumar, M.M.; Kanaka, T.S. Presence of Immunoreactive Beta-Endorphin in Human Brain Tumor Cyst Fluids. J. Neurol. Sci. 1983, 59, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Wang, X.Q.; Wang, C.H.; Lu, C.L. Effect of Dynorphin A1-13 on C6 Glioma Cells Swelling Induced by Glutamate. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2001, 17, 76–78. [Google Scholar] [PubMed]
- Bortsov, A.V.; Millikan, R.C.; Belfer, I.; Boortz-Marx, R.L.; Arora, H.; McLean, S.A. μ-Opioid Receptor Gene A118G Polymorphism Predicts Survival in Patients with Breast Cancer. Anesthesiology 2012, 116, 896–902. [Google Scholar] [CrossRef]
- Chatikhine, V.A.; Chevrier, A.; Chauzy, C.; Duval, C.; d’Anjou, J.; Girard, N.; Delpech, B. Expression of Opioid Peptides in Cells and Stroma of Human Breast Cancer and Adenofibromas. Cancer Lett. 1994, 77, 51–56. [Google Scholar] [CrossRef]
- Drell, T.L.; Joseph, J.; Lang, K.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Effects of Neurotransmitters on the Chemokinesis and Chemotaxis of MDA-MB-468 Human Breast Carcinoma Cells. Breast Cancer Res. Treat. 2003, 80, 63–70. [Google Scholar] [CrossRef]
- Melander, O.; Orho-Melander, M.; Manjer, J.; Svensson, T.; Almgren, P.; Nilsson, P.M.; Engström, G.; Hedblad, B.; Borgquist, S.; Hartmann, O.; et al. Stable Peptide of the Endogenous Opioid Enkephalin Precursor and Breast Cancer Risk. J. Clin. Oncol. 2015, 33, 2632–2638. [Google Scholar] [CrossRef]
- Zagon, I.S.; Porterfield, N.K.; McLaughlin, P.J. Opioid Growth Factor—Opioid Growth Factor Receptor Axis Inhibits Proliferation of Triple Negative Breast Cancer. Exp. Biol. Med. 2013, 238, 589–599. [Google Scholar] [CrossRef]
- Ramírez-Expósito, M.J.; Dueñas-Rodríguez, B.; Carrera-González, M.P.; Navarro-Cecilia, J.; Martínez-Martos, J.M. Circulating Levels of β-Endorphin and Cortisol in Breast Cancer. Compr. Psychoneuroendocrinol. 2021, 5, 100028. [Google Scholar] [CrossRef]
- Argueta, D.A.; Aich, A.; Lei, J.; Kiven, S.; Nguyen, A.; Wang, Y.; Gu, J.; Zhao, W.; Gupta, K. β-Endorphin at the Intersection of Pain and Cancer Progression: Preclinical Evidence. Neurosci. Lett. 2021, 744, 135601. [Google Scholar] [CrossRef]
- Heiny, B.M.; Albrecht, V.; Beuth, J. Correlation of Immune Cell Activities and Beta-Endorphin Release in Breast Carcinoma Patients Treated with Galactose-Specific Lectin Standardized Mistletoe Extract. Complement. Ther. Med. 1999, 7, 121. [Google Scholar] [CrossRef]
- Heiny, B.M.; Beuth, J. Mistletoe Extract Standardized for the Galactoside-Specific Lectin (ML-1) Induces Beta-Endorphin Release and Immunopotentiation in Breast Cancer Patients. Anticancer Res. 1994, 14, 1339–1342. [Google Scholar] [PubMed]
- Vaswani, K.K.; Tejwani, G.A.; Abou-Issa, H.M. Effect of 7,12-Dimethylbenz[a]Anthracene-Induced Mammary Carcinogenesis on the Opioid Peptide Levels in the Rat Central Nervous System. Cancer Lett. 1986, 31, 115–122. [Google Scholar] [CrossRef]
- Zhang, C.; Franklin, T.; Sarkar, D.K. Inhibition of Mammary Cancer Progression in Fetal Alcohol Exposed Rats by β -Endorphin Neurons. Alcohol. Clin. Exp. Res. 2016, 40, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.K.; Zhang, C. Beta-Endorphin Neuron Regulates Stress Response and Innate Immunity to Prevent Breast Cancer Growth and Progression. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2013; Volume 93, pp. 263–276. ISBN 978-0-12-416673-8. [Google Scholar]
- Sarkar, D.K.; Murugan, S.; Zhang, C.; Boyadjieva, N. Regulation of Cancer Progression by β-Endorphin Neuron. Cancer Res. 2012, 72, 836–840. [Google Scholar] [CrossRef]
- Faith, R.E.; Liang, H.J.; Murgo, A.J.; Plotnikoff, N.P. Neuroimmunomodulation with Enkephalins: Enhancement of Human Natural Killer (NK) Cell Activity in Vitro. Clin. Immunol. Immunopathol. 1984, 31, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Faith, R.E.; Liang, H.J.; Plotnikoff, N.P.; Murgo, A.J.; Nimeh, N.F. Neuroimmunomodulation with Enkephalins: In Vitro Enhancement of Natural Killer Cell Activity in Peripheral Blood Lymphocytes from Cancer Patients. Nat. Immun. Cell Growth Regul. 1987, 6, 88–98. [Google Scholar]
- Bryant, H.U.; Conroy, W.G.; Isom, G.E.; Malven, P.V.; Yim, G.K.W. Presence of Dynorphin-like Immunoreactivity but Not Opiate Binding in Walker-256 Tumors. Life Sci. 1985, 37, 155–160. [Google Scholar] [CrossRef]
- Qu, N.; Wang, R.; Meng, Y.; Liu, N.; Zhai, J.; Shan, F. Methionine Enkephalin Inhibited Cervical Carcinoma via Apoptosis Promotion and Reduction of Myeloid Derived Suppressor Cell Infiltrated in Tumor. Int. Immunopharmacol. 2022, 110, 108933. [Google Scholar] [CrossRef]
- Saraswati, W.; Wardani, R.; Suhatno, S.; Hartono, P.; Imandiri, A. The Effect of Electroacupuncture Therapy on Pain, Plasma β-Endorphin, and Quality of Life of Stage III Cervical Cancer Patients: A Randomized Control Trial. J. Acupunct. Meridian Stud. 2021, 14, 4–12. [Google Scholar] [CrossRef]
- Ma, M.; Wang, X.; Liu, N.; Shan, F.; Feng, Y. Low-Dose Naltrexone Inhibits Colorectal Cancer Progression and Promotes Apoptosis by Increasing M1-Type Macrophages and Activating the Bax/Bcl-2/Caspase-3/PARP Pathway. Int. Immunopharmacol. 2020, 83, 106388. [Google Scholar] [CrossRef] [PubMed]
- Alumets, J.; Sundler, F.; Alm, P.; Falkmer, S.; Ljungberg, O.; Håkanson, R.; Mårtensson, H.; Tibblin, S. Immunohistochemical Evidence of Peptide Hormones in Endocrine Tumors of the Rectum. Cancer 1981, 48, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Iishi, H.; Tatsuta, M.; Baba, M.; Okuda, S.; Taniguchi, H. Enhancement by Methionine Enkephalin of Colon Carcinogenesis Induced by Azoxymethane. Cancer Res. 1991, 51, 785–788. [Google Scholar] [PubMed]
- Wang, X.; Li, S.; Yan, S.; Shan, Y.; Wang, X.; Jingbo, Z.; Wang, Y.; Shan, F.; Griffin, N.; Sun, X. Methionine Enkephalin Inhibits Colorectal Cancer by Remodeling the Immune Status of the Tumor Microenvironment. Int. Immunopharmacol. 2022, 111, 109125. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Luo, Y.; Fujii, K.; Sasahira, T.; Moriwaka, Y.; Tatsumoto, N.; Sasaki, T.; Yamashita, Y.; Ohmori, H. CD10 Enhances Metastasis of Colorectal Cancer by Abrogating the Anti-Tumoural Effect of Methionine-Enkephalin in the Liver. Gut 2010, 59, 348–356. [Google Scholar] [CrossRef]
- Ogasawara, M.; Murata, J.; Ayukawa, K.; Saiki, I. Differential Effect of Intestinal Neuropeptides on Invasion and Migration of Colon Carcinoma Cells in Vitro. Cancer Lett. 1997, 119, 125–130. [Google Scholar] [CrossRef]
- Tari, A.; Miyachi, Y.; Sumii, K.; Haruma, K.; Yoshihara, M.; Kajiyama, G.; Miyoshi, A. Beta-Endorphin-like Immunoreactivity in Normal Mucosa, Muscle Layer, Adenocarcinoma, and Polyp of the Colon. Dig. Dis. Sci. 1988, 33, 429–434. [Google Scholar] [CrossRef]
- Hiramoto, K.; Yokoyama, S.; Yamate, Y. Ultraviolet A Eye Irradiation Ameliorates Colon Carcinoma Induced by Azoxymethane and Dextran Sodium Sulfate through β-Endorphin and Methionine-Enkephalin. Photodermatol. Photoimmunol. Photomed. 2017, 33, 84–91. [Google Scholar] [CrossRef]
- Murugan, S.; Dave, Y.; Rakhit, A.; Sarkar, D.K. Hypothalamic Beta-Endorphin Neurons Suppress Preneoplastic and Neoplastic Lesions Development in 1,2-Dimethylhydrazine Induced Rat Colon Cancer Model. J. Cancer 2017, 8, 3105–3113. [Google Scholar] [CrossRef]
- Simon, R.H.; Arbo, T.E.; Lundy, J. β-Endorphin Injected into the Nucleus of the Raphe Magnus Facilitates Metastatic Tumor Growth. Brain Res. Bull. 1984, 12, 487–491. [Google Scholar] [CrossRef]
- Karl, M.; Saviolakis, G.A.; Gravanis, A.; Chrousos, G.P.; Margioris, A.N. The PC12 Rat Pheochromocytoma Cell Line Expresses the Prodynorphin Gene and Secretes the 8 KDa Dynorphin Product. Regul. Pept. 1996, 61, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; McLaughlin, P.J. Opioids and the Apoptotic Pathway in Human Cancer Cells. Neuropeptides 2003, 37, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Shan, F.; Qu, N.; Huang, H.; Handley, M.; Griffin, N.; Zhang, S.; Cao, X. Regulatory Role of Methionine Enkephalin in Myeloid-Derived Suppressor Cells and Macrophages in Human Cutaneous Squamous Cell Carcinoma. Int. Immunopharmacol. 2021, 99, 107996. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Cao, X.; Qu, N.; Huang, H.; Handley, M.; Zhang, S.; Shan, F. Methionine Enkephalin Activates Autophagy and Stimulates Tumour Cell Immunogenicity in Human Cutaneous Squamous Cell Carcinoma. Int. Immunopharmacol. 2021, 96, 107733. [Google Scholar] [CrossRef]
- Wang, X.; Tian, J.; Jiao, X.; Geng, J.; Wang, R.; Liu, N.; Gao, X.; Griffin, N.; Gao, Y.; Shan, F. The Novel Mechanism of Anticancer Effect on Gastric Cancer through Inducing G0/G1 Cell Cycle Arrest and Caspase-Dependent Apoptosis in Vitro and in Vivo by Methionine Enkephalin. Cancer Manag. Res. 2018, 10, 4773–4787. [Google Scholar] [CrossRef]
- Wang, X.; Jiao, X.; Meng, Y.; Chen, H.; Griffin, N.; Gao, X.; Shan, F. Methionine Enkephalin (MENK) Inhibits Human Gastric Cancer through Regulating Tumor Associated Macrophages (TAMs) and PI3K/AKT/mTOR Signaling Pathway inside Cancer Cells. Int. Immunopharmacol. 2018, 65, 312–322. [Google Scholar] [CrossRef]
- Zhou, X.; Cao, S.-G.; Tan, X.-J.; Liu, X.-D.; Li, Z.-Q.; Kong, L.-X.; Tian, Y.-L.; Liu, D.; Shen, S.; Sun, Y.-Q.; et al. Effects of Transcutaneous Electrical Acupoint Stimulation (TEAS) on Postoperative Recovery in Patients with Gastric Cancer: A Randomized Controlled Trial. Cancer Manag. Res. 2021, 13, 1449–1458. [Google Scholar] [CrossRef]
- Tari, A.; Miyachi, Y.; Hide, M.; Sumii, K.; Kajiyama, G.; Tahara, E.; Tanaka, K.; Miyoshi, A. β-Endorphinlike Immunoreactivity and Somatostatinlike Immunoreactivity in Normal Gastric Mucosa, Muscle Layer, and Adenocarcinoma. Gastroenterology 1985, 88, 670–674. [Google Scholar] [CrossRef]
- Gorur, A.; Patiño, M.; Takahashi, H.; Corrales, G.; Pickering, C.R.; Gleber-Netto, F.O.; Myers, J.N.; Cata, J.P. Mu-Opioid Receptor Activation Promotes in Vitro and in Vivo Tumor Growth in Head and Neck Squamous Cell Carcinoma. Life Sci. 2021, 278, 119541. [Google Scholar] [CrossRef]
- Levin, R.J.; Wu, Y.; McLaughlin, P.J.; Zagon, I.S. Expression of the Opioid Growth Factor, [Met5]-Enkephalin, and the Zeta Opioid Receptor in Head and Neck Squamous Cell Carcinoma. Laryngoscope 1997, 107, 335–339. [Google Scholar] [CrossRef]
- McLaughlin, P.J.; Stucki, J.K.; Zagon, I.S. Modulation of the Opioid Growth Factor ([Met5]-Enkephalin)-Opioid Growth Factor Receptor Axis: Novel Therapies for Squamous Cell Carcinoma of the Head and Neck. Head Neck 2012, 34, 513–519. [Google Scholar] [CrossRef]
- Cheng, F.; Zagon, I.S.; Verderame, M.F.; McLaughlin, P.J. The Opioid Growth Factor (OGF)–OGF Receptor Axis Uses the P16 Pathway to Inhibit Head and Neck Cancer. Cancer Res. 2007, 67, 10511–10518. [Google Scholar] [CrossRef]
- McLaughlin, P.; Zagon, I. Progression of Squamous Cell Carcinoma of the Head and Neck Is Associated with Down-Regulation of the Opioid Growth Factor Receptor. Int. J. Oncol. 2006, 28, 1577–1583. [Google Scholar] [CrossRef]
- Warren, W.H.; Lee, I.; Gould, V.E.; Memoli, V.A.; Jao, W. Paragangliomas of the Head and Neck: Ultrastructural and Immunohistochemical Analysis. Ultrastruct. Pathol. 1985, 8, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.T.; Peterson, K.A. Beta Endorphin Enhances in Vitro Lymphokine Production in Patients with Squamous Carcinoma of the Head and Neck. Otolaryngol. Neck Surg. 1986, 94, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; Rahn, K.A.; McLaughlin, P.J. Opioids and Migration, Chemotaxis, Invasion, and Adhesion of Human Cancer Cells. Neuropeptides 2007, 41, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; McLaughlin, P.J. Opioids and Differentiation in Human Cancer Cells. Neuropeptides 2005, 39, 495–505. [Google Scholar] [CrossRef]
- Salim, S.A.; Milroy, C.; Rode, J.; Corrin, B.; Hamid, Q. Immunocytochemical Characterization of Neuroendocrine Tumours of the Larynx. Histopathology 1993, 23, 69–73. [Google Scholar] [CrossRef]
- Bishop, J.W.; Osamura, R.Y.; Tsutsumi, Y. Multiple Hormone Production in an Oat Cell Carcinoma of the Larynx. Pathol. Int. 1985, 35, 915–923. [Google Scholar] [CrossRef]
- Monstein, H.-J.; Folkesson, R.; Terenius, L. Proenkephalin A-like MRNA in Human Leukemia Leukocytes and CNS-Tissues. Life Sci. 1986, 39, 2237–2241. [Google Scholar] [CrossRef]
- Martin-Kleiner, I.; Gabrilovac, J.; Kušec, R.; Boranić, M. Methionine Enkephalin Suppresses Metabolic Activity of a Leukemic Cell Line (NALM-1) and Enhances CD10 Expression. Int. Immunopharmacol. 2003, 3, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Mernenko, O.A.; Blishchenko, E.Y.; Mirkina, I.I.; Karelin, A.A. Met-Enkephalin Induces Cytolytic Processes of Apoptotic Type in K562 Human Erythroid Leukemia Cells. FEBS Lett. 1996, 383, 230–232. [Google Scholar] [CrossRef]
- Heagy, W.; Duca, K.; Finberg, R.W. Enkephalins Stimulate Leukemia Cell Migration and Surface Expression of CD9. J. Clin. Investig. 1995, 96, 1366–1374. [Google Scholar] [CrossRef]
- Iannetti, P.; Fabbri, A.; Meloni, G.; Moleti, M.L.; Ulisse, S.; Mandelli, F.; Isidori, A.; Imperato, C. Immunoreactive Beta-Endorphin Levels in Cerebrospinal Fluid of Children with Acute Lymphoblastic Leukemia: Relationship with Glucocorticoid Therapy and Neurological Complications. J. Endocrinol. Investig. 1989, 12, 623–629. [Google Scholar] [CrossRef]
- Barni, S.; Lissoni, P.; Tancini, G.; Crispino, S.; Paolorossi, F.; Rovelli, F.; Fumagalli, G.; Ferri, L.; Esposti, D.; Esposti, G.; et al. Acute Effects of Various Chemotherapeutic Combinations on Hypophyseal and Pineal Hormone Secretions in Cancer Patients. Tumori J. 1987, 73, 181–185. [Google Scholar] [CrossRef]
- Makeshova, A.B.; Mamukova, L.I.; Levina, A.A.; Sysoeva, E.P.; Eralieva, M.O.; Savchenko, V.G. Dynamic Beta-Endorphin Determination in Hematologic Patients. Ter Arkh 2012, 84, 22–25. [Google Scholar]
- Malkova, N.V.; Krasnova, S.B.; Navolotskaya, E.V.; Zargarova, T.A.; Prasolov, V.S. Effect of Beta-Endorphin and Beta-Endorphin-like Peptide Immunorphin on the Growth of Human Leukemic Cells in Vitro. Russ J Immunol 2002, 7, 239–244. [Google Scholar]
- Shahabi, N.A.; Sharp, B.M. Activation of Protein Kinase C Rapidly Down- Regulates Naloxone-Resistant Receptors for Beta-Endorphin on U937 Cells. J Pharmacol Exp Ther 1993, 64, 276–281. [Google Scholar]
- Avella, D.M.; Kimchi, E.T.; Donahue, R.N.; Tagaram, H.R.S.; McLaughlin, P.J.; Zagon, I.S.; Staveley-O’Carroll, K.F. The Opioid Growth Factor-Opioid Growth Factor Receptor Axis Regulates Cell Proliferation of Human Hepatocellular Cancer. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2010, 298, R459–R466. [Google Scholar] [CrossRef]
- Rogosnitzky, M.; Finegold, M.J.; McLaughlin, P.J.; Zagon, I.S. Opioid Growth Factor (OGF) for Hepatoblastoma: A Novel Non-Toxic Treatment. Investig. New Drugs 2013, 31, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Murugan, S.; Boyadjieva, N.; Sarkar, D.K. Protective Effects of Hypothalamic Beta-Endorphin Neurons Against Alcohol-Induced Liver Injuries and Liver Cancers in Rat Animal Models. Alcohol. Clin. Exp. Res. 2014, 38, 2988–2997. [Google Scholar] [CrossRef] [PubMed]
- Krajnik, M.; Schäfer, M.; Sobanski, P.; Kowalewski, J.; Bloch-Boguslawska, E.; Zylicz, Z.; Mousa, S.A. Enkephalin, Its Precursor, Processing Enzymes, and Receptor as Part of a Local Opioid Network throughout the Respiratory System of Lung Cancer Patients. Hum. Pathol. 2010, 41, 632–642. [Google Scholar] [CrossRef]
- Gosney, J.R.; Gosney, M.A.; Lye, M. Serum Leucine-Enkephalin in Bronchial Carcinoma and Its Relation to Tumour Location. Thorax 1990, 45, 9–11. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ahn, H.J.; Kim, J.K.; Kim, J.; Lee, S.H.; Chae, H.B. Morphine Suppresses Lung Cancer Cell Proliferation Through the Interaction with Opioid Growth Factor Receptor: An In Vitro and Human Lung Tissue Study. Anesth. Analg. 2016, 123, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, H.; Handley, M.; Griffin, N.; Bai, X.; Shan, F. A Novel Mechanism of Lung Cancer Inhibition by Methionine Enkephalin through Remodeling the Immune Status of the Tumor Microenvironment. Int. Immunopharmacol. 2021, 99, 107999. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, N.; Ma, M.; Huang, H.; Handley, M.; Bai, X.; Shan, F. Methionine Enkephalin (MENK) Suppresses Lung Cancer by Regulating the Bcl-2/Bax/Caspase-3 Signaling Pathway and Enhancing Natural Killer Cell-Driven Tumor Immunity. Int. Immunopharmacol. 2021, 98, 107837. [Google Scholar] [CrossRef]
- Mathew, B.; Lennon, F.E.; Siegler, J.; Mirzapoiazova, T.; Mambetsariev, N.; Sammani, S.; Gerhold, L.M.; LaRiviere, P.J.; Chen, C.-T.; Garcia, J.G.N.; et al. The Novel Role of the Mu Opioid Receptor in Lung Cancer Progression: A Laboratory Investigation. Anesth. Analg. 2011, 112, 558–567. [Google Scholar] [CrossRef]
- Maneckjee, R.; Minna, J.D. Opioid and Nicotine Receptors Affect Growth Regulation of Human Lung Cancer Cell Lines. Proc. Natl. Acad. Sci. USA 1990, 87, 3294–3298. [Google Scholar] [CrossRef]
- Faith, R. Inhibition of Pulmonary Metastases and Enhancement of Natural Killer Cell Activity by Methionine-Enkephalin. Brain. Behav. Immun. 1988, 2, 114–122. [Google Scholar] [CrossRef]
- Calogero, A.E.; Polosa, R.; Neville, E.; D’Agata, R. Measurements of Hormonal Peptides in the Bronchoalveolar Fluid as Tumor Markers of Lung Cancer. J. Endocrinol. Investig. 1995, 18, 354–358. [Google Scholar] [CrossRef]
- Calogero, A.E.; Minacapilli, G.; Nicolosi, A.M.G.; Moncada, M.L.; Mistretta, A.; Latteri, S.F.; Polosa, P.; D’Agata, R. Limited Clinical Usefulness of Plasma Corticotropin-Releasing Hormone, Adrenocorticotropin and ß-Endorphin Measurements as Markers of Lung Cancer. J. Endocrinol. Investig. 1992, 15, 581–586. [Google Scholar] [CrossRef]
- Black, M.; Carey, F.A.; Farquharson, M.A.; Murray, G.D.; McNicol, A.M. Expression of the Pro-Opiomelanocortin Gene in Lung Neuroendocrine Tumours: In Situ Hybridization and Immunohistochemical Studies. J. Pathol. 1993, 169, 329–334. [Google Scholar] [CrossRef]
- Taylor, J.E. Human Small Cell Lung Cancer Cells Express High-Affinity Naloxone-Insensitive [125I]β-Endorphin Binding Sites. Life Sci. 1994, 56, PL97–PL102. [Google Scholar] [CrossRef]
- Melzig, M.F.; Nylander, I.; Vlaskovska, M.; Terenius, L. β-Endorphin Stimulates Proliferation of Small Cell Lung Carcinoma Cells in Vitro via Nonopioid Binding Sites. Exp. Cell Res. 1995, 219, 471–476. [Google Scholar] [CrossRef]
- Ruff, M.; Schiffmann, E.; Terranova, V.; Pert, C.B. Neuropeptides Are Chemoattractants for Human Tumor Cells and Monocytes: A Possible Mechanism for Metastasis. Clin. Immunol. Immunopathol. 1985, 37, 387–396. [Google Scholar] [CrossRef]
- Geijer, T.; Bergh, J.; Terenius, L. Expression of Preprodynorphin in Human Small Cell Lung Carcinoma Cell Lines. Regul. Pept. 1991, 34, 181–188. [Google Scholar] [CrossRef]
- Banerjee, J.; Papu John, A.M.S.; Schuller, H.M. Regulation of Nonsmall-Cell Lung Cancer Stem Cell like Cells by Neurotransmitters and Opioid Peptides: Neuronal Regulation of Lung Cancer Stem Cells. Int. J. Cancer 2015, 137, 2815–2824. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Krajnik, M.; Sobanski, P.; Kowaleswski, J.; Bloch-Boguslawska, E.; Zylicz, M. Dynorphin Expression, Processing and Receptors in the Alveolar Macrophages, Cancer Cells and Bronchial Epithelium of Lung Cancer Patients. Histol. Histopathol. 2010, 25, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-E.; Liu, L.-F.; Dusting, G.J.; Weng, W.-T.; Chen, S.-C.; Kung, M.-L.; Tee, R.; Liu, G.-S.; Tai, M.-H. Pro-Opiomelanocortin Gene Delivery Suppresses the Growth of Established Lewis Lung Carcinoma through a Melanocortin-1 Receptor-Independent Pathway: POMC Gene Therapy for Lung Cancer. J. Gene Med. 2012, 14, 44–53. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A.; Zbytek, B.; Brozyna, A.A.; Granese, J.; Pisarchik, A.; Szczesniewski, A.; Tobin, D.J. Regulated Proenkephalin Expression in Human Skin and Cultured Skin Cells. J. Investig. Dermatol. 2011, 131, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Wick, M.R.; Millns, J.L.; Sibley, R.K.; Pittelkow, M.R.; Winkelmann, R.K. Secondary Neuroendocrine Carcinomas of the Skin. J. Am. Acad. Dermatol. 1985, 13, 134–142. [Google Scholar] [CrossRef]
- Murgo, A.J. Inhibition of B16-BL6 Melanoma Growth in Mice by Methionine-Enkephalin2. JNCI J. Natl. Cancer Inst. 1985, 75, 341–344. [Google Scholar] [CrossRef]
- Wang, D.-M.; Jiao, X.; Plotnikoff, N.P.; Griffin, N.; Qi, R.-Q.; Gao, X.-H.; Shan, F.-P. Killing Effect of Methionine Enkephalin on Melanoma in Vivo and in Vitro. Oncol. Rep. 2017, 38, 2132–2140. [Google Scholar] [CrossRef]
- Wang, D.-M.; Wang, G.-C.; Yang, J.; Plotnikoff, N.P.; Griffin, N.; Han, Y.-M.; Qi, R.-Q.; Gao, X.-H.; Shan, F.-P. Inhibition of the Growth of Human Melanoma Cells by Methionine Enkephalin. Mol. Med. Rep. 2016, 14, 5521–5527. [Google Scholar] [CrossRef]
- Zagon, I.S.; Donahue, R.N.; Rogosnitzky, M.; Mclaughlin, P.J. Imiquimod Upregulates the Opioid Growth Factor Receptor to Inhibit Cell Proliferation Independent of Immune Function. Exp. Biol. Med. 2008, 233, 968–979. [Google Scholar] [CrossRef]
- O’Hern, K.; Chambers, M.; Ryan, C.; Chapman, M.S. In Lieu of Penectomy: Complete Resolution of Penile Melanoma in Situ with Topical Imiquimod and Tretinoin. Int. J. Dermatol. 2021, 60, e297–e299. [Google Scholar] [CrossRef]
- Nahm, W.J.; Gwillim, E.C.; Badiavas, E.V.; Nichols, A.J.; Kirsner, R.S.; Boggeln, L.H.; Shen, J.T. Treating Melanoma in Situ During a Pandemic with Telemedicine and a Combination of Imiquimod, 5-Fluorouracil, and Tretinoin. Dermatol. Ther. 2021, 11, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Iznardo, H.; Garcia-Melendo, C.; Yélamos, O. Lentigo Maligna: Clinical Presentation and Appropriate Management. Clin. Cosmet. Investig. Dermatol. 2020, 13, 837–855. [Google Scholar] [CrossRef] [PubMed]
- Lobo, Y.; Templeman, R. Conservative Treatment of Lentigo Maligna with Topical Imiquimod 5% Cream: A Case Report. Dermatol. Online J. 2020, 26. [Google Scholar] [CrossRef]
- Scarfì, F.; Patrizi, A.; Veronesi, G.; Lambertini, M.; Tartari, F.; Mussi, M.; Melotti, B.; Dika, E. The Role of Topical Imiquimod in Melanoma Cutaneous Metastases: A Critical Review of the Literature. Dermatol. Ther. 2020, 33, e14165. [Google Scholar] [CrossRef]
- Neil, S.M.; Johnson, S.K.; Bleehen, S.S.; Brown, B.L.; Tomlinson, S. Stimulation of the Adenylate Cyclase of a B16 Melanoma Cell Line by Pro-Opiocortin-Related Peptides—A Structure-Activity Study. Regul. Pept. 1981, 2, 193–200. [Google Scholar] [CrossRef]
- Nagahama, M.; Funasaka, Y.; Fernandez-Frez, M.L.; Ohashi, A.; Chakraborty, A.K.; Ueda, M.; Ichihashi, M. Immunoreactivity of Alpha-Melanocyte-Stimulating Hormone, Adrenocorticotrophic Hormone and Beta-Endorphin in Cutaneous Malignant Melanoma and Benign Melanocytic Naevi. Br. J. Dermatol. 1998, 138, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, S.; Hardt, K.; Schadendorf, D.; Henschler, R. Endogenous μ-Opioid Peptides Modulate Immune Response towards Malignant Melanoma. Exp. Dermatol. 2011, 20, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Fell, G.L.; Robinson, K.C.; Mao, J.; Woolf, C.J.; Fisher, D.E. Skin β-Endorphin Mediates Addiction to UV Light. Cell 2014, 157, 1527–1534. [Google Scholar] [CrossRef]
- Kerros, C.; Brood, I.; Sola, B.; Jauzac, P.; Allouche, S. Reduction of Cell Proliferation and Potentiation of Fas-Induced Apoptosis by the Selective Kappa-Opioid Receptor Agonist U50 488 in the Multiple Myeloma LP-1 Cells. J. Neuroimmunol. 2010, 220, 69–78. [Google Scholar] [CrossRef]
- Bamberger, A.-M.; Pu, L.-P.; Cool, D.R.; Loh, Y.P. The Neuro-2a Neuroblastoma Cell Line Expresses [Met]-Enkephalin and Vasopressin mRNA and Peptide. Mol. Cell. Endocrinol. 1995, 113, 155–163. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.J.; Zagon, I.S.; Skitzki, J. Human Neuroblastoma Cell Growth in Tissue Culture Is Regulated by Opioid Growth Factor. Int. J. Oncol. 1999, 14, 373–453. [Google Scholar] [CrossRef] [PubMed]
- Westphal, M.; Li, C.H. β-Endorphin: Demonstration of Binding Sites in Three Human Neuroblastoma Cell Lines Specific for the COOH-Terminal Segment of the Human Hormone. Biochem. Biophys. Res. Commun. 1984, 120, 873–878. [Google Scholar] [CrossRef]
- Zagon, I.S.; McLaughlin, P.J. Opioid Antagonist Modulation of Murine Neuroblastoma: A Profile of Cell Proliferation and Opioid Peptides and Receptors. Brain Res. 1989, 480, 16–28. [Google Scholar] [CrossRef]
- Zagon, I.S.; Goodman, S.R.; McLaughlin, P.J. Characterization of Opioid Binding Sites in Murine Neuroblastoma. Brain Res. 1988, 449, 80–88. [Google Scholar] [CrossRef]
- Satoh, M.; Yokosawa, H.; Ishii, S. Degradation of Dynorphin-(1–13) and Dynorphin-(1–17) by the Neuroblastoma Cell Membrane. Evidence for the Involvement of a Cysteine Protease. Biochem. Biophys. Res. Commun. 1986, 140, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-J.; Chen, W.-F.; Sung, C.-S.; Jean, Y.-H.; Hung, C.-H.; Chen, F.-A.; Hsieh, M.-H.; Wen, Z.-H. Isoflurane Attenuates Dynorphin-Induced Cytotoxicity and Downregulation of Bcl-2 Expression in Differentiated Neuroblastoma SH-SY5Y Cells: Isoflurane Attenuates Dynorphin Toxicity. Acta Anaesthesiol. Scand. 2009, 53, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Inui, A.; Sano, K.; Ueno, N.; Teranishi, A.; Hirosue, Y.; Nakajima, M.; Okita, M.; Togami, J.; Koshiya, K.; et al. Dynorphin Binds to Neuropeptide Y and Peptide YY Receptors in Human Neuroblastoma Cell Lines. Am. J. Physiol.-Endocrinol. Metab. 1994, 267, E702–E709. [Google Scholar] [CrossRef]
- Sporrong, B.; Falkmer, S.; Robboy, S.J.; Alumets, J.; Hakanson, R.; Ljungberg, O.; Sundler, F. Neurohormonal Peptides in Ovarian Carcinoids: An Immunohistochemical Study of 81 Primary Carcinoids and of Intraovarian Metastases from Six Mid-gut Carcinoids. Cancer 1982, 49, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S.; Donahue, R.; McLaughlin, P.J. Targeting the Opioid Growth Factor: Opioid Growth Factor Receptor Axis for Treatment of Human Ovarian Cancer. Exp. Biol. Med. 2013, 238, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Donahue, R.N.; McLaughlin, P.J.; Zagon, I.S. Low-Dose Naltrexone Suppresses Ovarian Cancer and Exhibits Enhanced Inhibition in Combination with Cisplatin. Exp. Biol. Med. 2011, 236, 883–895. [Google Scholar] [CrossRef]
- Omar, R.A.; Tabbakh, G.H. Immunoreactive Beta-Endorphin in Ovarian Sex Cord-Stromal Tumors. Arch. Pathol. Lab. Med. 1987, 111, 436–439. [Google Scholar]
- Schneider-Matyka, D.; Skwirczyńska, E.; Gaur, M.; Hukowska-Szematowicz, B.; Kwiatkowski, S.; Mikla, M.; Grochans, E.; Cymbaluk-Płoska, A. Evaluation of the Influence of Biological Factors during the Course of Treatment in Patients with Ovarian Cancer. Int. J. Environ. Res. Public Health 2022, 19, 10516. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Kita, T.; Miyauchi, M.; Iwano, I.; Kato, K. Inhibition of Human Ovarian Cancer Cell Proliferation in Vitro by Neuroendocrine Hormones. Gynecol. Oncol. 1989, 32, 60–64. [Google Scholar] [CrossRef]
- Gopalakrishnan, G.; Lepetre, S.; Maksimenko, A.; Mura, S.; Desmaële, D.; Couvreur, P. Lipid-Conjugation of Endogenous Neuropeptides: Improved Biotherapy against Human Pancreatic Cancer. Adv. Healthc. Mater. 2015, 4, 1015–1022. [Google Scholar] [CrossRef]
- Cheng, F.; McLaughlin, P.J.; Verderame, M.F.; Zagon, I.S. The OGF-OGFr Axis Utilizes the P21 Pathway to Restrict Progression of Human Pancreatic Cancer. Mol. Cancer 2008, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Zagon, I.S. Opioid Growth Factor Improves Clinical Benefit and Survival in Patients with Advanced Pancreatic Cancer. Open Access J. Clin. Trials 2010, 2, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Conter, R.L.; Demers, L.M.; McLaughlin, P.J.; Zagon, I.S. Elevated Levels of Opioid Growth Factor in the Plasma of Patients with Pancreatic Cancer. Pancreas 2000, 21, 158–164. [Google Scholar] [CrossRef]
- Vieau, D.; Seidah, N.G.; Day, R. Mouse Insulinoma Beta TC3 Cells Express Prodynorphin Messenger Ribonucleic Acid and Derived Peptides: A Unique Cellular Model for the Study of Prodynorphin Biosynthesis and Processing. Endocrinology 1995, 136, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Konoshita, T.; Gasc, J.M.; Villard, E.; Seidah, N.G.; Corvol, P.; Pinet, F. Co-Expression of PC2 and Proenkephalin in Human Tumoral Adrenal Medullary Tissues. Biochimie 1994, 76, 241–244. [Google Scholar] [CrossRef]
- Yanase, T.; Nawata, H.; Kato, K.; Ibayashi, H. Catecholamines and Opioid Peptides in Human Phaeochromocytomas. Acta Endocrinol. 1986, 113, 378–384. [Google Scholar] [CrossRef]
- Cesselin, F.; Pique, L.; Bertagna, X.; Benlot, C.; Antréassian, J.; Proeschel, M.F.; Girard, F.; Zogbi, F.; Legrand, J.C.; Luton, J.P.; et al. Simultaneous Evaluation of the Catecholamine Pathway and Three Opioid Peptide Producing Systems in Human Pheochromocytomas. Neuropeptides 1984, 4, 175–182. [Google Scholar] [CrossRef]
- Yoshimasa, T.; Nakao, K.; Ohtsuki, H.; Li, S.; Imura, H. Methionine-Enkephalin and Leucine-Enkephalin in Human Sympathoadrenal System and Pheochromocytoma. J. Clin. Investig. 1982, 69, 643–650. [Google Scholar] [CrossRef]
- Schroeder, J.O.; Asa, S.L.; Kovacs, K.; Killinger, D.; Hadley, G.L.; Volpé, R. Report of a Case of Pheochromocytoma Producing Immunoreactive ACTH and Beta-Endorphin. J. Endocrinol. Investig. 1984, 7, 117–122. [Google Scholar] [CrossRef]
- Bertagna, X.; Pique, L.; Ochoa, C.; Luton, J.P.; Bricaire, H.; Serin, D.; Girard, F.; Plouin, P.F.; Corvol, P.; Cesselin, F.; et al. Simultaneous Measurement of β-Endorphin, Lipotrophins, and Met-Enkephalin in Phaeochromocytomas. Acta Endocrinol. 1982, 101, 72–77. [Google Scholar] [CrossRef]
- Howlett, T.A.; Besser, G.M.; Rees, L.H. Characterization of Immunoreactive Dynorphin in Human Phaeochromocytomas. J. Endocrinol. 1988, 117, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Yanase, T.; Nawata, H.; Kato, K.; Ibayashi, H. Preproenkephalin B-Derived Opioid Peptides in Human Phaeochromocytomas. Acta Endocrinol. 1987, 114, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Tozawa, F.; Tachibana, S.; Demura, H.; Shizume, K.; Sasaki, A.; Mouri, T.; Miura, Y. Multiple Forms of Immunoreactive Dynorphin in Human Pituitary and Pheochromocytoma. Life Sci. 1983, 32, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Yoshimasa, T.; Nakao, K.; Oki, S.; Tanaka, I.; Nakai, Y.; Imura, H. Presence of Dynorphin-Like Immunoreactivity in Pheochromocytomas. J. Clin. Endocrinol. Metab. 1981, 53, 213–214. [Google Scholar] [CrossRef]
- Zhu, X.; Robertson, J.T.; Sacks, H.S.; Dohan, F.C.; Tseng, J.-L.; Desiderio, D.M. Opioid and Tachykinin Neuropeptides in Prolactin-Secreting Human Pituitary Adenomas. Peptides 1995, 16, 1097–1107. [Google Scholar] [CrossRef]
- Trouillas, J.; Girod, C.; Sassolas, G.; Vitte, P.A.; Claustrat, B.; Perrin, G.; Lhéritier, M.; Fischer, C.; Dubois, M.P. A Human β-Endorphin Pituitary Adenoma. J. Clin. Endocrinol. Metab. 1984, 58, 242–249. [Google Scholar] [CrossRef]
- Sharp, B.; Melmed, S.; Goldberg, R.; Carlson, H.E.; Refetoff, S.; Hershman, J.M. Radioimmunoassay Detection of Endorphins from Long-Term Culture of Human Pituitary Tumour Cells. Acta Endocrinol. 1982, 99, 174–178. [Google Scholar] [CrossRef]
- Scheithauer, B.W.; Jaap, A.J.; Horvath, E.; Kovacs, K.; Lloyd, R.V.; Meyer, F.B.; Laws, E.R.; Young, W.F. Clinically Silent Corticotroph Tumors of the Pituitary Gland. Neurosurgery 2000, 47, 723–730. [Google Scholar] [CrossRef]
- Low, M.J.; Liu, B.; Hammer, G.D.; Rubinstein, M.; Allen, R.G. Post-Translational Processing of Proopiomelanocortin (POMC) in Mouse Pituitary Melanotroph Tumors Induced by a POMC-Simian Virus 40 Large T Antigen Transgene. J. Biol. Chem. 1993, 268, 24967–24975. [Google Scholar] [CrossRef]
- Beaubien, B.C.; Herbert, E. Modulation of Beta-Endorphin Secretion from Mouse Pituitary Tumor Cells by Calmodulin Inhibitor W7. NIDA Res. Monogr. 1986, 414–417. [Google Scholar]
- Furui, T.; Kageyama, N.; Kuwayama, A.; Nakao, K.; Fukushima, M. Increase of β-Endorphin in Cerebrospinal Fluid after Removal of ACTH-Secreting Pituitary Adenomas. Pain 1981, 11, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Shuman, L.; Warrick, J.I.; Raman, J.D.; Degraff, D.J. Androgen Represses Opioid Growth Factor Receptor (OGFR) in Human Prostate Cancer LNCaP Cells and OGFR Expression in Human Prostate Cancer Tissue. Am. J. Clin. Exp. Urol. 2018, 6, 164–171. [Google Scholar] [PubMed]
- Zylla, D.; Gourley, B.L.; Vang, D.; Jackson, S.; Boatman, S.; Lindgren, B.; Kuskowski, M.A.; Le, C.; Gupta, K.; Gupta, P. Opioid Requirement, Opioid Receptor Expression, and Clinical Outcomes in Patients with Advanced Prostate Cancer: Opioids and Prostate Cancer Outcomes. Cancer 2013, 119, 4103–4110. [Google Scholar] [CrossRef]
- Kampa, M.; Bakogeorgou, E.; Hatzoglou, A.; Damianaki, A.; Martin, P.-M.; Castanas, E. Opioid Alkaloids, and Casomorphin Peptides Decrease the Proliferation of Prostatic Cancer Cell Lines (LNCaP, PC3, and DU145) through a Partial Interaction with Opioid Receptors. Eur. J. Pharmacol. 1997, 335, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, P.-A.; Wadström, L.B.; Alumets, J.; Falkmer, S.; Grimelius, L. Peptide-Hormone- And Serotonin-Immunoreactive Tumour Cells in Carcinoma of the Prostate. Pathol. Res. Pract. 1987, 182, 298–307. [Google Scholar] [CrossRef]
- Sarkar, D.K.; Boyadjieva, N.I.; Chen, C.P.; Ortigüela, M.; Reuhl, K.; Clement, E.M.; Kuhn, P.; Marano, J. Cyclic Adenosine Monophosphate Differentiated β-Endorphin Neurons Promote Immune Function and Prevent Prostate Cancer Growth. Proc. Natl. Acad. Sci. USA 2008, 105, 9105–9110. [Google Scholar] [CrossRef]
- Moon, T.D. The Effect of Opiates upon Prostatic Carcinoma Cell Growth. Biochem. Biophys. Res. Commun. 1988, 153, 722–727. [Google Scholar] [CrossRef]
- Zhang, R.-X.; Li, A.; Liu, B.; Wang, L.; Xin, J.; Ren, K.; Qiao, J.-T.; Berman, B.M.; Lao, L. Electroacupuncture Attenuates Bone Cancer-Induced Hyperalgesia and Inhibits Spinal Preprodynorphin Expression in a Rat Model. Eur. J. Pain 2008, 12, 870–878. [Google Scholar] [CrossRef]
- Shimoyama, M.; Tatsuoka, H.; Ohtori, S.; Tanaka, K.; Shimoyama, N. Change of Dorsal Horn Neurochemistry in a Mouse Model of Neuropathic Cancer Pain. Pain 2005, 114, 221–230. [Google Scholar] [CrossRef]
- Honore, P.; Rogers, S.D.; Schwei, M.J.; Salak-Johnson, J.L.; Luger, N.M.; Sabino, M.C.; Clohisy, D.R.; Mantyh, P.W. Murine Models of Inflammatory, Neuropathic and Cancer Pain Each Generates a Unique Set of Neurochemical Changes in the Spinal Cord and Sensory Neurons. Neuroscience 2000, 98, 585–598. [Google Scholar] [CrossRef]
- Bisignani, G.J.; Mclaughlin, P.J.; Ordille, S.D.; Beltz, M.S.; Jarowenko, M.V.; Zagon, I.S. Human Renal Cell Cancer Proliferation in Tissue Culture Is Tonically Inhibited by Opioid Growth Factor. J. Urol. 1999, 162, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Tarkkanen, A.; Tervo, T.; Tervo, K.; Panula, P. Immunohistochemical Evidence for Preproenkephalin A Synthesis in Human Retinoblastoma. Investig. Ophthalmol. Vis. Sci. 1984, 25, 1210–1212. [Google Scholar]
- Westphal, M.; Li, C.H. Human Retinoblastomas Have Binding Sites for the COOH-Terminal Segment of Human β-Endorphin. Int. J. Pept. Protein Res. 2009, 26, 557–559. [Google Scholar] [CrossRef] [PubMed]
- McMurray, C.T.; Devi, L.; Calavetta, L.; Douglass, J.O. Regulated Expression of the Prodynorphin Gene in the R2C Leydig Tumor Cell Line*. Endocrinology 1989, 124, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Holdaway, I.M.; Jagusch, M.; Kerr, A.R.; Donald, R.A.; Pullan, P.T. Ectopic Secretion of ACTH and Met-Enkephalin from a Thymic Carcinoid. J. Endocrinol. Investig. 1982, 5, 33–37. [Google Scholar] [CrossRef]
- Schweigerer, L.; Schmidt, W.; Teschemacher, H.; Gramsch, C. Beta-Endorphin: Surface Binding and Internalization in Thymoma Cells. Proc. Natl. Acad. Sci. USA 1985, 82, 5751–5755. [Google Scholar] [CrossRef]
- Yamaji, I.; Iimura, O.; Mito, T.; Yoshida, S.; Shimamoto, K.; Minase, T. An Ectopic, ACTH Producing, Oncocytic Carcinoid Tumor of the Thymus Report of a Case. Jpn. J. Med. 1984, 23, 62–66. [Google Scholar] [CrossRef]
- Csuhai, E.; Safavi, A.; Hersh, L.B. Purification and Characterization of a Secreted Arginine-Specific Dibasic Cleaving Enzyme from EL-4 Cells. Biochemistry 1995, 34, 12411–12419. [Google Scholar] [CrossRef]
- McLaughlin, P.J.; Zagon, I.S.; Park, S.S.; Conway, A.; Donahue, R.N.; Goldenberg, D. Growth Inhibition of Thyroid Follicular Cell-Derived Cancers by the Opioid Growth Factor (OGF)—Opioid Growth Factor Receptor (OGFr) Axis. BMC Cancer 2009, 9, 369. [Google Scholar] [CrossRef]
- Oosterom, R.; Verleun, T.; Bruining, H.A.; Hackeng, W.H.L.; Lamberts, S.W.J. Secretion of Adrenocorticotropin, β -Endorphin and Calcitonin by Cultured Medullary Thyroid Carcinoma Cells. Effects of Synthetic Corticotropin-Releasing Factor and Lysine Vasopressin. Acta Endocrinol. 1986, 113, 65–72. [Google Scholar] [CrossRef]
- Maneckjee, R.; Biswas, R.; Vonderhaar, B.K. Binding of Opioids to Human MCF-7 Breast Cancer Cells and Their Effects on Growth. Cancer Res. 1990, 50, 2234–2238. [Google Scholar] [PubMed]
- Wang, C.-Z.; Li, X.-L.; Sun, S.; Xie, J.-T.; Aung, H.H.; Tong, R.; McEntee, E.; Yuan, C.-S. Methylnaltrexone, a Peripherally Acting Opioid Receptor Antagonist, Enhances Tumoricidal Effects of 5-Fu on Human Carcinoma Cells. Anticancer Res. 2009, 29, 2927–2932. [Google Scholar] [PubMed]
- Suzuki, M.; Chiwaki, F.; Sawada, Y.; Ashikawa, M.; Aoyagi, K.; Fujita, T.; Yanagihara, K.; Komatsu, M.; Narita, M.; Suzuki, T.; et al. Peripheral Opioid Antagonist Enhances the Effect of Anti-Tumor Drug by Blocking a Cell Growth-Suppressive Pathway In Vivo. PLoS ONE 2015, 10, e0123407. [Google Scholar] [CrossRef]
- Singleton, P.A.; Lingen, M.W.; Fekete, M.J.; Garcia, J.G.N.; Moss, J. Methylnaltrexone Inhibits Opiate and VEGF-Induced Angiogenesis: Role of Receptor Transactivation. Microvasc. Res. 2006, 72, 3–11. [Google Scholar] [CrossRef]
- Szczepaniak, A.; Fichna, J.; Zielińska, M. Opioids in Cancer Development, Progression and Metastasis: Focus on Colorectal Cancer. Curr. Treat. Options Oncol. 2020, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Narayan, A.; Hunkele, A.; Xu, J.; Bassoni, D.L.; Pasternak, G.W.; Pan, Y.-X. Mu Opioids Induce Biased Signaling at the Full-Length Seven Transmembrane C-Terminal Splice Variants of the Mu Opioid Receptor Gene, Oprm1. Cell. Mol. Neurobiol. 2021, 41, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Vadhel, A.; Bashir, S.; Mir, A.H.; Girdhar, M.; Kumar, D.; Kumar, A.; Mohan, A.; Malik, T.; Mohan, A. Opium Alkaloids, Biosynthesis, Pharmacology and Association with Cancer Occurrence. Open Biol. 2023, 13, 220355. [Google Scholar] [CrossRef]
- Rashidian, H.; Hadji, M.; Gholipour, M.; Naghibzadeh-Tahami, A.; Marzban, M.; Mohebbi, E.; Safari-Faramani, R.; Bakhshi, M.; Seyyedsalehi, M.S.; Hosseini, B.; et al. Opium Use and Risk of Lung Cancer: A Multicenter Case-Control Study in Iran. Int. J. Cancer 2023, 152, 203–213. [Google Scholar] [CrossRef]
- Houston, M.G.; McMenamin, Ú.; Johnston, B.; McDowell, R.D.; Hughes, C.M.; Murchie, P.; Cardwell, C.R. Exposure to Weak Opioids and Risk of Gastrointestinal Tract Cancers: A Series of Nested Case-Control Studies. Br. J. Clin. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Ağagündüz, D.; Cocozza, E.; Cemali, Ö.; Bayazıt, A.D.; Nanì, M.F.; Cerqua, I.; Morgillo, F.; Saygılı, S.K.; Berni Canani, R.; Amero, P.; et al. Understanding the Role of the Gut Microbiome in Gastrointestinal Cancer: A Review. Front. Pharmacol. 2023, 14, 1130562. [Google Scholar] [CrossRef]
- Duque-Díaz, E.; Meléndez-Flórez, G.; Hurtado Giraldo, H.; Coveñas, R. Involvement of the Adrenocorticotropic Hormone in Cancer and Other Pathologies. In Advances in Health and Disease; Duncan, L.T., Ed.; Nova Science Publishers: New York, NY, USA, 2023; Volume 68, pp. 185–208. [Google Scholar]
- Li, G.; Li, B.; Song, J.; Wang, N.; Gao, Z. Endomorphin-2 Analog Inhibits the Growth of DLD-1 and RKO Human Colon Cancer Cells by Inducing Cell Apoptosis. Med. Sci. Monit. 2020, 26, e921251. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor | MET/LEU | References |
|---|---|---|
| Brain | Pro-enkephalin/MET in human tumors. Higher MET level, lower tumor degree. MET promotes apoptosis in rat C6 glioma cells and increases the activity of caspases 3, 8, and 9. MET-binding sites decrease with increasing malignancy of gliomas. A shift from mu-opioid receptors in low-grade gliomas to delta-opioid receptors in high-grade gliomas occurs. MET favors the growth of human U-373 MG astrocytoma cells. | [134,135,136,139] |
| Breast | MET/LEU expression in cells and stroma of human cancer samples. MET, but not LEU, stimulated the migration of MDA-MB-468 human carcinoma cells. A low-fasting plasma level of pro-enkephalin correlated with an increased risk of cancer development in postmenopausal/middle-aged women. MET/opioid growth factor receptor system blocked the proliferation of triple-negative breast cancer cell lines. | [144,145,146,147] |
| Cervical | MET blocked cervical carcinoma progression and induced apoptosis. MET decreased the number of myeloid-derived suppressor cells in both tumor and circulation. | [159] |
| Colorectal | Enkephalins in rectal carcinoids. MET/LEU did not affect tumor cell migration, chemotaxis, or invasion. MET did not alter the viability of tumor cells. MET enhanced colon carcinogenesis. MET exerts an antitumor action against colorectal tumors (MC38 cell line). MET augmented immunogenicity and recognition of tumor cells; downregulated Kras, Bcl2, and Bclxl; blocked the synthesis of inflammatory cytokines; and reduced immune checkpoints in tumor cells. Naltrexone blocked the antitumor action induced by MET. CD10 increased colorectal cancer cell metastasis by abrogating the antitumor action mediated by MET. MET decreased the phosphorylation of ERK/epidermal growth factor receptors and increased p38-dependent apoptosis. LEU decreased the invasive capacity of murine colon 26-L5 adenocarcinoma cells. | [162,163,164,165,166,172,186] |
| Cutaneous squamous cell carcinoma | MET blocked the cell growth of tumor cells by apoptosis. MET blocked the proliferation of A431 cells by apoptosis and promoted autophagy in tumor cells. MET decreased immunosuppression by reducing the number of myeloid-derived suppressor cells. | [173,206] |
| Gastric | MET blocked the growth of human tumor cells by apoptosis. MET upregulated the expression of the opioid growth factor receptor. Tumor cell apoptosis, mediated by MET, was blocked when the opioid receptor expression was in knockdown. | [175,176] |
| Head and neck | MET expression in human head and neck squamous cell carcinoma. MET/LEU did not affect tumor cell migration, chemotaxis, or invasion. MET did not affect either the differentiation or the viability of tumor cells. Reduced DNA synthesis and tumor weight/volume after treatment with MET. Downregulation of the opioid growth factor receptor favored tumor progression. LEU in head and neck paragangliomas. | [172,180,181,183,184,186,187] |
| Larynx | MET in neuroendocrime tumors (paragangliomas). | [188] |
| Leukemia | Pro-ENK A in leukocytes from patients with chronic lymphoblastic leukemia. MET inhibited the metabolic activity of the leukemic NALM-1 cell line. MET promoted apoptosis in K562 human erythroid leukemia cells. MET favored pre-B acute lymphoblastoid cell migration. | [190,191,192,193] |
| Liver | MET concentration was higher in metastasis-positive human livers than in normal livers. Proliferation of hepatocellular carcinoma cells: inhibited after treatment with MET. Silencing of the opioid growth factor receptor promoted the proliferation of hepatocellular carcinoma cells. Patients with hepatoblastoma are cured after surgical resection and treatment with naltrexone and MET. | [165,199,200] |
| Lung | Pro-enkephalin/MET expression. Serum LEU levels are higher in patients with bronchial carcinoma than in control individuals. MET exerted an antitumor effect against cancer cells: this was abolished in growth factor receptor knockdown. MET increased the expression of opioid growth factor receptors and promoted apoptosis in cancer cells. Methylnaltrexone counteracted Lewis lung carcinoma growth and decreased metastasis; morphine favored tumor growth. Nicotine partially/totally reversed opioid-induced growth suppression in cancer cells. MET blocked pulmonary metastasis and enhanced the activity of NK cells. | [202,203,205,206,207,208,209] |
| Melanoma | MET/LEU expressions decreased in melanocytic tumors. MET exerted an antitumor effect in mice xenografted with B16-BL6 cells; this was inhibited with naloxone. Tumor growth/cell dissemination was counteracted with MET; the peptide blocked A375 cell proliferation through apoptosis. Imiquimod upregulated the expression of the opioid growth factor receptor, increasing the MET antitumor effect. MET promotes cell cycle arrest and increases the expression of opioid receptors in B16 cells. MET decreased tumor weight/volume. MET promoted cell cycle arrest and favored apoptosis in human A375 cells. | [220,222,223,224,225] |
| Neuroblastoma | MET in mouse Neuro2a cells. MET arrested the growth of human SK-N-SH cells. MET decreased the tumor mitotic index, which was counteracted with naltrexone. | [236,237] |
| Ovarian | Enkephalins in ovarian carcinoids. MET/opioid growth factor receptor in human cancer cells. MET exerted an inhibitory proliferative effect on tumor cells. MET neutralization: cell proliferation and silencing of the opioid growth factor receptor favored tumor cell replication. MET delayed cells moving. Naltrexone inhibited tumor progression and, with cisplatin, exerted an enhanced inhibitory effect. MET blocked the proliferation of human ovarian KF cancer cells; this effect was counteracted with naloxone. | [16,244,245,246,249] |
| Pancreatic | High MET plasma level in patients with pancreatic cancer. MET/LEU did not affect the migration, chemotaxis, or invasion of tumor cells (MIA PaCa-2, PANC-1, BxPC-3) MET lipid conjugation: increased MET tumor-suppression activity in human pancreatic adenocarcinomas. MET/opioid growth factor receptor system attenuated tumor progression. MET ameliorated clinical symptoms/survival in patients with advanced pancreatic cancer. | [186,250,251,252,253] |
| Pheochromocytoma | MET/LEU, proprotein convertase 2 and pro-enkephalin in human pheochromocytomas. Nicotine promoted MET release from pheochromocytoma cells. MET/LEU ratio is higher in extramedullary than in medullary pheochromocytomas. | [255,256,257,258] |
| Pituitary | MET level increased in prolactin-releasing human pituitary adenomas. | [265] |
| Prostate | DAGO/DSLET blocked the proliferation of tumor cells; this was counteracted with diprenorphine. LEU in prostatic carcinomas. | [274,275] |
| Renal | MET blocked the proliferation of human cancer cells. | [281] |
| Retinoblastoma | MET expression in humans. | [282] |
| Thymic | MET in a thymic carcinoid. | [285] |
| Thyroid | Human anaplastic thyroid cancer cells: MET/opioid growth factor receptor expression. MET blocked cell replication. Naltrexone promoted tumor cell growth, and anti-MET antibodies counteracted the inhibitory action mediated by MET. | [289] |
| Tumor | Beta-Endorphin | References |
|---|---|---|
| Bone | Cinobufagin promoted beta-END mRNA and protein expressions in microglia. It upregulated the expressions of mu-opioid receptors and beta-END in the tumor and tissues placed close to the tumor. | [130,131] |
| Brain | Beta-END-binding sites in human glioblastoma cells. Beta-END in human brain tumor cyst fluids. | [140,141] |
| Breast | High plasma beta-END levels in healthy women were even higher in healthy postmenopausal women; lower in women with breast cancer. Chemotherapy improved beta-END levels in postmenopausal women. END did not affect the migratory capacity of human breast carcinoma cells. Beta-END expression in cells and stroma of human breast cancer samples. Beta-END activates survival/mitogenic signaling pathways in human cancer cells. Increasing plasma beta-END levels correlated with increasing tumor burden. A correlation between increased plasma beta-END levels and improved quality of life in breast cancer patients. Beta-END counteracts breast cancer development by favoring immune-mediated anticancer defenses. Beta-END alters the tumor microenvironment. | [144,145,149,151,155,156,157,177] |
| Cervical | Electroacupuncture increases plasma beta-END levels in humans. | [160] |
| Colorectal | Beta-END in adenocarcinomas derived from the colon mucosa; its expression is higher in adenocarcinomas than in the mucosal layer of normal colons. Beta-END in rectal carcinoids. Beta-END did not affect tumor cell migration, viability, chemotaxis, or invasion. Colon carcinoma symptoms decreased after ultraviolet A eye irradiation; these effects were reduced when beta-END inhibitors were administered and totally disappeared with naltrexone. Animals with hypothalamic beta-END neuronal transplants failed to develop tumors and showed a lesser adenoma development. Beta-END administration into the raphe magnus nucleus: favored metastasis inhibited with naloxone, and when this nucleus was electrically stimulated, metastasis was attenuated. | [162,167,168,169,170,172,186] |
| Gastric | Beta-END in adenocarcinomas derived from the antral mucosa. Plasma beta-END level decreased in patients after transcutaneous electrical stimulation. | [177,178] |
| Head and neck | Beta-END did not affect the migration, chemotaxis, or invasion of squamous carcinoma cells. Beta-END did not affect either the differentiation or the viability of tumor cells. Beta-END increased the production of leukocyte migration inhibitory factor, reaching almost normal levels in patients with squamous carcinoma. | [172,185,186,187] |
| Larynx | Beta-END in tumor cells. | [189] |
| Leukemia | Beta-END in the cerebrospinal fluid of children with acute lymphoblastic leukemia: highest levels observed at the end of the intensification chemotherapy; glucocorticoid treatment decreased beta-END levels. Plasma beta-END levels decreased in patients with solid tumors after treatment with a chemotherapeutic drug. Plasma beta-END levels are higher in patients with acute leukemia than in healthy individuals. Beta-END promoted the growth of T-lymphoblastoid cells. | [194,195,196,197] |
| Liver | Neurons expressing beta-END transplanted into the hypothalamus prevented hepatocellular carcinoma formation. This strategy inhibited carcinogen-induced liver histopathologies and augmented the concentration of NK cell cytotoxic agents. | [201] |
| Lung | Beta-END in the bronchoalveolar lavage fluid/plasma of patients. Beta-END in lung small-cell carcinomas and carcinoid tumors. U1,690 cell line expresses beta-END, and the peptide promotes its proliferation through non-opioid binding sites. Beta-END acts as a chemoattractant for small-cell lung carcinoma cells favoring migration and metastasis. | [210,211,212,214,215] |
| Melanoma | Beta-END in human samples. Beta-END in secondary neuroendocrine carcinomas of the skin but not in primary carcinomas. B16 melanoma cells synthesize and release beta-END. B16 cells decreased tumor growth in mu-opioid receptor-deficient animals. A correlation occurs between beta-END levels and tumor progression. Beta-END immunoreactivity: lower in benign melanocytic naevi than in metastatic and advanced melanomas. | [221,232,233] |
| Neuroblastoma | Beta-END-binding sites expression. MET decreased the tumor mitotic index, which was counteracted with naltrexone. | [238,239] |
| Ovarian | Beta-END in ovarian sex cord-stromal tumors and ovarian carcinoids. A positive correlation between patients’ survival time/disease-free time and plasma beta-END levels. Lower beta-END concentrations were observed in patients with recurrence Beta-END blocked the proliferation of human ovarian KF cancer cells; this effect was counteracted with naloxone. | [244,247,248,249] |
| Pancreatic | Beta-END did not affect tumor cell migration, chemotaxis, or invasion (MIA PaCa-2, PANC-1, BxPC-3). | [186] |
| Pheochromocytoma | Beta-END expression and release. | [257,259,260] |
| Pituitary | Beta-END in pituitary adenomas. Beta-END released from human cancer cells. Beta-END presence in clinically silent pituitary corticotroph adenomas. Beta-END/beta-END1-27 in extracts of pituitary melanotroph tumors transplanted subcutaneously. W7 potentiated the release of beta-END from cancer cells. | [266,267,268,269,270] |
| Prostate | Beta-END in prostatic carcinomas. Rats with transplanted neurons expressing beta-END into the hypothalamic paraventricular nucleus showed a protective effect against prostate cancer development. | [275,276] |
| Retinoblastoma | Beta-END-binding sites in human cell lines. | [283] |
| Thymic | Beta-END binds to non-opioid binding sites expressed in thymoma cells. Beta-END in an oncocytic carcinoid tumor of the thymus. | [286,287] |
| Thyroid | Presence of opioid peptides derived from the three opioid precursors in human thyroid medullary carcinomas. Beta-END released from cultured medullary thyroid carcinoma cells. | [11,290] |
| Tumor | DYN | References |
|---|---|---|
| Brain | DYN A1-13 decreased water content, mediated by glutamate, in C6 glioma cells. | [142] |
| Breast | DYN A1-17/DYN A1-8 in Walker 256 tumors. | [158] |
| Colorectal | DYN A1-8 did not affect tumor cell migration, viability, chemotaxis, or invasion. | [172,186] |
| Head and neck | DYN A1-8 did not affect the migration, chemotaxis, or invasion of squamous carcinoma cells. DYN A1-8 did not affect tumor cells’ differentiation or viability. | [172,186,187] |
| Lung | Pre-pro-DYN mRNA in small-cell lung carcinomas. Serum DYN A/B levels increased in a non-small-cell lung cancer cell xenograft stress reduction mouse model. DYN B blocked cAMP formation in non-small-cell lung cancer cells. Lung tumor cells co-expressed pro-DYN and DYN, prohormone convertase 1, prohormone convertase 2, or carboxypeptidase E. DYN was observed in cancer cells infiltrating human lung tissues and nerve fibers in the bronchial submucosa. Lung cancer cells express opioid receptors and contain several combinations of opioid peptides; opioid receptor agonists blocked the growth of tumor cells. Pro-opiomelanocortin gene delivery blocked the growth of tumor cells and vasculature. | [208,216,217,218,219] |
| Myeloma | U50,488 decreased tumor cell proliferation. | [235] |
| Neuroblastoma | Pre-pro-DYN mRNA and pre-pro-ENK in tumor cells. Detection of opioid-binding sites in tumor cells. DYN promoted apoptosis in tumor cells; this was inhibited with isoflurane. | [216,240,242] |
| Pancreatic | DYN A1-8 did not affect tumor cell migration, chemotaxis, or invasion (MIA PaCa-2, PANC-1, BxPC-3). Mouse insulinoma beta TC3 cells: high expression of pro-DYN mRNA and derived peptides. | [186,254] |
| Pheochromocytoma | Pro-DYN gene expression and DYN release. DYN in humans. Nicotine favored the release of DYN from tumor cells. DYN A1-17 was the major component in pheochromocytomas, whereas DYN A1-13/DYN A1-12 were minor. | [171,257,261,262,263,264] |
| Prostate | DYN A, DYN A1-13, and DYN A1-7 promote tumor cell growth. Naloxone blocked the effect mediated by DYN A and increased the growth of tumor cells at high concentrations. | [277,278] |
| Testicular | Pro-DYN mRNA and its derived peptides in tumor cells. | [284] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, M.L.; Rodríguez, F.D.; Coveñas, R. Involvement of the Opioid Peptide Family in Cancer Progression. Biomedicines 2023, 11, 1993. https://doi.org/10.3390/biomedicines11071993
Sánchez ML, Rodríguez FD, Coveñas R. Involvement of the Opioid Peptide Family in Cancer Progression. Biomedicines. 2023; 11(7):1993. https://doi.org/10.3390/biomedicines11071993
Chicago/Turabian StyleSánchez, Manuel Lisardo, Francisco D. Rodríguez, and Rafael Coveñas. 2023. "Involvement of the Opioid Peptide Family in Cancer Progression" Biomedicines 11, no. 7: 1993. https://doi.org/10.3390/biomedicines11071993
APA StyleSánchez, M. L., Rodríguez, F. D., & Coveñas, R. (2023). Involvement of the Opioid Peptide Family in Cancer Progression. Biomedicines, 11(7), 1993. https://doi.org/10.3390/biomedicines11071993