
Sertoli Cells Express Accommodation, Survival, and Immunoregulatory Factors When Exposed to Normal Human Serum

 ,
,  ,
,
Abstract
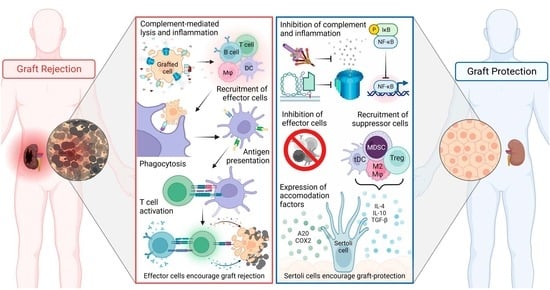
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Sertoli Cell Isolation
2.3. RNA Sequencing
2.4. PCR for Genes of Interest
2.5. Protein Collection and Quantification by Western Blot and ELISA
2.6. Bioinformatic Analyses
2.7. Statistical Analyses
3. Results
3.1. DEG Identified by RNA Sequencing
3.2. Gene Expression Analysis
3.3. Protein Expression Analysis of A20 (TNFAIP3) and CCL2
3.4. GO and Pathway Analyses
3.5. Functional Pathway Analyses of Immune-Related Genes
3.5.1. Innate Immune Signaling Pathways
3.5.2. Cytokine Signaling
3.5.3. T Cell Regulation
4. Discussion
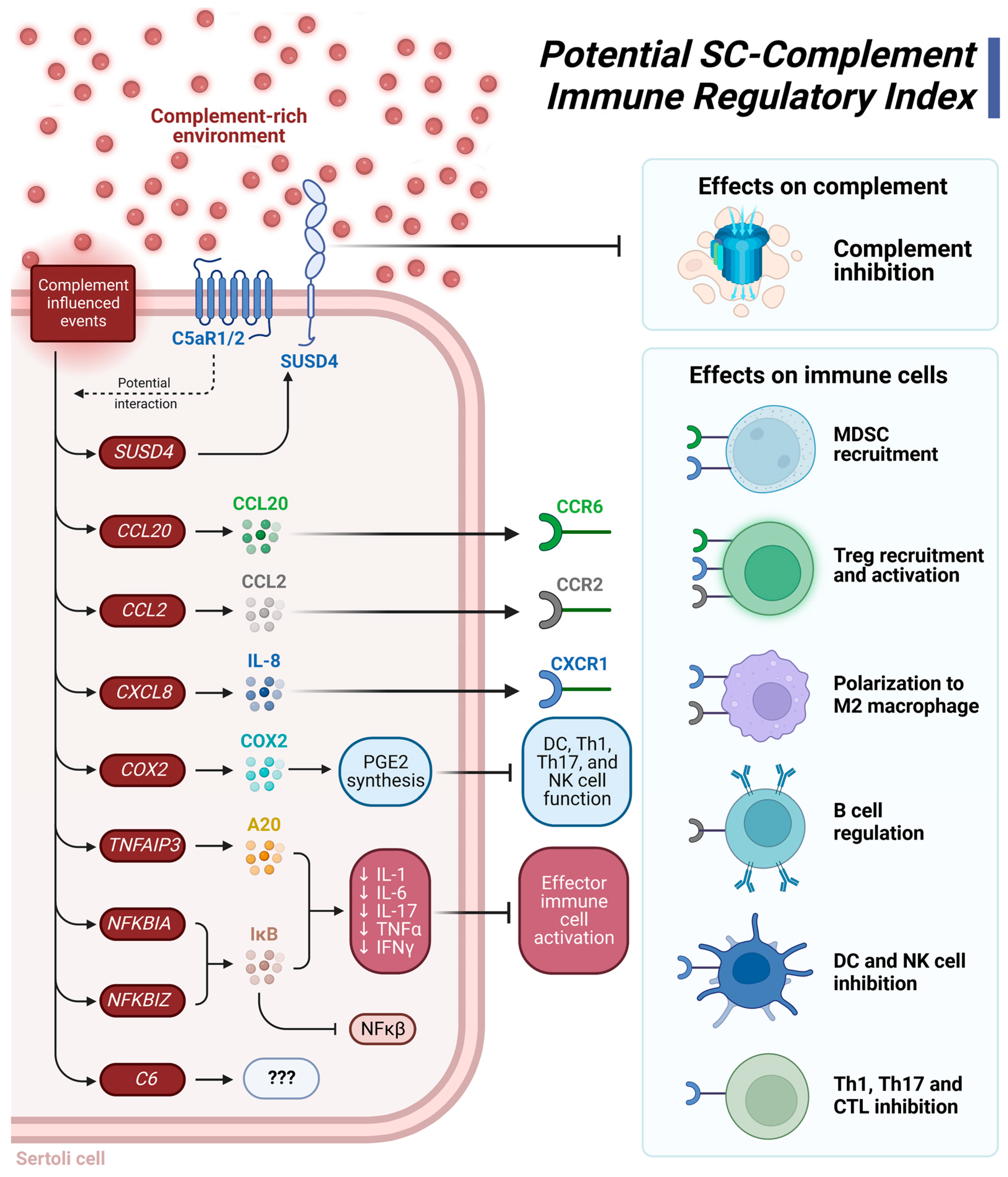
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cooper, D.K.C. Genetically engineered pig kidney transplantation in a brain-dead human subject. Xenotransplantation 2021, 28, e12718. [Google Scholar] [CrossRef]
- Griffith, B.P.; Goerlich, C.E.; Singh, A.K.; Rothblatt, M.; Lau, C.L.; Shah, A.; Lorber, M.; Grazioli, A.; Saharia, K.K.; Hong, S.N.; et al. Genetically Modified Porcine-to-Human Cardiac Xenotransplantation. N. Engl. J. Med. 2022, 387, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.M.; Dass, B.; Halley, K.R.; Korbutt, G.S.; Dixon, D.E.; Rajotte, R.V. Sertoli cell line lacks the immunoprotective properties associated with primary Sertoli cells. Cell Transplant. 2008, 17, 525–534. [Google Scholar] [CrossRef]
- Dufour, J.M.; Rajotte, R.V.; Seeberger, K.; Kin, T.; Korbutt, G.S. Long-term survival of neonatal porcine Sertoli cells in non-immunosuppressed rats. Xenotransplantation 2003, 10, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.M.; Lord, S.J.; Kin, T.; Rayat, G.R.; Dixon, D.E.; Bleackley, R.C.; Korbutt, G.S.; Rajotte, R.V. Comparison of Successful and Unsuccessful Islet/Sertoli Cell Cotransplant Grafts in Streptozotocin-Induced Diabetic Mice. Cell Transplant. 2007, 16, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.M.; Rajotte, R.V.; Kin, T.; Korbutt, G.S. Immunoprotection of rat islet xenografts by cotransplantation with sertoli cells and a single injection of antilymphocyte serum. Transplantation 2003, 75, 1594–1596. [Google Scholar] [CrossRef]
- Korbutt, G.S.; Elliott, J.F.; Rajotte, R.V. Cotransplantation of allogeneic islets with allogeneic testicular cell aggregates allows long-term graft survival without systemic immunosuppression. Diabetes 1997, 46, 317–322. [Google Scholar] [CrossRef]
- Stites, E.; Le Quintrec, M.; Thurman, J.M. The Complement System and Antibody-Mediated Transplant Rejection. J. Immunol. 2015, 195, 5525–5531. [Google Scholar] [CrossRef]
- Tatapudi, V.S.; Montgomery, R.A. Therapeutic Modulation of the Complement System in Kidney Transplantation: Clinical Indications and Emerging Drug Leads. Front. Immunol. 2019, 10, 2306. [Google Scholar] [CrossRef]
- Dufour, J.M.; Hamilton, M.; Rajotte, R.V.; Korbutt, G.S. Neonatal porcine Sertoli cells inhibit human natural antibody-mediated lysis. Biol. Reprod. 2005, 72, 1224–1231. [Google Scholar] [CrossRef]
- Washburn, R.L.; Martinez-Marin, D.; Korać, K.; Sniegowski, T.; Rodriguez, A.R.; Chilton, B.S.; Hibler, T.; Pruitt, K.; Bhutia, Y.D.; Dufour, J.M. The Sertoli cell complement signature: A suspected mechanism in xenograft survival. Int. J. Mol. Sci. 2023, 25, 1890. [Google Scholar] [CrossRef] [PubMed]
- Arbore, G.; Kemper, C.; Kolev, M. Intracellular complement—The complosome—In immune cell regulation. Mol. Immunol. 2017, 89, 2–9. [Google Scholar] [CrossRef] [PubMed]
- West, E.E.; Kolev, M.; Kemper, C. Complement and the Regulation of T Cell Responses. Annu. Rev. Immunol. 2018, 36, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef]
- Tian, H.; Wang, G.; Wang, Q.; Zhang, B.; Jiang, G.; Li, H.; Chai, D.; Fang, L.; Wang, M.; Zheng, J. Complement C1q binding protein regulates T cells’ mitochondrial fitness to affect their survival, proliferation, and anti-tumor immune function. Cancer Sci. 2022, 113, 875–890. [Google Scholar] [CrossRef]
- Kouser, L.; Madhukaran, S.P.; Shastri, A.; Saraon, A.; Ferluga, J.; Al-Mozaini, M.; Kishore, U. Emerging and Novel Functions of Complement Protein C1q. Front. Immunol. 2015, 6, 317. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Draghici, S.; Khatri, P.; Tarca, A.L.; Amin, K.; Done, A.; Voichita, C.; Georgescu, C.; Romero, R. A systems biology approach for pathway level analysis. Genome Res. 2007, 17, 1537–1545. [Google Scholar] [CrossRef]
- Donato, M.; Xu, Z.; Tomoiaga, A.; Granneman, J.G.; Mackenzie, R.G.; Bao, R.; Than, N.G.; Westfall, P.H.; Romero, R.; Draghici, S. Analysis and correction of crosstalk effects in pathway analysis. Genome Res. 2013, 23, 1885–1893. [Google Scholar] [CrossRef]
- Ortutay, C.; Vihinen, M. Immunome knowledge base (IKB): An integrated service for immunome research. BMC Immunol. 2009, 10, 3. [Google Scholar] [CrossRef]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.; Brinkman, F.S.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef] [PubMed]
- Zammit, N.W.; Walters, S.N.; Seeberger, K.L.; O’Connell, P.J.; Korbutt, G.S.; Grey, S.T. A20 as an immune tolerance factor can determine islet transplant outcomes. JCI Insight 2019, 4, e131028. [Google Scholar] [CrossRef] [PubMed]
- Mondini, M.; Loyher, P.L.; Hamon, P.; Gerbé de Thoré, M.; Laviron, M.; Berthelot, K.; Clémenson, C.; Salomon, B.L.; Combadière, C.; Deutsch, E.; et al. CCR2-Dependent Recruitment of Tregs and Monocytes Following Radiotherapy Is Associated with TNFα-Mediated Resistance. Cancer Immunol. Res. 2019, 7, 376–387. [Google Scholar] [CrossRef]
- Comerford, I.; Bunting, M.; Fenix, K.; Haylock-Jacobs, S.; Litchfield, W.; Harata-Lee, Y.; Turvey, M.; Brazzatti, J.; Gregor, C.; Nguyen, P.; et al. An immune paradox: How can the same chemokine axis regulate both immune tolerance and activation?: CCR6/CCL20: A chemokine axis balancing immunological tolerance and inflammation in autoimmune disease. Bioessays 2010, 32, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Lin, J.; Xu, A.; Lou, J.; Qian, C.; Li, X.; Wang, Y.; Yu, W.; Tao, H. CCL2: An Important Mediator Between Tumor Cells and Host Cells in Tumor Microenvironment. Front. Oncol. 2021, 11, 722916. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Wright, K.; Mital, P.; Hibler, T.; Miranda, J.M.; Thompson, L.A.; Halley, K.; Dufour, J.M. Neonatal Pig Sertoli Cells Survive Xenotransplantation by Creating an Immune Modulatory Environment Involving CD4 and CD8 Regulatory T Cells. Cell Transplant. 2020, 29, 963689720947102. [Google Scholar] [CrossRef]
- Brown, G.R.; Hem, V.; Katz, K.S.; Ovetsky, M.; Wallin, C.; Ermolaeva, O.; Tolstoy, I.; Tatusova, T.; Pruitt, K.D.; Maglott, D.R.; et al. Gene: A gene-centered information resource at NCBI. Nucleic Acids Res. 2015, 43, D36–D42. [Google Scholar] [CrossRef]
- Ortutay, C.; Siermala, M.; Vihinen, M. Molecular characterization of the immune system: Emergence of proteins, processes, and domains. Immunogenetics 2007, 59, 333–348. [Google Scholar] [CrossRef]
- Wills-Karp, M. Complement activation pathways: A bridge between innate and adaptive immune responses in asthma. Proc. Am. Thorac. Soc. 2007, 4, 247–251. [Google Scholar] [CrossRef]
- Jang, J.H.; Shin, H.W.; Lee, J.M.; Lee, H.W.; Kim, E.C.; Park, S.H. An Overview of Pathogen Recognition Receptors for Innate Immunity in Dental Pulp. Mediat. Inflamm. 2015, 2015, 794143. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Washburn, R.L.; Hibler, T.; Kaur, G.; Dufour, J.M. Sertoli Cell Immune Regulation: A Double-Edged Sword. Front. Immunol. 2022, 13, 913502. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Yeretssian, G. NOD-Like Receptors: Master Regulators of Inflammation and Cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Fischereder, M. Chemokines and chemokine receptors in renal transplantation—From bench to bedside. Acta Physiol. Hung. 2007, 94, 67–81. [Google Scholar] [CrossRef]
- Campbell, D.J.; Kim, C.H.; Butcher, E.C. Chemokines in the systemic organization of immunity. Immunol. Rev. 2003, 195, 58–71. [Google Scholar] [CrossRef]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontology 2000 2015, 69, 142–159. [Google Scholar] [CrossRef]
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 cells in human disease. Immunol. Rev. 2008, 223, 87–113. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Gadina, M.; Siegel, R.M. 9—Cytokines and Cytokine Receptors. In Clinical Immunology, 5th ed.; Rich, R.R., Fleisher, T.A., Shearer, W.T., Schroeder, H.W., Frew, A.J., Weyand, C.M., Eds.; Elsevier: London, UK, 2019; pp. 127–155.e1. [Google Scholar]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Ang, R.L.; Ting, A.T. Tumor necrosis factor-driven cell death in donor organ as a barrier to immunological tolerance. Curr. Opin. Organ Transplant. 2019, 24, 12–19. [Google Scholar] [CrossRef]
- Sharp, L.L.; Schwarz, D.A.; Bott, C.M.; Marshall, C.J.; Hedrick, S.M. The Influence of the MAPK Pathway on T Cell Lineage Commitment. Immunity 1997, 7, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.S.; Plain, K.M.; Bishop, G.A. Role of IL-4 and Th2 responses in allograft rejection and tolerance. Curr. Opin. Organ Transplant. 2009, 14, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Illigens, B.M.; Yamada, A.; Anosova, N.; Dong, V.M.; Sayegh, M.H.; Benichou, G. Dual effects of the alloresponse by Th1 and Th2 cells on acute and chronic rejection of allotransplants. Eur. J. Immunol. 2009, 39, 3000–3009. [Google Scholar] [CrossRef]
- Lin, Y.; Vandeputte, M.; Waer, M. Accommodation and T-independent B cell tolerance in rats with long term surviving hamster heart xenografts. J. Immunol. 1998, 160, 369–375. [Google Scholar] [CrossRef]
- Heidt, S.; Segundo, D.S.; Chadha, R.; Wood, K.J. The impact of Th17 cells on transplant rejection and the induction of tolerance. Curr. Opin. Organ Transplant. 2010, 15, 456–461. [Google Scholar] [CrossRef]
- Hanidziar, D.; Koulmanda, M. Inflammation and the balance of Treg and Th17 cells in transplant rejection and tolerance. Curr. Opin. Organ Transplant. 2010, 15, 411–415. [Google Scholar] [CrossRef]
- Maturu, P.; Jones, D.; Ruteshouser, E.C.; Hu, Q.; Reynolds, J.M.; Hicks, J.; Putluri, N.; Ekmekcioglu, S.; Grimm, E.A.; Dong, C.; et al. Role of Cyclooxygenase-2 Pathway in Creating an Immunosuppressive Microenvironment and in Initiation and Progression of Wilms’ Tumor. Neoplasia 2017, 19, 237–249. [Google Scholar] [CrossRef]
- Wang, D.; Yang, L.; Yue, D.; Cao, L.; Li, L.; Wang, D.; Ping, Y.; Shen, Z.; Zheng, Y.; Wang, L.; et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019, 452, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Yang, X.; Du, Y.; Wang, G.; Chan, D.W.; Wu, D.; Xu, P.; Ni, P.; Xu, D.; Hu, Y. CXCL8 Associated Dendritic Cell Activation Marker Expression and Recruitment as Indicators of Favorable Outcomes in Colorectal Cancer. Front. Immunol. 2021, 12, 667177. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Himmel, M.E.; Crome, S.Q.; Ivison, S.; Piccirillo, C.; Steiner, T.S.; Levings, M.K. Human CD4+ FOXP3+ regulatory T cells produce CXCL8 and recruit neutrophils. Eur. J. Immunol. 2011, 41, 306–312. [Google Scholar] [CrossRef]
- Verhasselt, V.; Goldman, M.; Willems, F. Oxidative stress up-regulates IL-8 and TNF-alpha synthesis by human dendritic cells. Eur. J. Immunol. 1998, 28, 3886–3890. [Google Scholar] [CrossRef]
- Ren, G.; Zhao, X.; Zhang, L.; Zhang, J.; L’Huillier, A.; Ling, W.; Roberts, A.I.; Le, A.D.; Shi, S.; Shao, C.; et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef]
- Kramer, K.; Thye, T.; Treszl, A.; Peine, S.; Koch, M.; Sterneck, M.; Nashan, B.; Thude, H. Polymorphism in NFKBIA gene is associated with recurrent acute rejections in liver transplant recipients. Tissue Antigens 2014, 84, 370–377. [Google Scholar] [CrossRef]
- Saba, N.F.; Choi, M.; Muller, S.; Shin, H.J.; Tighiouart, M.; Papadimitrakopoulou, V.A.; El-Naggar, A.K.; Khuri, F.R.; Chen, Z.G.; Shin, D.M. Role of cyclooxygenase-2 in tumor progression and survival of head and neck squamous cell carcinoma. Cancer Prev. Res. 2009, 2, 823–829. [Google Scholar] [CrossRef]
- Mital, P.; Kaur, G.; Dufour, J.M. Immunoprotective sertoli cells: Making allogeneic and xenogeneic transplantation feasible. Reproduction 2010, 139, 495–504. [Google Scholar] [CrossRef]
- Kaur, G. Primary Sertoli cells survive allotransplantation by modifying the cellular-immune response and promoting T regulatory cells at graft site. In Immune-Privileged Sertoli Cells Survive Allotransplantation by Inhibiting Adaptive Immune Response; Texas Tech University Libraries: Lubbock, TX, USA, 2012; pp. 145–188. [Google Scholar]
- Cao, Q.; Wang, Y.; Zheng, D.; Sun, Y.; Wang, Y.; Lee, V.W.; Zheng, G.; Tan, T.K.; Ince, J.; Alexander, S.I.; et al. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J. Am. Soc. Nephrol. 2010, 21, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | NPSC Ensembl Number | Human GeneID | LFC | p-Adj |
|---|---|---|---|---|
| CCL20 | ENSSSCG00000016254 | 6364 | 3.613 | 1.00 × 10−6 |
| PTGS2 | ENSSSCG00000015579 | 5743 | 3.529 | 1.00 × 10−6 |
| KERA | ENSSSCG00000000917 | 11081 | 3.371 | 1.61 × 10−4 |
| NR4A3 | ENSSSCG00000005385 | 8013 | 2.967 | 1.00 × 10−6 |
| ssc-mir-212 * | ENSSSCG00000019896 | N/A | 2.416 | 2.17 × 10−3 |
| ssc-mir-221 * | ENSSSCG00000018698 | N/A | 2.412 | 5.41 × 10−3 |
| BTG2 * | ENSSSCG00000028322 | 7832 | 2.278 | 2.41 × 10−13 |
| F2Z5K9 * | ENSSSCG00000026982 | N/A | 2.237 | 5.42 × 10−3 |
| EGR3 | ENSSSCG00000009630 | 1960 | 2.048 | 1.00 × 10−6 |
| EGR1 | ENSSSCG00000014336 | 1958 | 2.018 | 1.00 × 10−6 |
| SELE | ENSSSCG00000006286 | 6401 | 1.986 | 3.46 × 10−6 |
| SORCS3 | ENSSSCG00000010617 | 22986 | 1.869 | 1.50 × 10−2 |
| CXCL2 * | ENSSSCG00000008959 | 2920 | 1.782 | 6.32 × 10−7 |
| C6 | ENSSSCG00000016861 | 729 | 1.759 | 3.20 × 10−2 |
| BDKRB1 * | ENSSSCG00000002501 | 623 | 1.666 | 1.30 × 10−3 |
| CSF3 * | ENSSSCG00000017482 | 2438 | 1.664 | 2.43 × 10−2 |
| FOXR1 | ENSSSCG00000015101 | 283150 | 1.599 | 9.00 × 10−3 |
| MSC | ENSSSCG00000006187 | 9242 | 1.548 | 1.00 × 10−6 |
| ATG10 * | ENSSSCG00000029605 | 83734 | 1.538 | 6.25 × 10−11 |
| IER3 | ENSSSCG00000027607 | 8870 | 1.499 | 1.62 × 10−5 |
| LMCD1 | ENSSSCG00000011538 | 29995 | 1.498 | 2.02 × 10−6 |
| JUNB | ENSSSCG00000013735 | 3726 | 1.424 | 1.00 × 10−6 |
| MYOC | ENSSSCG00000015481 | 4653 | 1.405 | 4.68 × 10−2 |
| BHLHE40 * | ENSSSCG00000011534 | 8553 | 1.399 | 5.44 × 10−4 |
| NFKBIZ | ENSSSCG00000011951 | 64332 | 1.355 | 4.00 × 10−3 |
| MAP3K8 | ENSSSCG00000020705 | 1326 | 1.334 | 2.18 × 10−4 |
| AK7 | ENSSSCG00000002504 | 122481 | 1.328 | 3.80 × 10−2 |
| TGM7 | ENSSSCG00000004712 | 116179 | 1.325 | 4.00 × 10−2 |
| NFKBIA | ENSSSCG00000001952 | 4792 | 1.323 | 1.00 × 10−3 |
| GEM | ENSSSCG00000006105 | 2669 | 1.293 | 2.00 × 10−3 |
| GPC3 | ENSSSCG00000012680 | 2719 | 1.276 | 2.10 × 10−2 |
| NFKB1 | ENSSSCG00000030957 | 4790 | 1.271 | 2.40 × 10−2 |
| SUSD4 | ENSSSCG00000011176 | 55061 | 1.265 | 2.20 × 10−2 |
| ADAM7 | ENSSSCG00000009646 | 8756 | 1.251 | 1.20 × 10−2 |
| VEGFA | ENSSSCG00000001695 | 7422 | 1.249 | 1.62 × 10−5 |
| VCAM1 | ENSSSCG00000006862 | 7412 | 1.240 | 2.00 × 10−3 |
| SHAS2 | ENSSSCG00000005992 | 3037 | 1.215 | 8.00 × 10−3 |
| SNARK * | ENSSSCG00000027243 | 81788 | 1.206 | 2.17 × 10−3 |
| NR4A2 | ENSSSCG00000015871 | 4929 | 1.206 | 1.00 × 10−3 |
| DUSP10 | ENSSSCG00000010831 | 11221 | 1.200 | 1.69 × 10−4 |
| LOC110259249 * | ENSSSCG00000052816 | N/A | 1.152 | 4.00 × 10−3 |
| TNFAIP3 | ENSSSCG00000004154 | 7128 | 1.120 | 1.80 × 10−2 |
| SLCO5A1 | ENSSSCG00000006196 | 81796 | 1.109 | 8.00 × 10−3 |
| LIF * | ENSSSCG00000009996 | 3976 | 1.105 | 1.26 × 10−3 |
| KDM6B | ENSSSCG00000017962 | 23135 | 1.100 | 8.00 × 10−3 |
| SERPINB2 | ENSSSCG00000004890 | 5055 | 1.088 | 3.70 × 10−2 |
| F3 | ENSSSCG00000022447 | 2152 | 1.053 | 5.00 × 10−3 |
| GADD45G * | ENSSSCG00000009585 | N/A | 1.048 | 8.80 × 10−3 |
| NFATC2 | ENSSSCG00000007477 | 4773 | 1.027 | 4.00 × 10−2 |
| PER1 ** | ENSSSCG00000017983 | 5187 | 0.979 | 8.00 × 10−3 |
| CXCL8 ** | ENSSSCG00000008953 | 3576 | 0.97 | 4.60 × 10−2 |
| CCL2 ** | ENSSSCG00000017723 | 6347 | 0.946 | 2.00 × 10−3 |
| DUSP1 ** | ENSSSCG00000016991 | 1843 | 0.856 | 3.70 × 10−2 |
| MAP3K21 ** | ENSSSCG00000010164 | 84451 | −0.724 | 4.70 × 10−2 |
| PSAP ** | ENSSSCG00000010281 | 5660 | −0.795 | 4.80 × 10−2 |
| GATA6 ** | ENSSSCG00000003702 | 2627 | −0.805 | 4.00 × 10−2 |
| TMEM245 ** | ENSSSCG00000005444 | 23731 | −0.861 | 2.20 × 10−2 |
| OXTR ** | ENSSSCG00000021585 | 5021 | −0.864 | 2.80 × 10−2 |
| IER5L | ENSSSCG00000005673 | 389792 | −1.272 | 3.46 × 10−6 |
| CYP1A1 | ENSSSCG00000001906 | 1543 | −1.394 | 4.50 × 10−5 |
| CCL8 ** | ENSSSCG00000017721 | N6355A | −2.257 | 0.026123 |
| LYZ | ENSSSCG00000000492 | 4069 | −2.312 | 3.46 × 10−6 |
| Gene | Name | Immune Function |
|---|---|---|
| ATG10 | Autophagy-related 10 | Autophagosome formation |
| BDKRB1 | Bradykinin receptor B1 | Induces inflammatory responses |
| BTG2 | B cell translocation gene 2 | Antiproliferative functions |
| C6 ** | Complement C6 | Formation of MAC |
| CCL2 ** | C-C motif ligand 2 | Induces monocyte trafficking |
| CCL20 ** | C-C motif ligand 20 | Antimicrobial cytokine |
| CCL8 | C-C motif ligand 8 | Antimicrobial cytokine that recruits leukocytes to inflammatory site |
| CSF3 | Colony stimulating factor 3 | Regulates granulocyte production, function, and differentiation |
| CXCL2 | C-X-C motif ligand 2 | Recruitment of neutrophils |
| CXCL8 ** | C-X-C motif ligand 8 | Angiogenesis regulation, infiltration of leukocytes, cell motility and survival |
| DUSP1 | Dual specificity phosphatase 1 | Decreases TLR signaling and inhibits MAPK induction of IL-6 and IL-8 |
| EGR1 ** | Early growth response 1 | Cell differentiation and mitogenesis |
| EGR3 | Early growth response 3 | Lymphocyte development |
| F3 ** | Coagulation factor III (thromboplastin) | Initiates blood coagulation cascades |
| IER3 | Immediate early response 3 | Protects against Fas or TNF-α apoptosis |
| LIF | Leukemia inhibitory factor | Differentiation of myeloid cells, immune tolerance |
| LYZ ** | Lysozyme | Antimicrobial, cleavage of peptidoglycans in bacterial cell walls |
| MAP3K8 | Mitogen-activated protein kinase kinase kinase 8 | Regulates IL-1β and TNF production in macrophages and dendritic cells |
| MSC | Musculin | B cell receptor signaling target |
| NFATC2 | Nuclear factor of activated T cells, cytoplasmic, calcineurin-dependent 2 | Activates IL-2 transcription through interaction with JUN in T cells |
| NFKB1 ** | Nuclear factor of kappa light polypeptide gene enhancer in B cells 1 | Regulation of immune and inflammatory response genes; regulation of cell growth |
| NFKBIA ** | Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha | Inhibition of NFκB |
| NFKBIZ ** | Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor zeta | Inhibits NFκB1 binding to DNA; regulates IL-6 production, regulates TLR and NOD-like receptor inflammation |
| NR4A3 | Nuclear receptor subfamily 4, A3 | Induces repressive effects in dendritic cells |
| PTGS2 ** | Prostaglandin-endoperoxidase synthase 2 | Prostaglandin biosynthesis important in inflammation and mitogenesis |
| SELE | Selectin E | Accumulates leukocytes to inflammatory cites; allows for adhesion of leukocytes to endothelium |
| SUSD4 | Sushi domain containing 4 | Membrane-bound inhibition of alternative and classical complement activation |
| TNFAIP3 ** | Tumor necrosis factor, alpha-induced protein 3 (A20) | Restricts TLR, NOD, and RIG-I signaling; negatively regulates NF-κB signaling pathway |
| VCAM1 ** | Vascular cell adhesion molecule 1 | Mediates leukocyte adhesion to endothelium |
| Pathway Name | #DEG/TG | p-Value | Genes |
|---|---|---|---|
| TNF signaling * | 10/77 | 0.00001 | ↑CCL20↑PTGS2 ↑SELE ↑JUNB ↑MAP3K8↑NFKBIA↑NFKB1↑VCAM1↑TNFAIP3↑CCL2 |
| AGE-RAGE signaling in diabetes | 8/73 | 0.000001 | ↑EGR1 ↑SELE↑NFKB1 ↑VEGFA ↑VCAM1 ↑F3↑CXCL8↑CCL2 |
| IL-17 signaling * | 7/55 | 0.00001 | ↑CCL20↑PTGS2↑NFKBIA↑NFKB1↑TNFAIP3↑CXCL8↑CCL2 |
| NF-κB signaling * | 6/69 | 0.000003 | ↑PTGS2↑NFKBIA↑NFKB1 ↑VCAM1↑TNFAIP3↑CXCL8 |
| Fluid shear stress and atherosclerosis | 6/90 | 0.000005 | ↑SELE↑NFKB1↑VEGFA↑VCAM1↑CCL2↑DUSP1 |
| C-type lectin receptor signaling * | 5/68 | 0.00002 | ↑PTGS2 ↑EGR3↑NFKBIA↑NFKB1 ↑NFATC2 |
| Chemokine signaling * | 5/102 | 0.00005 | ↑CCL20↑NFKBIA↑NFKB1↑CXCL8↑CCL2 |
| NOD-like receptor signaling * | 5/90 | 0.0003 | ↑NFKBIA↑NFKB1↑TNFAIP3↑CXCL8↑CCL2 |
| MAPK signaling pathway * | 5/196 | 0.01 | ↑MAP3KIB↑NFKB1 ↑VEGFA ↑DUSP10 ↑DUSP1 |
| Pathways in cancer | 5/339 | 0.014 | ↑PTGS2↑NFKBIA↑NFKB1 ↑VEGFA↑CXCL8 |
| TLR signaling * | 4/57 | 0.0005 | ↑MAP3KIB↑NFKBIA↑NFKB1↑CXCL8 |
| Osteoclast differentiation | 4/72 | 0.001 | ↑JUNB↑NFKBIA↑NFKB1 ↑NFATC2 |
| T cell receptor signaling * | 4/67 | 0.001 | ↑MAP3K8↑NFKBIA↑NFKB1 ↑NFATC2 |
| Complement and coagulation cascades * | 4/47 | 0.007 | ↑C6 ↑SERPINB2 ↑F3↑SUSD4 |
| B cell receptor signaling | 3/46 | 0.002 | ↑NFKBIA↑NFKB1 ↑NFATC2 |
| RIG-I-like receptor signaling * | 3/38 | 0.002 | ↑NFKBIA↑NFKB1↑CXCL8 |
| Cytokine-cytokine receptor interaction * | 3/125 | 0.002 | ↑CCL20↑CXCL8↑CCL2 |
| VEGF signaling | 3/38 | 0.004 | ↑PTGS2 ↑VEGFA ↑NFATC2 |
| Cellular senescence | 3/98 | 0.005 | ↑NFKB1 ↑NFATC2↑CXCL8 |
| Th17 cell differentiation * | 3/72 | 0.006 | ↑NFKBIA↑NFKB1 ↑NFATC2 |
| Th1 and Th2 cell differentiation * | 3/59 | 0.007 | ↑NFKBIA↑NFKB1 ↑NFATC2 |
| Relaxin signaling | 3/87 | 0.014 | ↑NFKBIA↑NFKB1 ↑VEGFA |
| Oxytocin signaling | 3/101 | 0.038 | ↑PTGS2 ↑NFATC2 ↓OXTR |
| Seratonergic synapse | 2/61 | 0.015 | ↑PTGS2 ↑DUSP1 |
| Neurotrophin signaling | 2/80 | 0.031 | ↑NFKBIA↑NFKB1 |
| Parathyroid hormone synthesis, secretion, and action | 2/78 | 0.035 | ↑EGR1 ↑NR4A2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Washburn, R.L.; Martinez-Marin, D.; Sniegowski, T.; Korać, K.; Rodriguez, A.R.; Miranda, J.M.; Chilton, B.S.; Bright, R.K.; Pruitt, K.; Bhutia, Y.D.; et al. Sertoli Cells Express Accommodation, Survival, and Immunoregulatory Factors When Exposed to Normal Human Serum. Biomedicines 2023, 11, 1650. https://doi.org/10.3390/biomedicines11061650
Washburn RL, Martinez-Marin D, Sniegowski T, Korać K, Rodriguez AR, Miranda JM, Chilton BS, Bright RK, Pruitt K, Bhutia YD, et al. Sertoli Cells Express Accommodation, Survival, and Immunoregulatory Factors When Exposed to Normal Human Serum. Biomedicines. 2023; 11(6):1650. https://doi.org/10.3390/biomedicines11061650
Chicago/Turabian StyleWashburn, Rachel L., Dalia Martinez-Marin, Tyler Sniegowski, Ksenija Korać, Alexis R. Rodriguez, Jonathan M. Miranda, Beverly S. Chilton, Robert K. Bright, Kevin Pruitt, Yangzom D. Bhutia, and et al. 2023. "Sertoli Cells Express Accommodation, Survival, and Immunoregulatory Factors When Exposed to Normal Human Serum" Biomedicines 11, no. 6: 1650. https://doi.org/10.3390/biomedicines11061650
APA StyleWashburn, R. L., Martinez-Marin, D., Sniegowski, T., Korać, K., Rodriguez, A. R., Miranda, J. M., Chilton, B. S., Bright, R. K., Pruitt, K., Bhutia, Y. D., & Dufour, J. M. (2023). Sertoli Cells Express Accommodation, Survival, and Immunoregulatory Factors When Exposed to Normal Human Serum. Biomedicines, 11(6), 1650. https://doi.org/10.3390/biomedicines11061650