Abstract
Activating mutations and fusions of the ALK oncogene have been identified as drivers in a number of malignancies. Crizotinib and subsequent ALK tyrosine kinase inhibitors have improved treatment outcomes for these patients. In this paper, we discuss the case of an adolescent patient with acute myeloid leukemia, who was identified to have an activating ALK fusion, which is a rare finding and has never been reported in cases of AML without monosomy 7. Crizotinib was added to this patient’s frontline therapy and was well tolerated. In cases of more common gene alterations, existing data supports the use of targeted agents as post-HSCT maintenance therapy; however, crizotinib was not able to be used post-HSCT for this patient due to the inability to obtain insurance coverage.
1. Introduction
The ALK oncogene encodes ALK, a membrane-bound receptor tyrosine kinase, which activates multiple downstream signaling pathways that are keys in cell proliferation and survival [1]. It was first identified in 1994 as a fusion partner in a t(2;5) chromosomal translocation in anaplastic large cell lymphoma, which is how it got its name (Anaplastic Lymphoma Kinase) [2]. Many other fusion partners and activating gene alterations (point mutations, deletions, and rearrangements) have since been identified as oncogenic drivers in a variety of malignancies, including non-small-cell lung cancer (NSCLC), inflammatory myofibroblastic tumor (IMT), neuroblastoma, and renal cancers [3,4,5]. In the past two decades, the landscape of treatment for these tumors has been changed by targeted therapy with small-molecule ALK tyrosine kinase inhibitors (TKI), of which several are currently FDA approved for use in adults, and one (crizotinib) for use in children [6,7].
More recently, oncogenic ALK fusions have been identified in a small subset of patients with acute myeloid leukemia (AML) and other myeloid-lineage hematologic malignancies (Table 1). Recent advances in genomic and transcriptomic technologies have allowed us to perform risk stratification and adjust treatment for patients with AML based on cytogenetic and molecular features, but certain alterations continue to confer poor prognoses despite treatment intensification. Patients with the monosomy 7 subtype AML, a subset with a poor prognosis, were recently retrospectively evaluated via RNA sequencing by Ries et al. (2020), and 14% of the patients were found to have cryptic ALK fusions [8,9]. ALK fusions have been described in adults with the monosomy 7 subtype of AML as well [10,11,12]. Importantly, cells transformed by ALK fusions demonstrate cytokine-independent proliferation and are sensitive in vitro to crizotinib [9].

Table 1.
Characteristics of patients reported in the literature to have myeloid-lineage hematologic malignancies with ALK fusions. Abbreviations: MDS, myelodysplastic syndrome; JMML, juvenile myelomonocytic leukemia; WBC, white blood cells; BM, bone marrow; ISCN, International System for Human Cytogenomic Nomenclature; RT-PCR, reverse transcription polymerase chain reaction.
In this paper, we describe the case of an 18-year-old female with AML, who was found to have a somatic RANBP2::ALK fusion. This is the first reported case of AML with this ALK fusion without monosomy 7. We describe her treatment and clinical course, including the use of crizotinib. Just as patients with FLT3-mutated AML have shown improved overall survival from the addition of targeted therapy (sorafenib and other kinase inhibitors), both with upfront chemotherapy and as post-HSCT maintenance therapy [14,15], we believe crizotinib used at these same timepoints will benefit these rare patients with ALK-fusion AML.
2. Case Presentation
An 18-year-old female presented to the emergency room at our hospital with one week of progressive fatigue and pallor. Her medical history included irregular menstrual periods and psychiatric diagnoses for which she took concomitant medications. She initially presented to a mobile clinic where a CBC was drawn, showing severe anemia. She was referred to the nearest pediatric emergency center, where a repeat CBC was significant, for a total WBC of 24.37 × 103/µL with a monocytic predominance (45%) and no blasts present by automatic differential; hemoglobin of 5.8 g/dL; and platelet count of 30 × 103/µL. An initial packed red blood cell (PRBC) transfusion was given with no significant increase in Hgb (post-transfusion Hgb of 6.0 g/dL). Her reticulocyte count, mean corpuscular volume (MCV), unconjugated bilirubin, and lactate dehydrogenase (LDH) were elevated, suggestive of possible acute hemolysis. The leading differential diagnosis at the time was acute leukemia, an autoimmune etiology causing destructive cytopenias, or a viral/infectious process. A flow cytometry analysis of peripheral blood identified a large population of monocytes, but without significant immunophenotypic aberrancies. Three days after admission, a bone marrow biopsy and aspirate were performed. The anatomic pathology revealed 20.5% blasts with features of abnormal monoblasts, which appeared more immature with prominent nuclear folds and gray to basophilic cytoplasm (Figure 1). The flow cytometry showed increased monocytes (46%) with immunophenotypic aberrancies, including CD4 (brighter), CD16+56 (partial, bright), CD117 (partial), and CD2 (partial), and a loss of CD11b (Figure 2). These results were consistent with acute myeloid leukemia with monocytic differentiation.
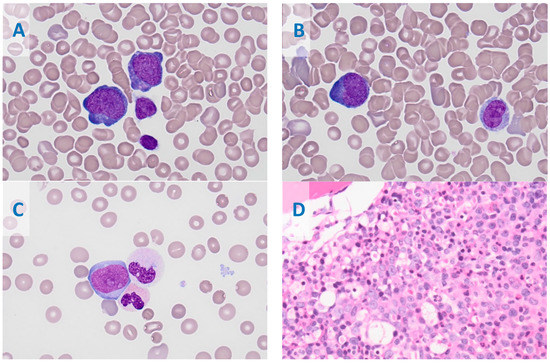
Figure 1.
Bone marrow aspirate smears show increased leukemic blasts (20.5%). The blasts are medium to large sized with high N:C ratio, with round to irregular to slightly convoluted nuclear contours, and have a finely dispersed chromatin pattern and visible nucleoli with variably basophilic and rarely azurophilic granules; overall, the blasts have features of monoblasts (A) and promonocytes (blast equivalents (B). Occasional neutrophils are dysplastic with abnormal nuclear lobation and hypogranulation (C). The core biopsy shows leukemic blast infiltration (D); (A–C), Wright–Giemsa Stain, at 1000×; (D), Hematoxylin and Eosin stain, with a magnification of 400×.
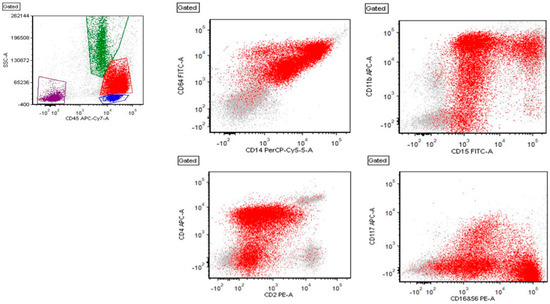
Figure 2.
Flow cytometry showing red: leukemic blasts, green: granulocytes, blue: lymphocytes, and burgundy: debris. The blasts are positive for CD45, CD64, CD14, CD15, CD11b (partial), CD2 (partial), CD4, CD16+56 (partial, bright), and CD117 (partial) (shown here). The blasts are also positive for (not shown here) CD13, CD33, HLA-DR, CD52 (partial), CD99, CD58, CD38, and myeloperoxidase (partial).
The patient was transferred from the satellite community campus to the main academic campus and was enrolled into the active Children’s Oncology Group (COG) study for AML, AAML1831. She was randomized to Arm B with CPX-351 (Vyxeos; 60 mg/m2 daunorubicin equivalents IV on days 1, 3, and 5) and gemtuzumab ozogamicin (4.5 mg on day 6). A lumbar puncture on day 8 was consistent with the CNS1 status. On day 10 of therapy, the local cytogenetics studies with chromosome analysis showed an abnormal clone in 18 out of 20 cells characterized by a pericentric inversion of chromosome 2 at bands 2p23-q13. A fluorescence in situ hybridization (FISH) evaluation was performed using 13 probe sets for AML and MDS and revealed no evidence of abnormal signal patterns. At the same time, the results of her centralized molecular studies from the COG AAML1831 study showed no CEBPα or NPM1 mutation, and a FLT3 internal tandem duplication (FLT3-ITD) with an allelic ratio of 0.62. Since the gilteritinib arm of the COG AAML1831 study was temporarily closed, she was taken off the protocol therapy the following day and started sorafenib at 400 mg PO daily on days 11–31. On day 27 of Induction 1, the patient’s primary oncologist was informed that the central lab’s FLT3-ITD result was an error due to sample mix-up. At the end of Induction 1 (day 45), an institutional next-generation DNA/RNA sequencing panel run on the diagnostic bone marrow aspirate was performed, confirming no FLT3-ITD mutation. The sequencing panel showed a fusion between exon 18 of RAN-binding protein 2 (RANB2) on chromosome 2 (2q13) and exon 20 of ALK receptor tyrosine kinase (ALK) on chromosome 2 (2p23.2-2p23.1). (Figure 3) This RANBP2::ALK fusion, which was presumed to retain the tyrosine kinase domain of ALK, was confirmed using RT-PCR and Sanger sequencing, and is consistent with the pericentric inversion of chromosome 2 noted in the initial diagnostic cytogenetic report.
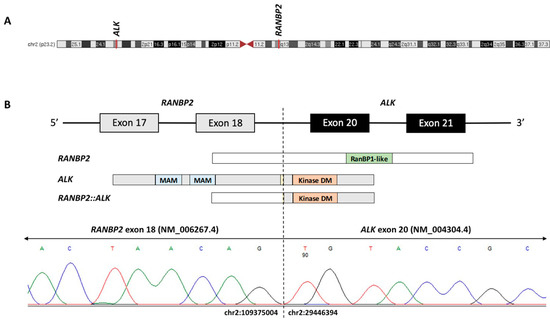
Figure 3.
(A) Schematic of chromosome 2 with red lines denoting the chromosomal positions of ALK [ALK receptor tyrosine kinase] and RANPB2 [RAN-binding protein 2] on chromosomes 2p23.1 and 2q13, respectively. (B) The RANBP2::ALK fusion protein is predicted to maintain an intact 3′ ALK tyrosine kinase domain (Kinase DM) by joining exon 18 of RANBP2 to exon 20 of ALK. The RT-PCR and Sanger sequencing confirmed the fusion breakpoint at chr2:109375004 (RANBP2, NM_006267.4, exon 18) and chr2:29446394 (ALK, NM_004304.4, exon 20). Domain abbreviations: MAM, meprin/A5/mu; RanBP1-like, Ran-binding protein RanBP1-like.
A bone marrow evaluation at the end of Induction 1 showed a minimal residual disease (MRD) positive state with 0.1% residual acute myeloid leukemia via flow cytometry. Chromosome analysis was not able to be performed at this timepoint due to a markedly hypocellular (10%) marrow, resulting in inadequate sample volume being obtained. Based on this finding, the patient was considered high risk and therefore eligible for HSCT at first remission. She began Induction 2 and was treated according to an institutional practice standard with cytarabine (100 mg/m2/dose IV every 12 h on days 1–8) and daunorubicin (50 mg/m2/dose on days 1, 3, and 5). On day 10 of Induction 2, once systemic chemotherapy was completed, crizotinib at 250 mg twice daily was started as a targeted agent against the known oncogenic activating ALK fusion. A bone marrow evaluation at the end of Induction 2 demonstrated MRD-negative remission. She then received Intensification 1 as per the institutional practice standard of high-dose cytarabine (1000 mg/m2/dose every 12 h on days 1–5) and etoposide (150 mg/m2/dose on days 1–5), again with the addition of crizotinib beginning on day 7 of the cycle. The MRD evaluation remained negative after Intensification 1.
The patient tolerated the chemotherapy and crizotinib well. She experienced nausea that was managed with scheduled antiemetics; an erythematous rash of lower extremities that was likely secondary to CPX-351 and was managed with topical steroid and emollient; and episodes of febrile neutropenia during hospitalizations, but never with any bacterial or fungal organisms isolated. While on crizotinib, QTc was monitored closely and remained within the safe range to continue the medication.
Following three cycles of chemotherapy, she underwent an allogeneic hematopoietic stem cell transplant with a 10/10 HLA-matched unrelated donor. A conditioning regimen of alemtuzumab, busulfan, and cyclophosphamide was used. Her post-transplant course was complicated by severe shock with generalized fluid overload on day +12 in the setting of engraftment syndrome, as well as concomitant hepatic veno-occlusive disease/sinusoidal obstruction syndrome. As a result, she developed acute kidney injury that initially required continuous renal replacement therapy and then intermittent hemodialysis (last session on day +29). She was treated with 21 days of defibrotide (days +12 to +32). She engrafted on day +16 post-transplant. A day +30 disease evaluation of both bone marrow and CSF was performed, showing no evidence of leukemia cells based on flow cytometry, no evidence of ALK gene rearrangement based on FISH probe, and 100% donor cells. She was discharged from the hospital on day +39. A day +106 evaluation of bone marrow again showed no evidence of leukemia, no ALK gene rearrangement, and 100% donor cells. The initial plan was to begin crizotinib as a maintenance therapy once all cell lines recovered, estimated to be around day +100; however, its coverage was denied by her private insurance despite multiple appeals. She developed mixed chimerism initially noted during peripheral blood surveillance on day +168. Serial monitoring of donor chimerism status via peripheral blood using cytogenetics for the sex chromosome and short tandem repeat (STR)-PCR showed stable mixed chimera with >95% donor cells and without requiring additional interventions. The FISH evaluation of peripheral blood showed no evidence of ALK gene rearrangement despite mixed chimerism.
At the most recent follow-up visit, the patient was day +197 post-transplant and remains in remission. She had intermittent neutropenia requiring granulocyte colony stimulating factor (GCSF) in the setting of a viral respiratory infection which has resolved. She has a recent fluctuating low-level Epstein Barr virus (EBV) viremia not requiring treatment at this time. Her kidney function has improved. She has had no evidence of graft versus host disease, is now off immune-suppressive agents, and remains transfusion independent with stable, low-level mixed chimerism.
3. Discussion
The discovery and implementation of targeted cancer-directed therapy has thrust the field of oncology forward and improved survival for patients with various malignancies. In the treatment of AML, this has been best demonstrated by treatment of patients with FMS-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD), an alteration present in about 30% of adult AML patients and 15% of pediatric AML patients. FLT3-ITD confers an especially poor prognosis with a high likelihood of relapse even after allogeneic stem cell transplant. However, the addition of TKIs that target FLT3, such as sorafenib, midostaurin, or gilteritinib, into frontline treatment in combination with conventional chemotherapy has reduced relapse rates and improved survival [16,17]. With this success, which is thought to be related to improved disease control before transplant, clinical practice has shifted toward also including a FLT3-targeting TKI as post-transplant maintenance therapy with the goal of reducing post-transplant relapses. While there is not yet a clear best practice or guideline for utilizing TKIs as post-transplant maintenance therapy in patients with FLT3-ITD+AML, many studies have shown survival benefits with tolerable toxicity profiles and costs [18]. The current Children’s Oncology Group frontline AML study, AAML1831, includes TKI (gilteritinib) maintenance therapy for one year after HSCT for patients with FLT3-ITD+AML.
Using this rationale, we hypothesized that adding the ALK-targeting TKI crizotinib to frontline treatment and as post-transplant maintenance therapy would benefit our patient. During upfront therapy, crizotinib was well tolerated and potentially contributed to the patient achieving and maintaining MRD negativity prior to HSCT. Unfortunately, because of the rarity of ALK fusions in AML, there are no existing prospective trial data to support prophylactic post-HSCT maintenance therapy in a MRD negative state for AML with ALK fusions, which has led to roadblocks in achieving insurance coverage of the medication.
In summary, this case study draws attention to a rare but targetable oncogenic fusion in a patient with AML, and it is the first reported case of an ALK fusion in AML without monosomy 7. Most institutional AML FISH probe sets do not include ALK; thus, we must rely on DNA and RNA sequencing as a crucial diagnostic tool to identify these and other rare, potentially actionable gene alterations. Our case study also highlights the challenges of our third-party payor health care system, which have substantial impact on physicians’ ability to use relevant, albeit limited, data to offer targeted agents off-label to patients with rare conditions.
Author Contributions
Conceptualization, M.S.R. and M.S.; writing—original draft preparation, M.S. and G.L.C.; writing—review and editing, M.S.R.; visualization and formal analysis, J.N.P., C.V.C. and K.E.F. All authors have read and agreed to the published version of the manuscript.
Funding
This report received no external funding.
Institutional Review Board Statement
This case study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of Baylor College of Medicine/Texas Children’s Hospital H-51145, approved on 15 February 2022.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We would like to thank all those involved in the clinical care of this patient, as well as all laboratory staff who were involved in generating and reporting the results that allowed us to best treat this patient.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Benharroch, D.; Meguerian-Bedoyan, Z.; Lamant, L.; Amin, C.; Brugières, L.; Terrier-Lacombe, M.J.; Delsol, G. ALK-positive lymphoma: A single disease with a broad spectrum of morphology. Blood J. Am. Soc. Hematol. 1998, 91, 2076–2084. [Google Scholar]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.A.; Hawkins, A.L.; Dvorak, C.; Henkle, C.; Ellingham, T.; Perlman, E. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999, 59, 2776–2780. [Google Scholar] [PubMed]
- Carén, H.; Abel, F.; Kogner, P.; Martinsson, T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J. 2008, 416, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.-H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children’s Oncology Group Study. J. Clin. Oncol. 2017, 35, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- Ries, R.E.; Triche, T.J.; Smith, J.L.; Leonti, A.R.; Alonzo, T.A.; Farrar, J.E.; Chen, X.; Liu, Y.; Shaw, T.; Huang, B.J.; et al. Genome and Transcriptome Profiling of Monosomy 7 AML Defines Novel Risk and Therapeutic Cohorts. Blood 2020, 136 (Suppl. S1), 20–21. [Google Scholar] [CrossRef]
- Manselle, M.K.; Ries, R.E.; Hylkema, T.; Leonti, A.; Kirkey, D.C.; Furlan, S.N.; Meshinchi, S. Functional consequence and therapeutic targeting of cryptic ALK fusions in monosomy 7 acute myeloid leukemia. Pediatr. Blood Cancer 2023, 70, e30180. [Google Scholar] [CrossRef] [PubMed]
- Maesako, Y.; Izumi, K.; Okamori, S.; Takeoka, K.; Kishimori, C.; Okumura, A.; Ohno, H. inv (2)(p23q13)/RAN-binding protein 2 (RANBP2)–ALK fusion gene in myeloid leukemia that developed in an elderly woman. Int. J. Hematol. 2014, 99, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Takeoka, K.; Okumura, A.; Maesako, Y.; Akasaka, T.; Ohno, H. Crizotinib resistance in acute myeloid leukemia with inv (2)(p23q13)/RAN binding protein 2 (RANBP2) anaplastic lymphoma kinase (ALK) fusion and monosomy 7. Cancer Genet. 2015, 208, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.; Jang, S.; Park, C.-J.; Cho, Y.-U.; Lee, J.-H.; Lee, K.-H.; Lee, J.-O.; Shin, J.-Y.; Kim, J.-I.; Huh, J.; et al. RANBP2-ALK fusion combined with monosomy 7 in acute myelomonocytic leukemia. Cancer Genet. 2014, 207, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Röttgers, S.; Gombert, M.; Teigler-Schlegel, A.; Busch, K.; Gamerdinger, U.; Slany, R.; Harbott, J.; Borkhardt, A. ALK fusion genes in children with atypical myeloproliferative leukemia. Leukemia 2010, 24, 1197–1200. [Google Scholar] [CrossRef]
- Bazarbachi, A.; Labopin, M.; Battipaglia, G.; Djabali, A.; Forcade, E.; Arcese, W.; Socié, G.; Blaise, D.; Passweg, J.R.; Cornelissen, J.J.; et al. Post-Transplant Sorafenib Improves Overall Survival in FLT3 Mutated AML: A Report from the EBMT Acute Leukemia Working Party. Blood 2018, 132 (Suppl. S1), 708. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Gerbing, R.; Brown, P.; Fox, E.; Choi, J.; Aplenc, R. Sorafenib in combination with standard chemotherapy for children with high allelic ratio FLT3/ITD+ acute myeloid leukemia: A report from the children’s oncology group protocol AAML1031. J. Clin. Oncol. 2022, 40, 2023–2035. [Google Scholar] [CrossRef] [PubMed]
- Röllig, C.; Serve, H.; Hüttmann, A.; Noppeney, R.; Müller-Tidow, C.; Krug, U.; Ehninger, G. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Röllig, C.; Serve, H.; Noppeney, R.; Hanoun, M.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; Krämer, A.; et al. Sorafenib or placebo in patients with newly diagnosed acute myeloid leukaemia: Long-term follow-up of the randomized controlled SORAML trial. Leukemia 2021, 35, 2517–2525. [Google Scholar] [CrossRef]
- Gagelmann, N.; Wolschke, C.; Klyuchnikov, E.; Christopeit, M.; Ayuk, F.; Kröger, N. TKI maintenance after stem-cell transplantation for FLT3-ITD positive acute myeloid leukemia: A systematic review and meta-analysis. Front. Immunol. 2021, 12, 630429. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).