
The Role of NMDA Receptor Partial Antagonist, Carbamathione, as a Therapeutic Agent for Transient Global Ischemia

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. In Vitro Study
2.2.1. PC-12 Cell Line Culture
2.2.2. Glutamate Toxicity
2.2.3. Hypoxia/Reoxygenation
2.2.4. Carb Concentration
2.2.5. Measurement of Cell Viability Using the ATP Assay
2.2.6. Electrophysiological Recording
2.3. In Vivo Study
2.3.1. Animal Preparation
2.3.2. Animal Care, Feeding, Housing, Monitoring, and Enrichment
2.3.3. Bilateral Common Carotid Artery Occlusion (BCAO)
2.3.4. Corner Tests
2.3.5. Locomotion (Force-Plate Actometer) Test
2.3.6. Mouse Groups and Treatment Schedules
2.3.7. Details of Euthanasia Method(s) Used
2.3.8. Criteria Established for Euthanizing Animals Prior to the Planned End of the Experiment and Whether This Was Needed
2.3.9. Assessment of Lesion Size Using 2,3,5-Triphenyltetrazolium Chloride (TTC)
2.3.10. Fate of the Surviving Animals at the Conclusion of the Experiment
2.4. Sample Collection for the Western Blot Analysis
2.5. Dosage of Carbamathione
2.6. Data and Statistical Analysis
3. Results
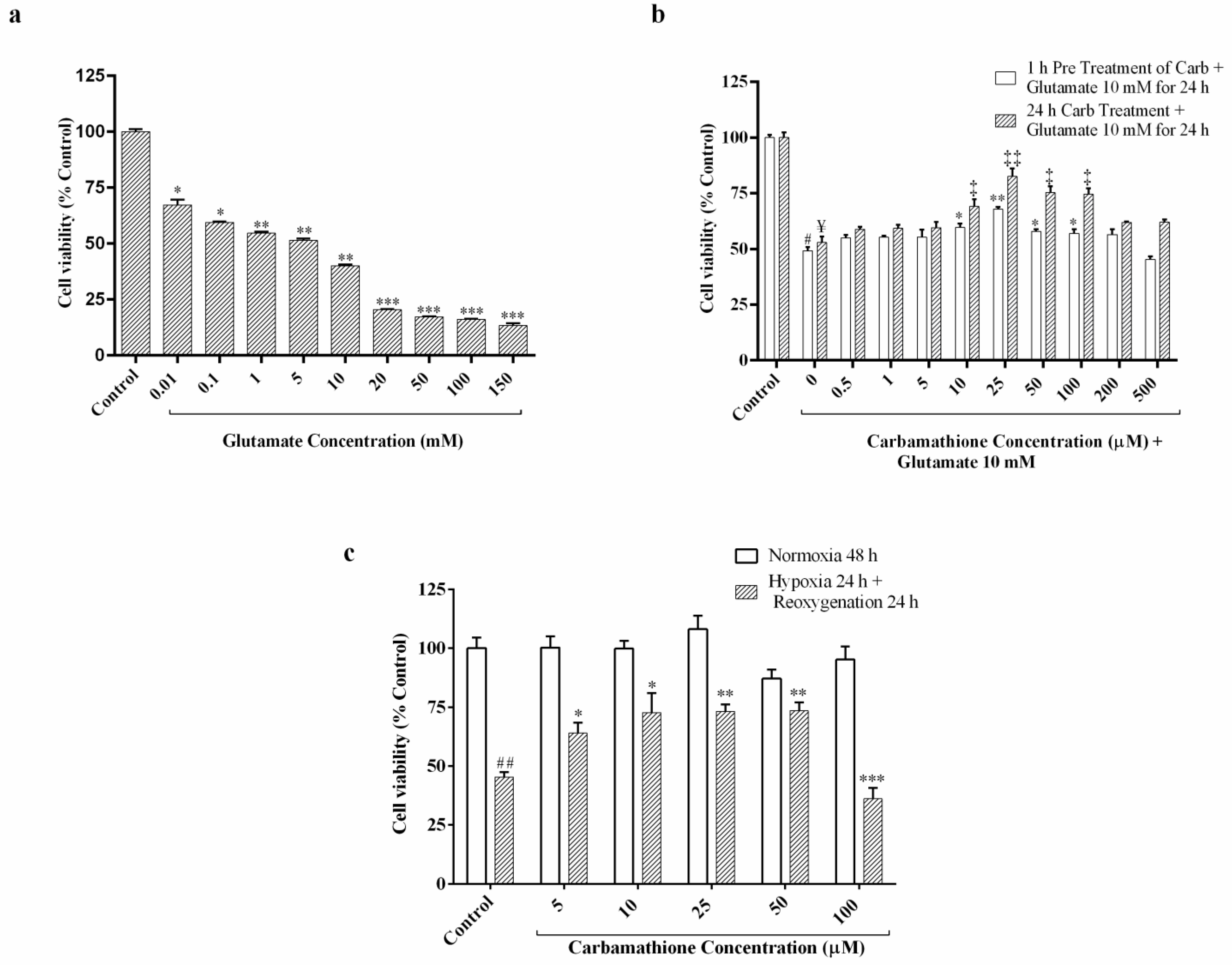
3.1. Glutamate Excitotoxicity Is Dose-Dependent in PC-12 Cell Culture
3.2. Carbamathione Protects PC-12 Cells against Glutamate-Induced Excitotoxicity
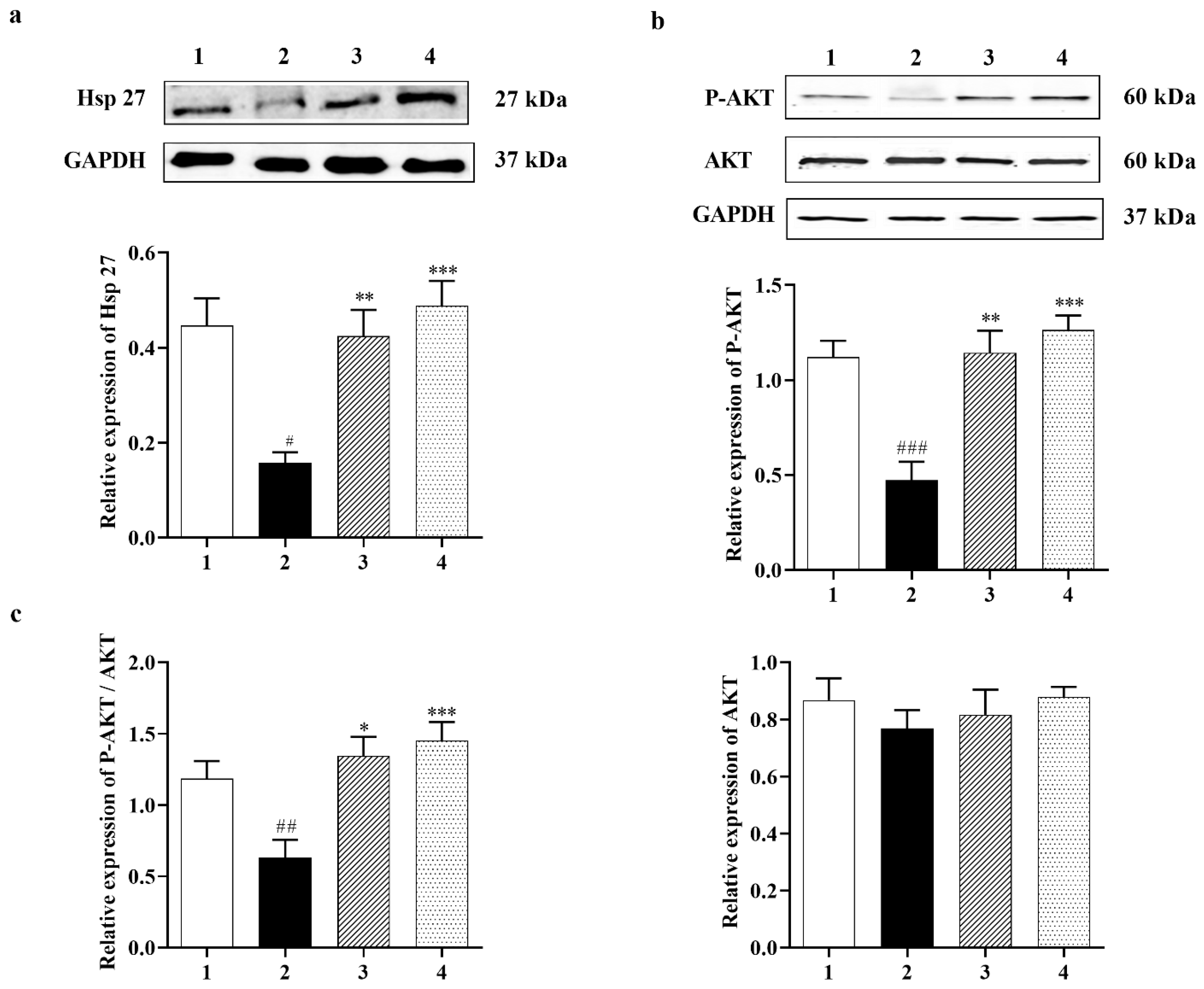
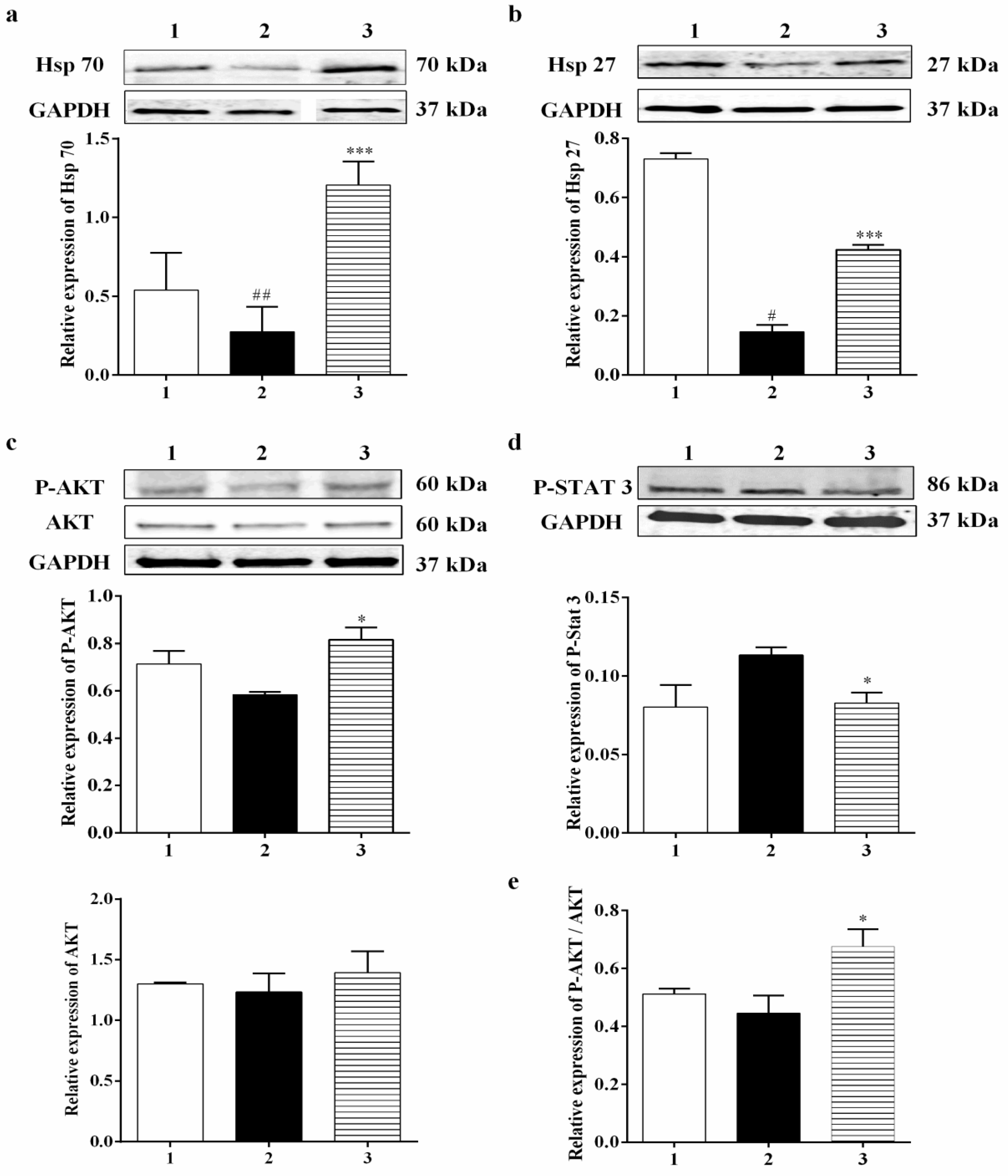
3.2.1. Carbamathione Modulates Expression of Heat Shock Protein (Hsp) and AKT Induced by Glutamate Toxicity
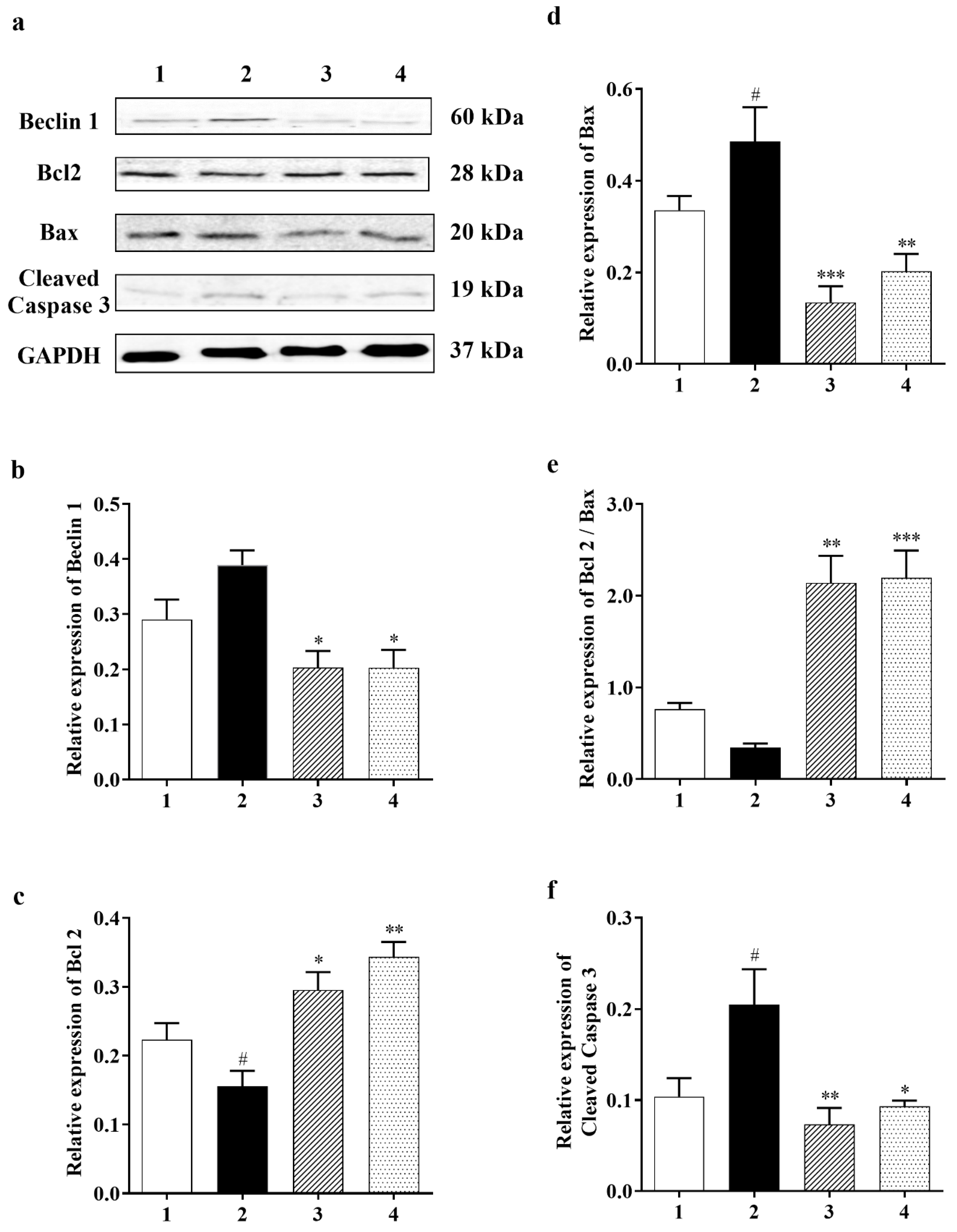
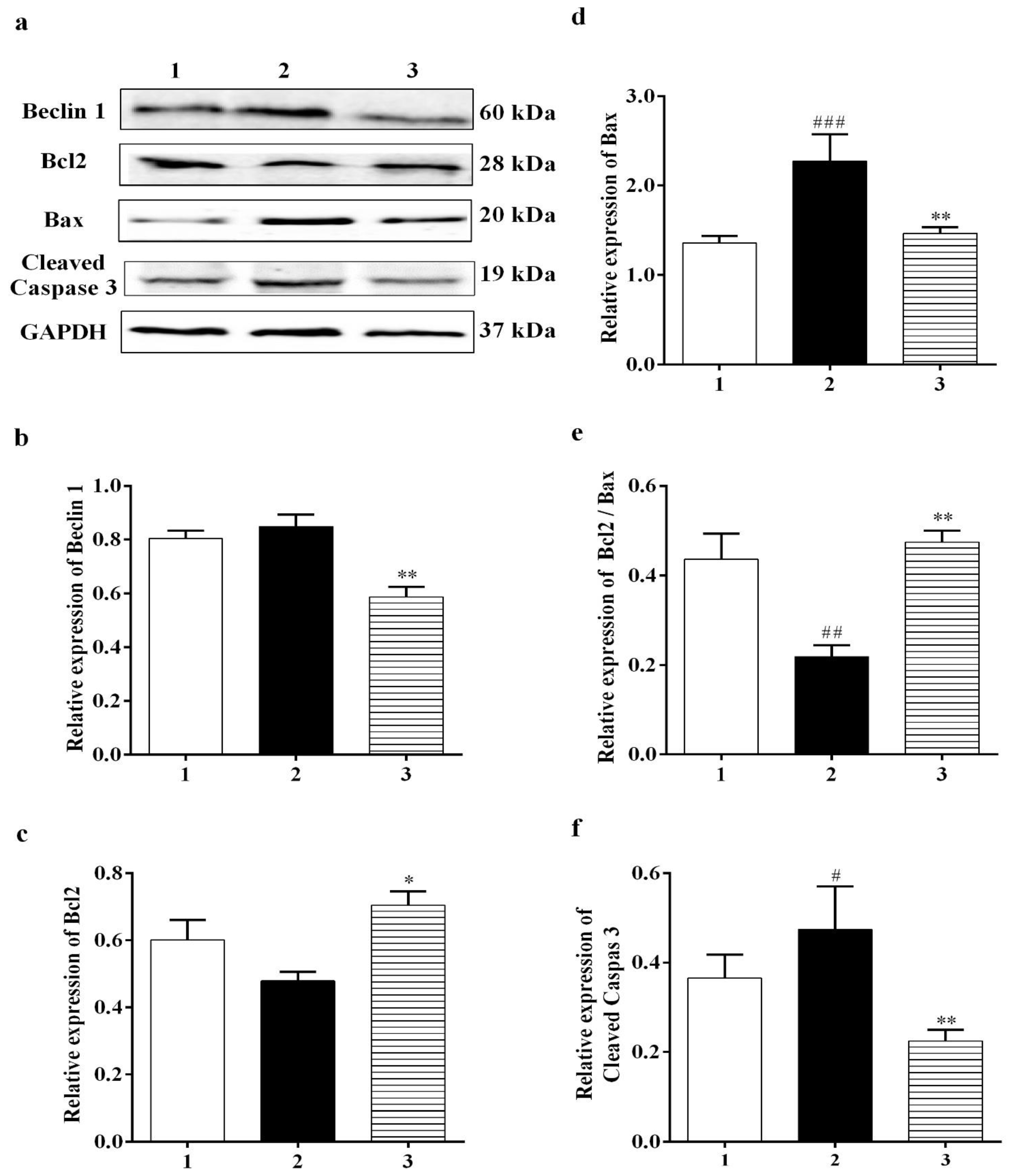
3.2.2. Effect of Carbamathione on the Expression Beclin 1, a Marker for Autophagy
3.2.3. Carbamathione Can Decrease Apoptosis by the Down-Regulation of Apoptotic Markers
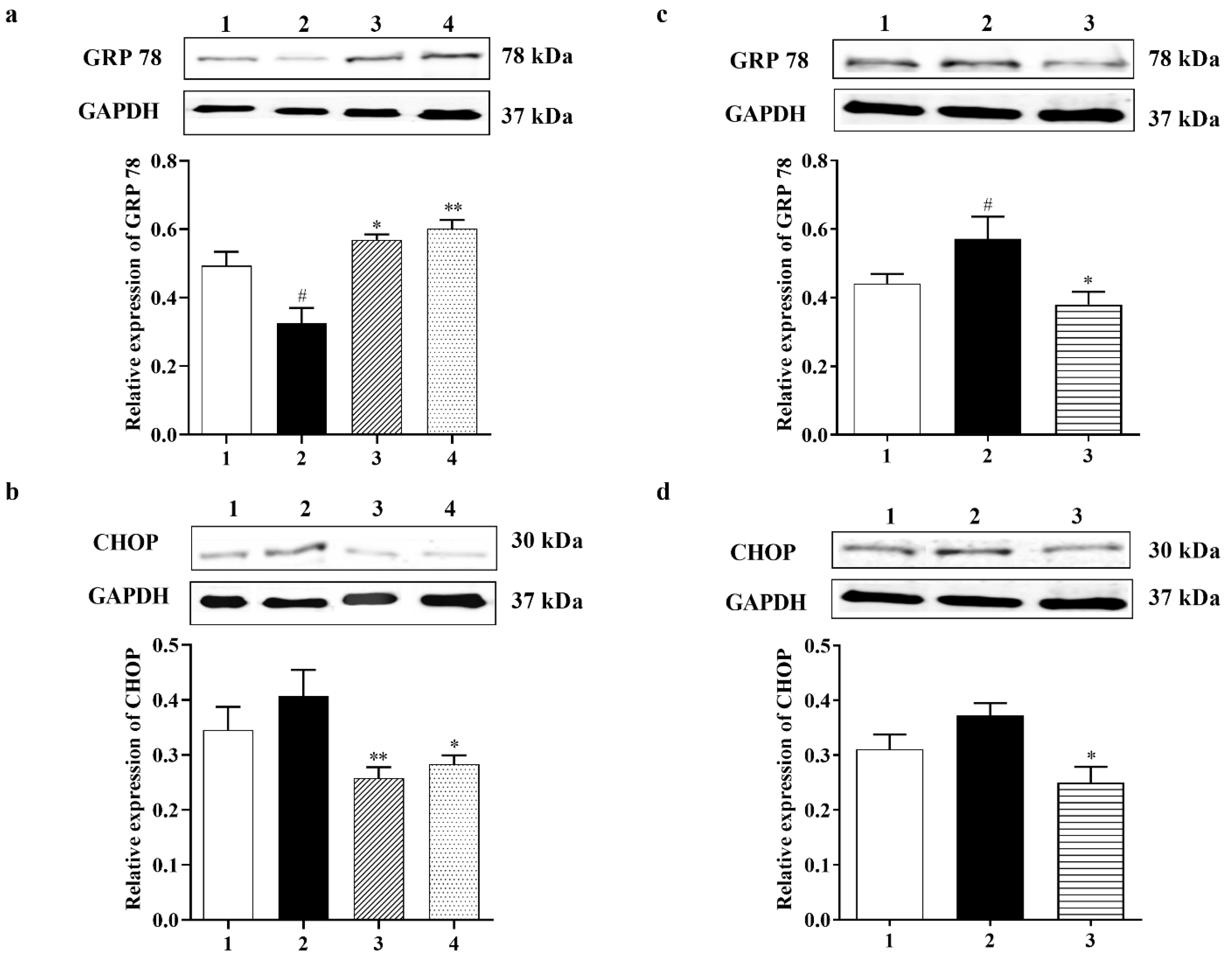
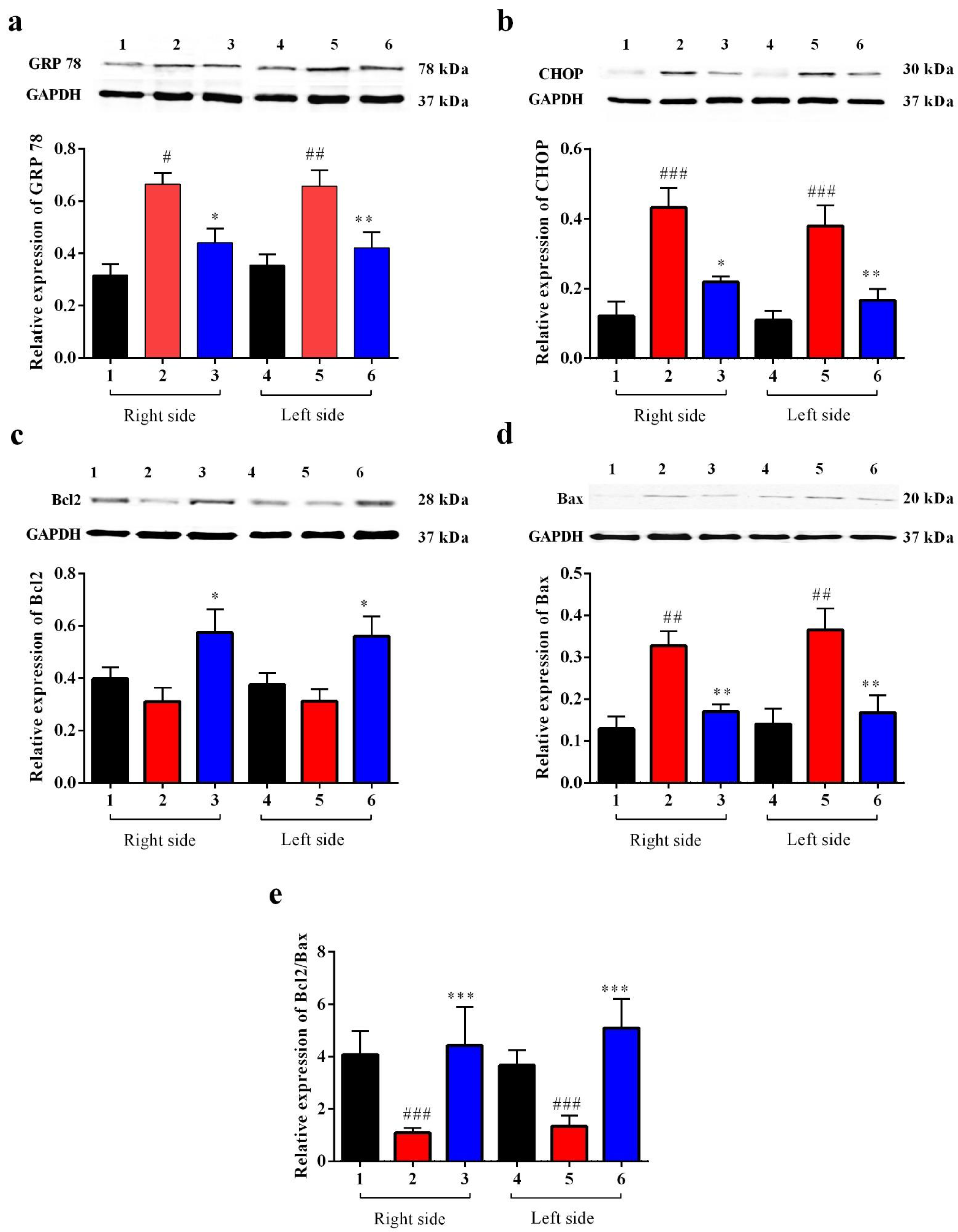
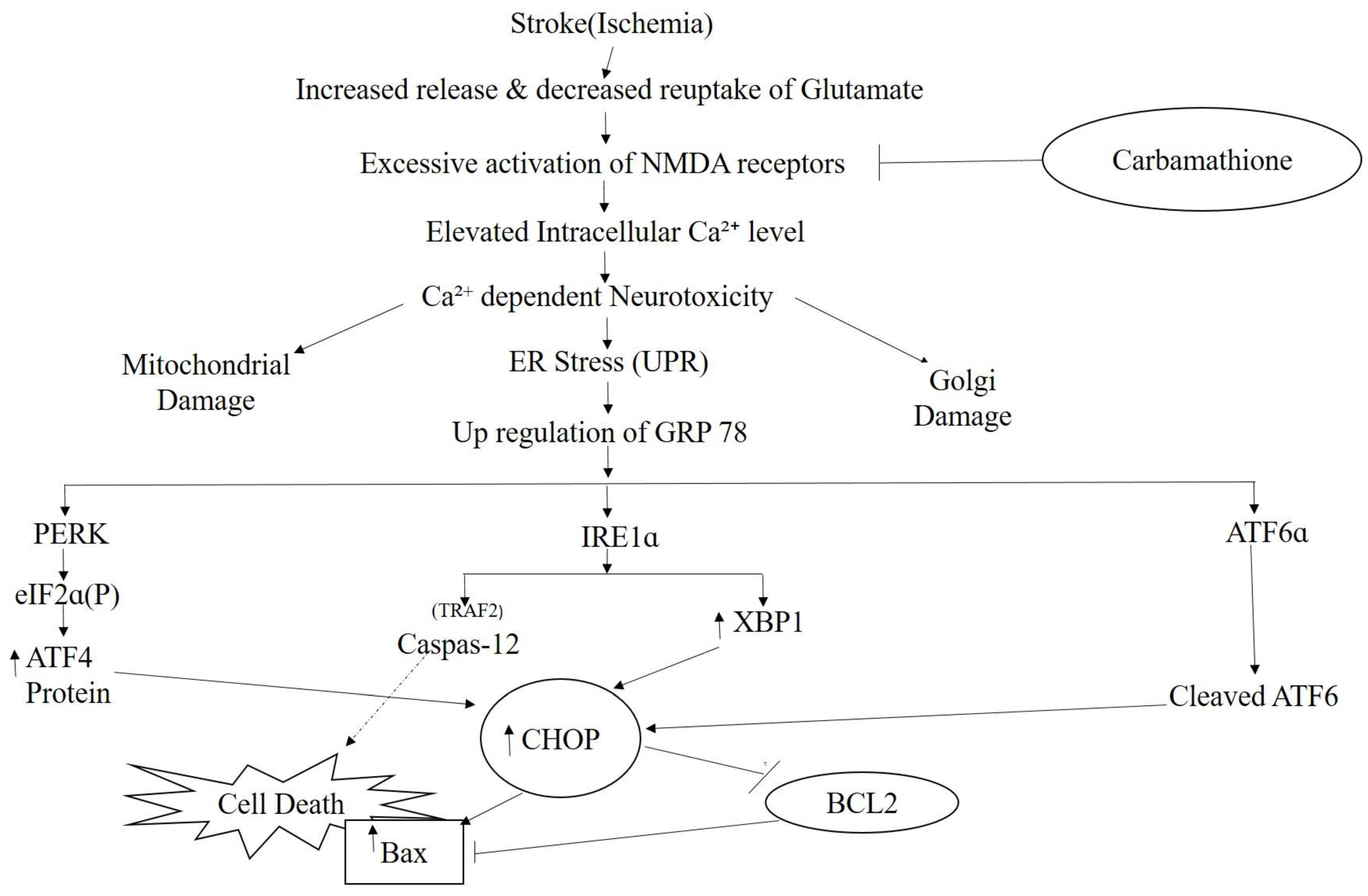
3.2.4. Carbamathione Protects Neuronal Cells against Glutamate Excitotoxicity by Suppressing the Expression of GRP 78 and CHOP
3.3. Carbamathione Demonstrates Robust Protective Activity against Hypoxia/Reoxygenation in PC-12 Cell Cultures
3.3.1. Effect of Carbamathione on the Expression of Heat Shock Protein, AKT, and P-STAT 3 Induced by Hypoxia/Reoxygenation
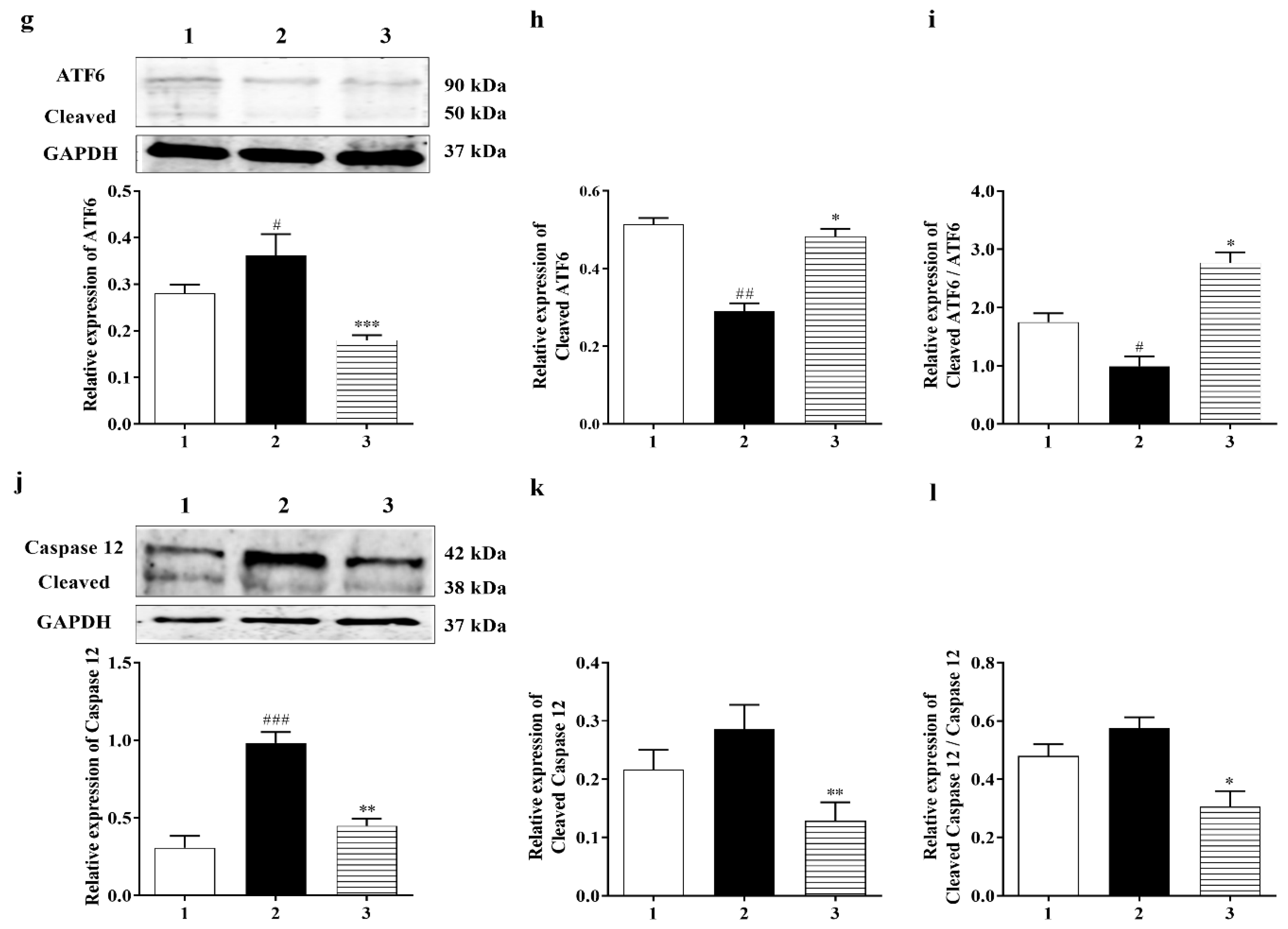
3.3.2. Carbamathione Inhibits the Expression of GRP 78, CHOP, and Caspase-12 Induced by Hypoxia/Reoxygenation
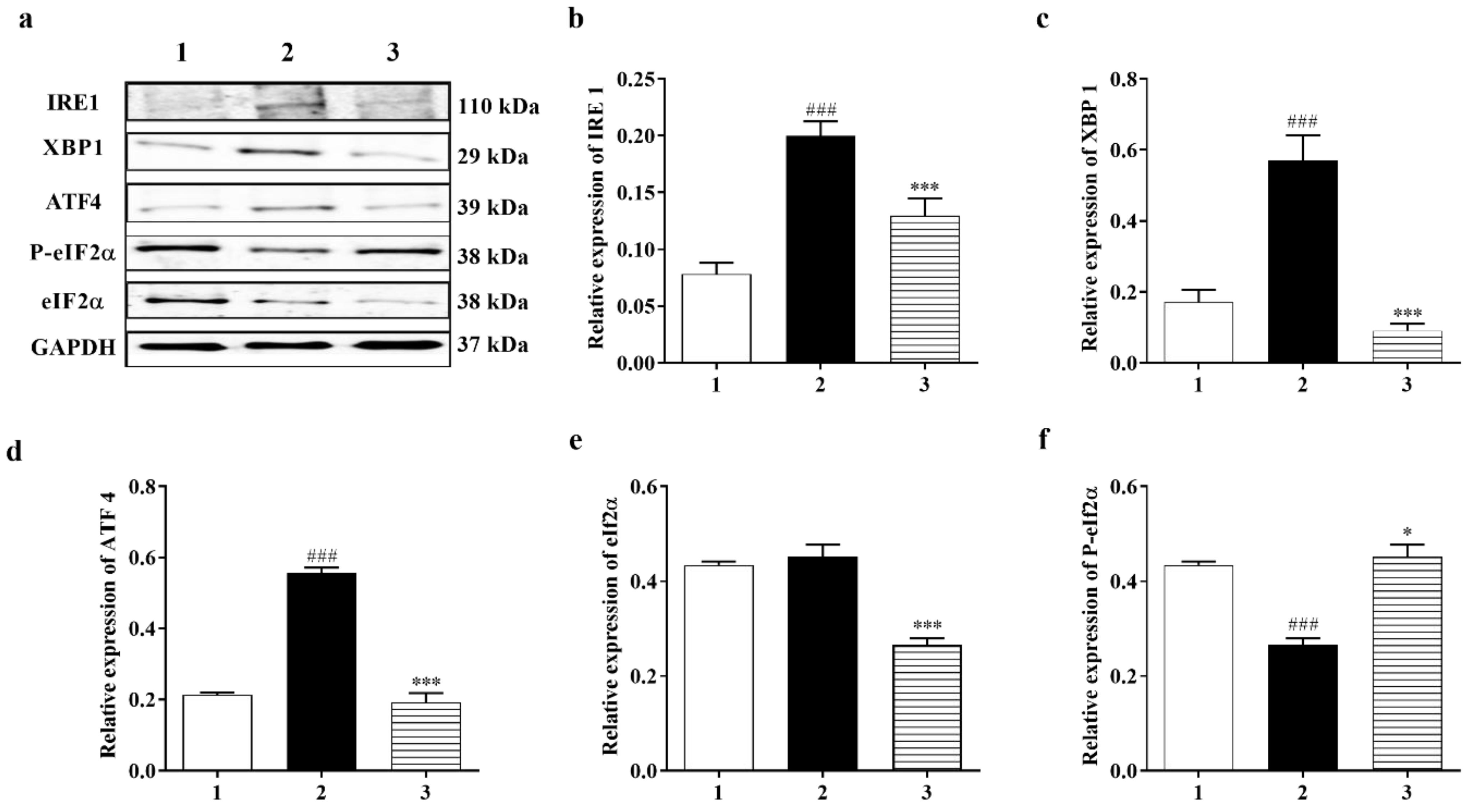
3.3.3. PERK and IRE1 Pathways Were Inhibited by Carbamathione under Hypoxia/Reoxygenation, although the ATF-6 Pathway Was Activated
3.3.4. Effect of Carbamathione on the Hypoxia/Reoxygenation-Induced Change in Beclin 1, Bcl2, Bax, and Caspase-3 Expression
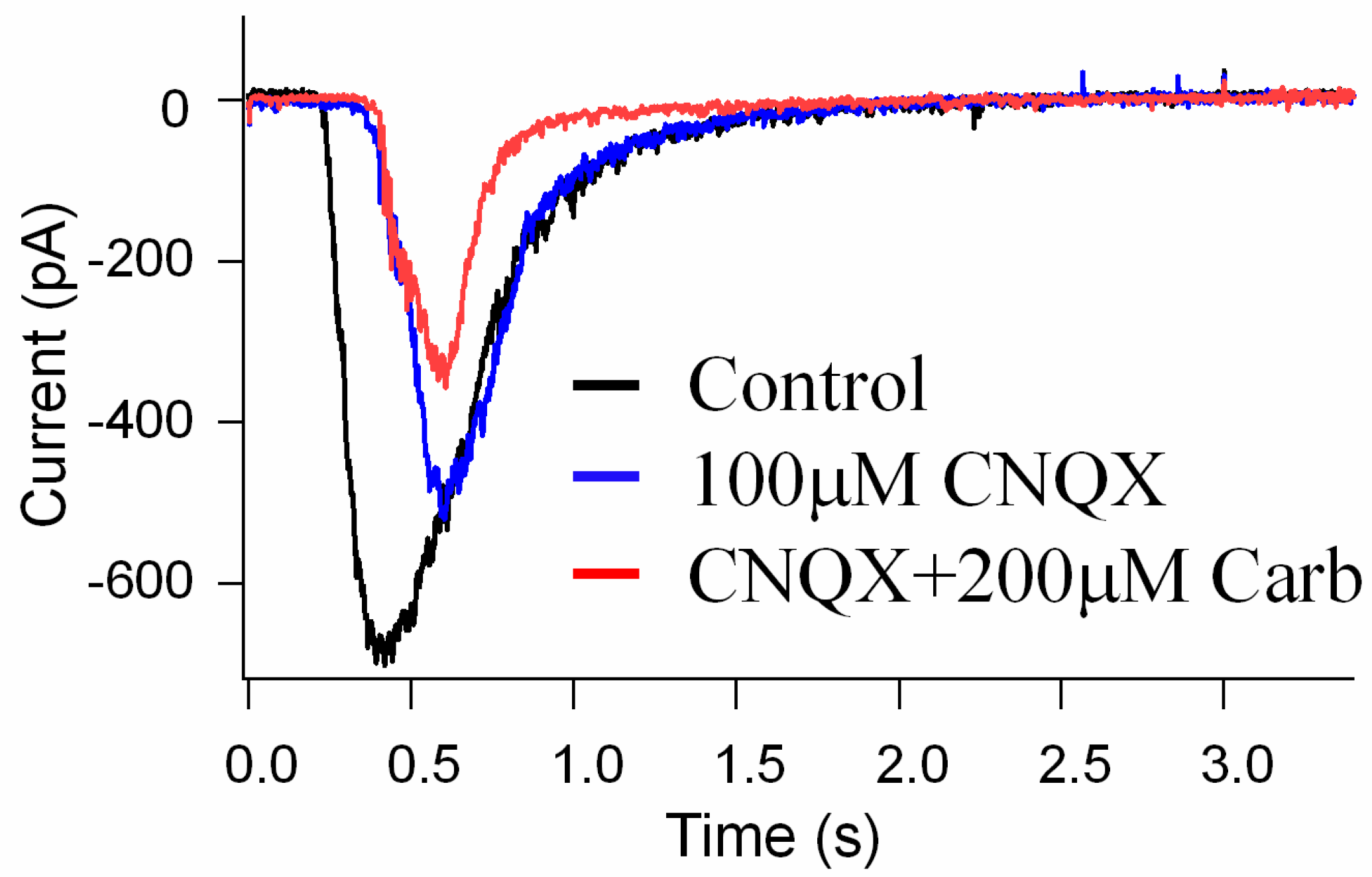
3.4. The Effects of Carb on the Suppression of NMDA Glutamate Receptor Activation in Retinal Neurons
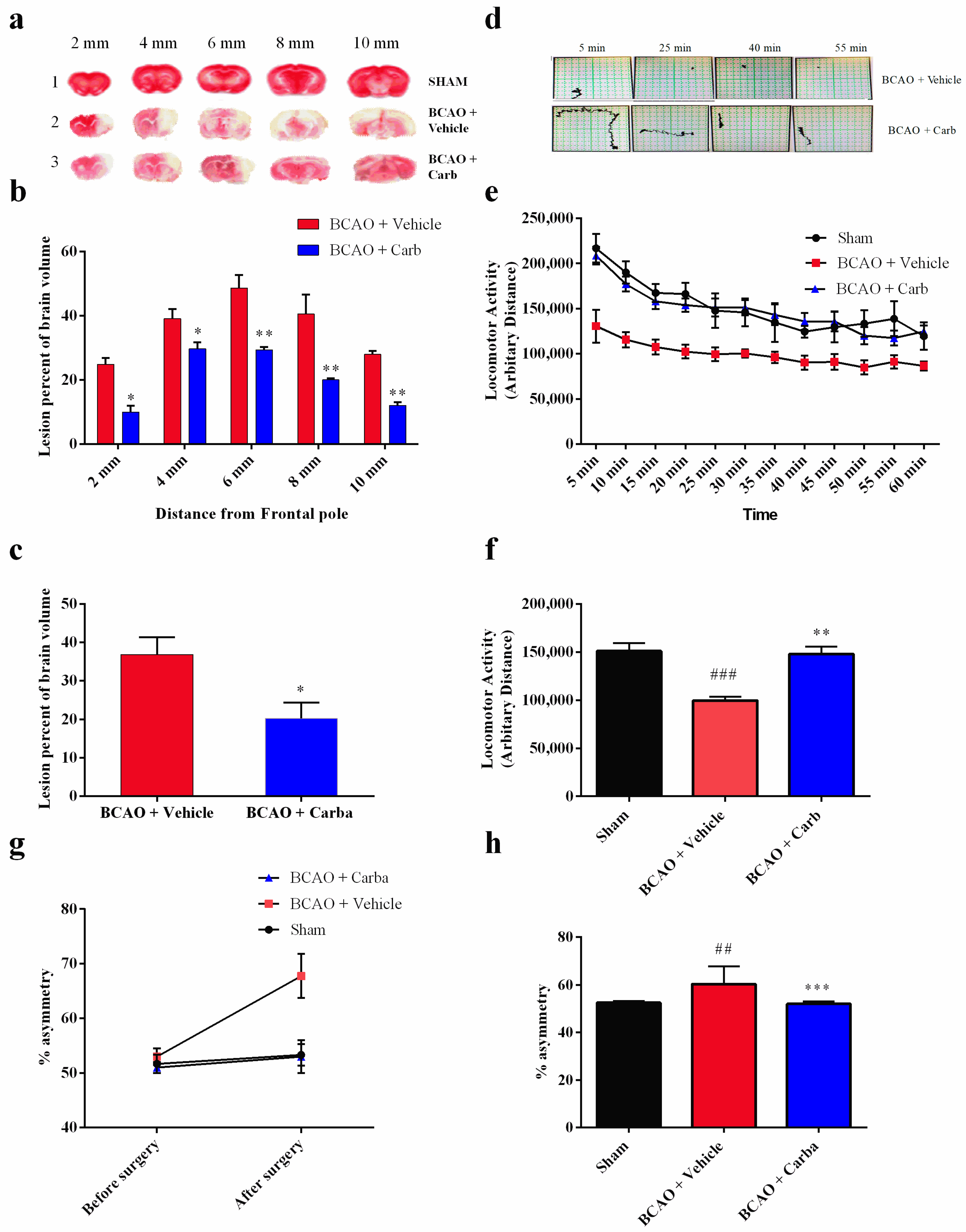
3.5. Carbamathione Treatment Attenuates Infarction Volume and Neurological Deficits
Carbamathione Can Modulate the Unfolded Protein Response and Decrease Apoptosis by Down-Regulating Apoptotic Markers
4. Discussion
Study Limitations and Future Work
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT/PKB | protein kinase B |
| ALDH2 | aldehyde dehydrogenase |
| AMPA | amino-3-hydroxy-5-methyl-4-isoxazolepropionate |
| ANOVA | analysis of variance |
| APAF1 | apoptotic protease activity factor 1 |
| ATF4 | activating transcription factor 4 |
| ATF6 | activating transcription factor 6 |
| ATP | adenosine 5 triphosphate |
| BAK | Bcl-2 antagonist/killer |
| BCAO | bilateral common Carotid Artery Occlusion |
| BAX | Bcl-2 associated protein X |
| BCL2 | B cell lymphoma 2 |
| BCL2-xL | B cell lymphoma 2 extra long |
| BDNF | brain-derived neurotrophic factor |
| BFGF | basic fibroblast growth factor |
| BH | Bcl-2 homology |
| BH3 | Bcl-2 homology domain 3 |
| BiP | binding immunoglobulin protein |
| Carb | Carbamathione |
| Caspase | cysteine aspartic acid protease |
| CHOP | C/EBP homologous protein |
| Cyt-c | cytochrome-c |
| DA | dopamine |
| DETC-MeSO | S-methyl N: N-diethythiolcarbamate sulfoxide |
| DSF | Disulfiram |
| EIF2α | eukaryotic translation initiation factor 2 alpha |
| ER | endoplasmic reticulum |
| ERAD | endoplasmic reticulum-associated degradation |
| ERAD | ER-associated protein degradation |
| FBS | fetal bovine serum |
| GABA | gamma (γ)-aminobutyric acid |
| GADD153 | growth arrest and DNA damage-inducible 153 |
| GAPDH | glyceraldehayde-3-phosphade dehydrogenase |
| GRP 78 | glucose-regulated protein 78 |
| GSH | reduced glutathione |
| H | horse serum |
| HSP | heat shock protein |
| Hyp | hypoxia |
| IRE1α | inositol-requiring protein-1alpha |
| JNK | cJUN NH2-terminal kinase |
| KA | kainic acid |
| mPFC | medial prefrontal cortex |
| NAc | nucleus accumbens |
| NMDA | N-methyl-D-aspartate |
| NOS | nitric oxide synthase |
| OMM | outer mitochondrial membrane |
| P38MAPK | mitogen-activated protein kinase |
| PC | pheochromocytoma |
| PD | Parkinson’s disease |
| PERK | protein kinase RNA (PKR)-like ER kinase |
| PI3K | phosphatidylinositol-3-kinase |
| PMDs | protein misfolding disorders |
| PUMA | p53-upregulated modulator of apoptosis |
| STAT3 | signal transducer and activator of transcription |
| TRAF2 | tumor necrosis factor-α receptor-associated factor 2 |
| TTC | 2,3,5-Triphenyl tetrazolium chloride |
| UPR | unfolded protein response |
| VOCC | voltage operated calcium channel |
| XBP-1 | X-Box-binding protein |
References
- Xu, J.; Kochanek, K.D.; Murphy, S.L.; Tejada-Vera, B. National Vital Statistics Reports; Centers for Disease Control and Prevention National Center for Health Statistics: Hyattsville, MD, USA, 2007; Volume 58, Number 19 (05/20/2010).
- Roffe, C. Hypoxaemia and stroke. Rev. Clin. Gerontol. 2001, 11, 323–335. [Google Scholar] [CrossRef]
- Prentice, H.; Modi, J.P.; Wu, J.-Y. Mechanisms of Neuronal Protection against Excitotoxicity, Endoplasmic Reticulum Stress, and Mitochondrial Dysfunction in Stroke and Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2015, 2015, 964518. [Google Scholar] [CrossRef] [PubMed]
- Dávalos, A.; Shuaib, A.; Wahlgren, N.G. Neurotransmitters and pathophysiology of stroke: Evidence for the release of glutamate and other transmitters/mediators in animals and humans. J. Stroke Cerebrovasc. Dis. 2000, 9, 2–8. [Google Scholar] [CrossRef]
- Pan, C.; Gupta, A.; Prentice, H.; Wu, J.-Y. Protection of taurine and granulocyte colony-stimulating factor against excitotoxicity induced by glutamate in primary cortical neurons. J. Biomed. Sci. 2010, 17 (Suppl. S1), S18. [Google Scholar] [CrossRef] [PubMed]
- Mohammad-Gharibani, P.; Modi, J.; Menzie, J.; Genova, R.; Ma, Z.; Tao, R.; Prentice, H.; Wu, J.Y. Mode of Action of S-Methyl-N, N-Diethylthiocarbamate Sulfoxide (DETC-MeSO) as a Novel Therapy for Stroke in a Rat Model. Mol. Neurobiol. 2014, 50, 655–672. [Google Scholar] [CrossRef]
- Virtanen, T.; Sivenius, J.; Jolkkonen, J. Dehydration and Stress Do Not Explain Severe Weight Loss after Experimental Stroke in Rats. J. Anim. Vet. Adv. 2003, 2, 247–252. [Google Scholar]
- Azfer, A.; Niu, J.; Rogers, L.M.; Adamski, F.M.; Kolattukudy, P.E. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1411–H1420. [Google Scholar] [CrossRef]
- DeGracia, D.J.; Montie, H.L. Cerebral ischemia and the unfolded protein response. J. Neurochem. 2004, 91, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Pan, C.; Giraldo, G.S.; Prentice, H.; Wu, J.-Y. Taurine protection of PC12 cells against endoplasmic reticulum stress induced by oxidative stress. J. Biomed. Sci. 2010, 17 (Suppl. S1), S17. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Cai, Q.; Zhang, C.; Yu, Q.; Jiang, Y.; Lee, J.H.; Hawke, D.; Wang, Y.; Xia, Y.; et al. Phosphoglycerate Kinase 1 Phosphorylates Beclin1 to Induce Autophagy. Mol. Cell 2017, 65, 917–931.e6. [Google Scholar] [CrossRef]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef]
- Hald, J.; Jacobson, E. A drug sensitizing the organism to ethyl alcohol. Lancet 1948, 2, 1001–1004. [Google Scholar] [CrossRef]
- Nagendra, S.N.; Faiman, M.D.; Davis, K.; Wu, J.Y.; Newby, X.; Schloss, J. V Carbamoylation of brain glutamate receptors by a disulfiram metabolite. J. Biol. Chem. 1997, 272, 24247–24251. [Google Scholar] [CrossRef]
- Hart, B.W.; Faiman, M.D. In vitro and in vivo inhibition of rat liver aldehyde dehydrogenase by S-methyl N,N-diethylthiolcarbamate sulfoxide, a new metabolite of disulfiram. Biochem. Pharmacol. 1992, 43, 403–406. [Google Scholar] [CrossRef]
- Madan, A.; Parkinson, A.; Faiman, M.D. Identification of the human and rat P450 enzymes responsible for the sulfoxidation of S-methyl N,N-diethylthiolcarbamate (DETC-ME). The terminal step in the bioactivation of disulfiram. Drug Metab. Dispos. 1995, 23, 1153–1162. [Google Scholar]
- Kaul, S.; Williams, T.D.; Lunte, C.E.; Faiman, M.D. LC-MS/MS determination of carbamathione in microdialysis samples from rat brain and plasma. J. Pharm. Biomed. Anal. 2010, 51, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Cano-Cebrián, M.J.; Zornoza-Sabina, T.; Guerri, C.; Polache, A.; Granero, L. Local acamprosate modulates dopamine release in the rat nucleus accumbens through NMDA receptors: An in vivo microdialysis study. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2003, 367, 119–125. [Google Scholar] [CrossRef]
- Faiman, M.D.; Kaul, S.; Latif, S.A.; Williams, T.D.; Lunte, C.E. S-(N, N-diethylcarbamoyl)glutathione (carbamathione), a disulfiram metabolite and its effect on nucleus accumbens and prefrontal cortex dopamine, GABA, and glutamate: A microdialysis study. Neuropharmacology 2013, 75, 95–105. [Google Scholar] [CrossRef]
- Ma, S.; Liu, H.; Jiao, H.; Wang, L.; Chen, L.; Liang, J.; Zhao, M.; Zhang, X. Neuroprotective effect of ginkgolide K on glutamate-induced cytotoxicity in PC 12 cells via inhibition of ROS generation and Ca2+ influx. Neurotoxicology 2012, 33, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Prentice, H.; Price, A.L.; Wu, J.-Y. Beneficial effect of taurine on hypoxia- and glutamate-induced endoplasmic reticulum stress pathways in primary neuronal culture. Amino Acids 2012, 43, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Modi, J.; Altamimi, A.; Morrell, A.; Chou, H.; Menzie, J.; Weiss, A.; Marshall, M.L.; Li, A.; Prentice, H.; Wu, J.-Y. Protective functions of AEURA in Cell Based Model of Stroke and Alzheimer disease. J. Neurosci. Neurol. Disord. 2017, 1, 016–023. [Google Scholar]
- Jiang, Z.; Yang, J.; Purpura, L.A.; Liu, Y.; Ripps, H.; Shen, W. Glycinergic feedback enhances synaptic gain in the distal retina. J. Physiol. 2014, 592, 1479–1492. [Google Scholar] [CrossRef]
- Ren, J.; Chen, Y.I.; Liu, C.H.; Chen, P.-C.; Prentice, H.; Wu, J.-Y.; Liu, P.K. Noninvasive tracking of gene transcript and neuroprotection after gene therapy. Gene Ther. 2016, 23, 1–9. [Google Scholar] [CrossRef]
- Tao, R.; Shokry, I.M.; Callanan, J.J.; Adams, H.D.; Ma, Z. Mechanisms and environmental factors that underlying the intensification of 3,4-methylenedioxymethamphetamine (MDMA, Ecstasy)-induced serotonin syndrome in rats. Psychopharmacology 2015, 232, 1245–1260. [Google Scholar] [CrossRef]
- Fowler, S.C.; Birkestrand, B.R.; Chen, R.; Moss, S.J.; Vorontsova, E.; Wang, G.; Zarcone, T.J. A force-plate actometer for quantitating rodent behaviors: Illustrative data on locomotion, rotation, spatial patterning, stereotypies, and tremor. J. Neurosci. Methods 2001, 107, 107–124. [Google Scholar] [CrossRef]
- Li, F.; Omae, T.; Fisher, M. Spontaneous hyperthermia and its mechanism in the intraluminal suture middle cerebral artery occlusion model of rats. Stroke 1999, 30, 2464–2470, discussion 2470–2471. [Google Scholar] [CrossRef]
- Bederson, J.B.; Pitts, L.H.; Germano, S.M.; Nishimura, M.C.; Davis, R.L.; Bartkowski, H.M. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke 1986, 17, 1304–1308. [Google Scholar] [CrossRef]
- Rich, P.R.; Mischis, L.A.; Purton, S.; Wiskich, J.T. The sites of interaction of triphenyltetrazolium chloride with mitochondrial respiratory chains. FEMS Microbiol. Lett. 2001, 202, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Irie, K.; Anwer, M.S.; Fisher, M. Delayed triphenyltetrazolium chloride staining remains useful for evaluating cerebral infarct volume in a rat stroke model. J. Cereb. Blood Flow Metab. 1997, 17, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Modi, J.P.; Gharibani, P.M.; Ma, Z.; Tao, R.; Menzie, J.; Prentice, H.; Wu, J.-Y. Protective mechanism of sulindac in an animal model of ischemic stroke. Brain Res. 2014, 1576, 91–99. [Google Scholar] [CrossRef]
- Mohammad-Gharibani, P.; Tiraihi, T.; Delshad, A.; Arabkheradmand, J.; Taheri, T. Improvement of contusive spinal cord injury in rats by co-transplantation of gamma-aminobutyric acid-ergic cells and bone marrow stromal cells. Cytotherapy 2013, 15, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Erie, C.; Sacino, M.; Houle, L.; Lu, M.L.; Wei, J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur. J. Neurosci. 2015, 42, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Buddhala, C.; Suarez, M.; Modi, J.; Prentice, H.; Ma, Z.; Tao, R.; Wu, J.Y. Calpain cleavage of brain glutamic acid decarboxylase 65 is pathological and impairs GABA neurotransmission. PLoS ONE 2012, 7, e33002. [Google Scholar] [CrossRef]
- Rane, M.J.; Pan, Y.; Singh, S.; Powell, D.W.; Wu, R.; Cummins, T.; Chen, Q.; McLeish, K.R.; Klein, J.B. Heat shock protein 27 controls apoptosis by regulating Akt activation. J. Biol. Chem. 2003, 278, 27828–27835. [Google Scholar] [CrossRef]
- Broughton, B.R.S.; Reutens, D.C.; Sobey, C.G. Apoptotic Mechanisms After Cerebral Ischemia. Stroke 2009, 40, e331–e339. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef]
- Stetler, R.A.; Cao, G.; Gao, Y.; Zhang, F.; Wang, S.; Weng, Z.; Vosler, P.; Zhang, L.; Signore, A.; Graham, S.H.; et al. Hsp27 Protects against Ischemic Brain Injury via Attenuation of a Novel Stress-Response Cascade Upstream of Mitochondrial Cell Death Signaling. J. Neurosci. 2008, 28, 13038. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.-R.; Wang, X.; Hu, W.-W.; et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.T.; Wolter, K.G.; Youle, R.J. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3668–3672. [Google Scholar] [CrossRef] [PubMed]
- Korsmeyer, S.J.; Gross, A.; Harada, H.; Zha, J.; Wang, K.; Yin, X.M.; Wei, M.; Zinkel, S. Death and survival signals determine active/inactive conformations of pro-apoptotic BAX, BAD, and BID molecules. Cold Spring Harb. Symp. Quant. Biol. 1999, 64, 343–350. [Google Scholar] [CrossRef]
- Leon, R.; Wu, H.; Jin, Y.; Wei, J.; Buddhala, C.; Prentice, H.; Wu, J.Y. Protective function of taurine in glutamate-induced apoptosis in cultured neurons. J. Neurosci. Res. 2009, 87, 1185–1194. [Google Scholar] [CrossRef]
- Lee, R.M.K.W. Morphology of cerebral arteries. Pharmacol. Ther. 1995, 66, 149–173. [Google Scholar] [CrossRef]
- Sommer, C.J. Ischemic stroke: Experimental models and reality. Acta Neuropathol. 2017, 133, 245. [Google Scholar] [CrossRef]
- Paschen, W.; Mengesdorf, T. Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium 2005, 38, 409–415. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflammation 2019, 16, 1–24. [Google Scholar] [CrossRef]
- Emelda, M. Difference between In Vivo and In Vitro. 2011. Available online: http://www.differencebetween.net/science/difference-between-in-vivo-and-in-vitro/ (accessed on 16 October 2017).
- Hainsworth, A.H.; Markus, H.S. Do in vivo Experimental Models Reflect Human Cerebral Small Vessel Disease? a Systematic Review. J. Cereb. Blood Flow Metab. 2008, 28, 1877–1891. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, Y.; Fu, X.; Li, L.; Hao, X.; Li, S. Nonhuman primate models of focal cerebral ischemia. Neural Regen. Res. 2017, 12, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Gharibani, P.; Modi, J.; Menzie, J.; Alexandrescu, A.; Ma, Z.; Tao, R.; Prentice, H.; Wu, J.-Y. Comparison between single and combined post-treatment with S-Methyl-N,N-diethylthiolcarbamate sulfoxide and taurine following transient focal cerebral ischemia in rat brain. Neuroscience 2015, 300, 460–473. [Google Scholar] [CrossRef]
- Gharibani, P.M.; Modi, J.; Pan, C.; Menzie, J.; Ma, Z.; Chen, P.-C.; Tao, R.; Prentice, H.; Wu, J.-Y. The mechanism of taurine protection against endoplasmic reticulum stress in an animal stroke model of cerebral artery occlusion and stroke-related conditions in primary neuronal cell culture. Adv. Exp. Med. Biol. 2013, 776, 241–258. [Google Scholar] [CrossRef]
- Zhang, L.; Schallert, T.; Zhang, Z.G.; Jiang, Q.; Arniego, P.; Li, Q.; Lu, M.; Chopp, M. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J. Neurosci. Methods 2002, 117, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Ningaraj, N.S.; Chen, W.; Schloss, J.V.; Faiman, M.D.; Wu, J.-Y. S-methyl-N,N-diethylthiocarbamate sulfoxide elicits neuroprotective effect against N-methyl-D-aspartate receptor-mediated neurotoxicity. J. Biomed. Sci. 2001, 8, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Gessner, T.; Jakubowski, M. Diethyldithiocarbamic acid methyl ester. A metabolite of disulfiram. Biochem. Pharmacol. 1972, 21, 219–230. [Google Scholar] [CrossRef]
- Walton, H.; Dodd, P. Glutamate–glutamine cycling in Alzheimer’s disease. Neurochem. Int. 2007, 50, 1052–1066. [Google Scholar] [CrossRef]
- Portera-Cailliau, C.; Hedreen, J.C.; Price, D.L.; Koliatsos, V.E. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J. Neurosci. 1995, 15, 3775–3787. [Google Scholar] [CrossRef]
- Hazell, A. Excitotoxic mechanisms in stroke: An update of concepts and treatment strategies. Neurochem. Int. 2007, 50, 941–953. [Google Scholar] [CrossRef]
- Manev, H.; Favaron, M.; Guidotti, A.; Costa, E. Delayed increase of Ca2+ influx elicited by glutamate: Role in neuronal death. Mol. Pharmacol. 1989, 36, 106–112. [Google Scholar]
- Waxman, E.A.; Lynch, D.R. N-methyl-D-aspartate receptor subtypes: Multiple roles in excitotoxicity and neurological disease. Neuroscientist 2005, 11, 37–49. [Google Scholar] [CrossRef]
- Nakka, V.P.; Gusain, A.; Mehta, S.L.; Raghubir, R. Molecular mechanisms of apoptosis in cerebral ischemia: Multiple neuroprotective opportunities. Mol. Neurobiol. 2008, 37, 7–38. [Google Scholar] [CrossRef]
- Valentim, L.; Laurence, K.M.; Townsend, P.A.; Carroll, C.J.; Soond, S.; Scarabelli, T.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2006, 40, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, I.A.; Goemans, G.C.; Skepper, J.N.; Tolkovsky, A.M. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene 2009, 28, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- Feder, M.E.; Hofmann, G.E. Heat- shock proteins, Molecular chaperons, and the stress response: Evolutionary and Ecological Physiology. Annu. Rev. Physiol. 1999, 61, 243–282. [Google Scholar] [CrossRef]
- Badin, R.A.; Lythgoe, M.F.; van der Weerd, L.; Thomas, D.L.; Gadian, D.G.; Latchman, D.S. Neuroprotective effects of virally delivered HSPs in experimental stroke. J. Cereb. Blood Flow Metab. 2006, 26, 371–381. [Google Scholar] [CrossRef]
- Yenari, M.A. Heat shock proteins and neuroprotection. Adv. Exp. Med. Biol. 2002, 513, 281–299. [Google Scholar]
- Whitlock, N.A.; Lindsey, K.; Agarwal, N.; Crosson, C.E.; Ma, J.-X. Heat shock protein 27 delays Ca2+-induced cell death in a caspase-dependent and -independent manner in rat retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Kilic, E.; Kilic, U.; Soliz, J.; Bassetti, C.L.; Gassmann, M.; Hermann, D.M. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J. 2005, 19, 2026–2028. [Google Scholar] [CrossRef]
- Pap, M.; Cooper, G.M. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 1998, 273, 19929–19932. [Google Scholar] [CrossRef]
- Schulz, I.; Engel, C.; Niestroj, A.J.; Zeitschel, U.; Menge, K.; Kehlen, A.; Meyer, A.; Roßner, S.; Demuth, H.U. Heteroarylketones inhibit astroglial interleukin-6 expression via a STAT3/NF-κB signaling pathway. J. Neuroinflam. 2011, 8, 86. [Google Scholar] [CrossRef]
- Song, S.Y.; Jung, Y.Y.; Hwang, C.J.; Lee, H.P.; Sok, C.H.; Kim, J.H.; Lee, S.M.; Seo, H.O.; Hyun, B.K.; Choi, D.Y.; et al. Inhibitory effect of ent-Sauchinone on amyloidogenesis via inhibition of STAT3-mediated NF-κB activation in cultured astrocytes and microglial BV-2 cells. J. Neuroinflam. 2014, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Albrecht, U.; Häussinger, D.; Heinrich, P.C.; Schaper, F. Hepatic acute phase proteins—Regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur. J. Cell Biol. 2012, 91, 496–505. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. The unfolding tale of the unfolded protein response. Cell 2001, 107, 827–830. [Google Scholar] [CrossRef]
- Hayashi, T.; Saito, A.; Okuno, S.; Ferrand-Drake, M.; Chan, P.H. Induction of GRP78 by Ischemic Preconditioning Reduces Endoplasmic Reticulum Stress and Prevents Delayed Neuronal Cell Death. J. Cereb. Blood Flow Metab. 2003, 23, 949–961. [Google Scholar] [CrossRef]
- Kumar, R.; Azam, S.; Sullivan, J.M.; Owen, C.; Cavener, D.R.; Zhang, P.; Ron, D.; Harding, H.P.; Chen, J.J.; Han, A.; et al. Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J. Neurochem. 2001, 77, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Prentice, H.; Modi, J.; Menzie, J.; Chou, H.; Weiss, A.; Marshall, L.; Wu, J.; Atlantic, F.; Raton, B.; Raton, B.; et al. Neuroprotective mechanisms of action of DETC-MeSO, GCSF, Sulindac, Taurine and AEURA. SciTz Neurol. Neurosci. 2016, 1, 1004. [Google Scholar]
- Ye, R.; Jung, D.Y.; Jun, J.Y.; Li, J.; Luo, S.; Ko, H.J.; Kim, J.K.; Lee, A.S. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes 2010, 59, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Rothe, M.; Sarma, V.; Dixit, V.; Goeddel, D.; Chung, P.; Harding, H.P.; Ron, D. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science 1995, 269, 1424–1427. [Google Scholar] [CrossRef]
- Szegezdi, E.; Fitzgerald, U.; Samali, A. Caspase-12 and ER-Stress-Mediated Apoptosis. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Modi, J.P.; Shen, W.; Menzie-Suderam, J.; Xu, H.; Lin, C.-H.; Tao, R.; Prentice, H.M.; Schloss, J.; Wu, J.-Y. The Role of NMDA Receptor Partial Antagonist, Carbamathione, as a Therapeutic Agent for Transient Global Ischemia. Biomedicines 2023, 11, 1885. https://doi.org/10.3390/biomedicines11071885
Modi JP, Shen W, Menzie-Suderam J, Xu H, Lin C-H, Tao R, Prentice HM, Schloss J, Wu J-Y. The Role of NMDA Receptor Partial Antagonist, Carbamathione, as a Therapeutic Agent for Transient Global Ischemia. Biomedicines. 2023; 11(7):1885. https://doi.org/10.3390/biomedicines11071885
Chicago/Turabian StyleModi, Jigar Pravinchandra, Wen Shen, Janet Menzie-Suderam, Hongyuan Xu, Chun-Hua Lin, Rui Tao, Howard M. Prentice, John Schloss, and Jang-Yen Wu. 2023. "The Role of NMDA Receptor Partial Antagonist, Carbamathione, as a Therapeutic Agent for Transient Global Ischemia" Biomedicines 11, no. 7: 1885. https://doi.org/10.3390/biomedicines11071885
APA StyleModi, J. P., Shen, W., Menzie-Suderam, J., Xu, H., Lin, C.-H., Tao, R., Prentice, H. M., Schloss, J., & Wu, J.-Y. (2023). The Role of NMDA Receptor Partial Antagonist, Carbamathione, as a Therapeutic Agent for Transient Global Ischemia. Biomedicines, 11(7), 1885. https://doi.org/10.3390/biomedicines11071885