

Pharmacokinetics of Biopharmaceuticals: Their Critical Role in Molecular Design

Abstract
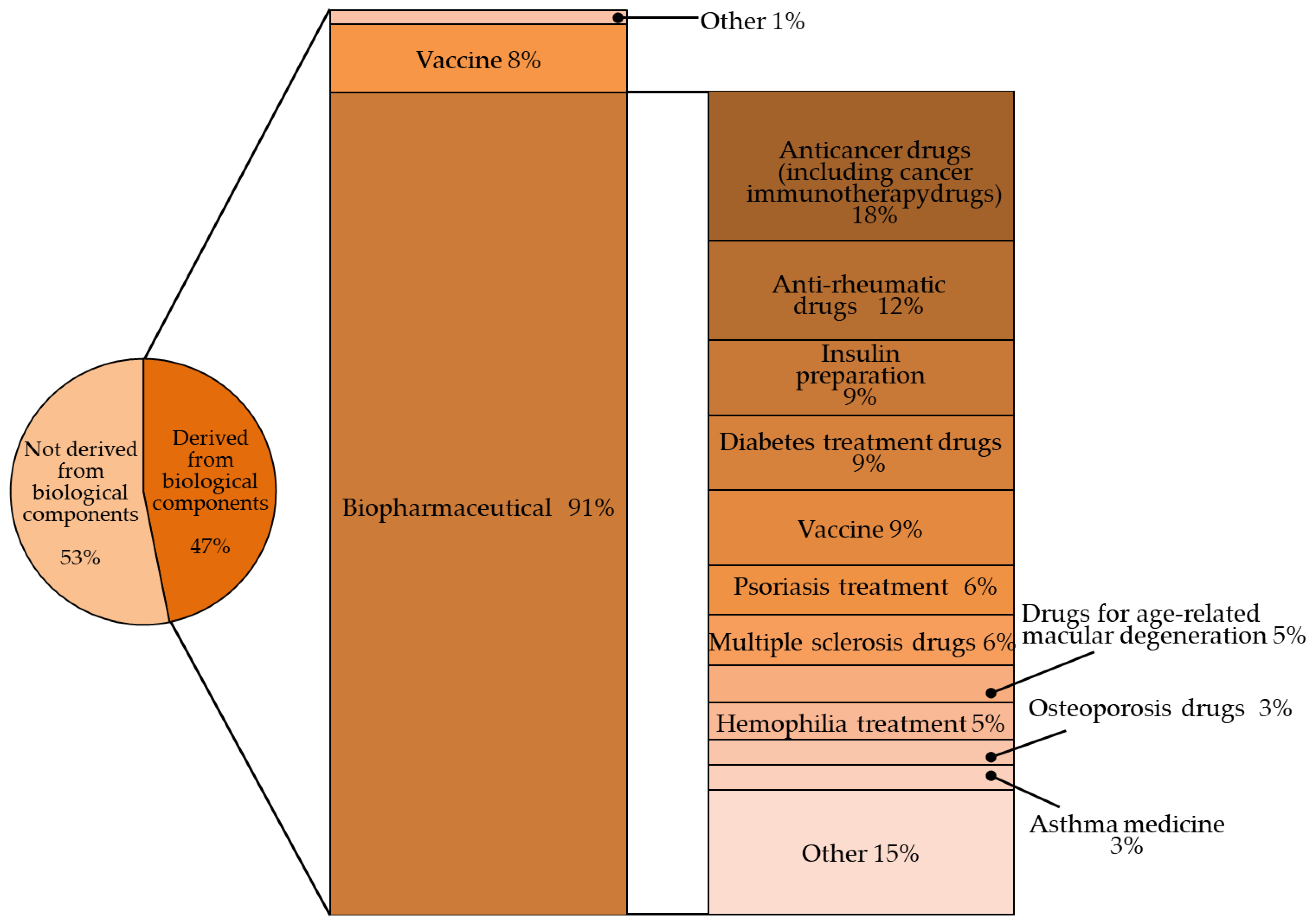
1. Introduction
2. Kinetics of Pharmaceuticals Derived from Biological Components
2.1. Absorption
2.2. Distribution
2.3. Metabolism
2.4. Elimination
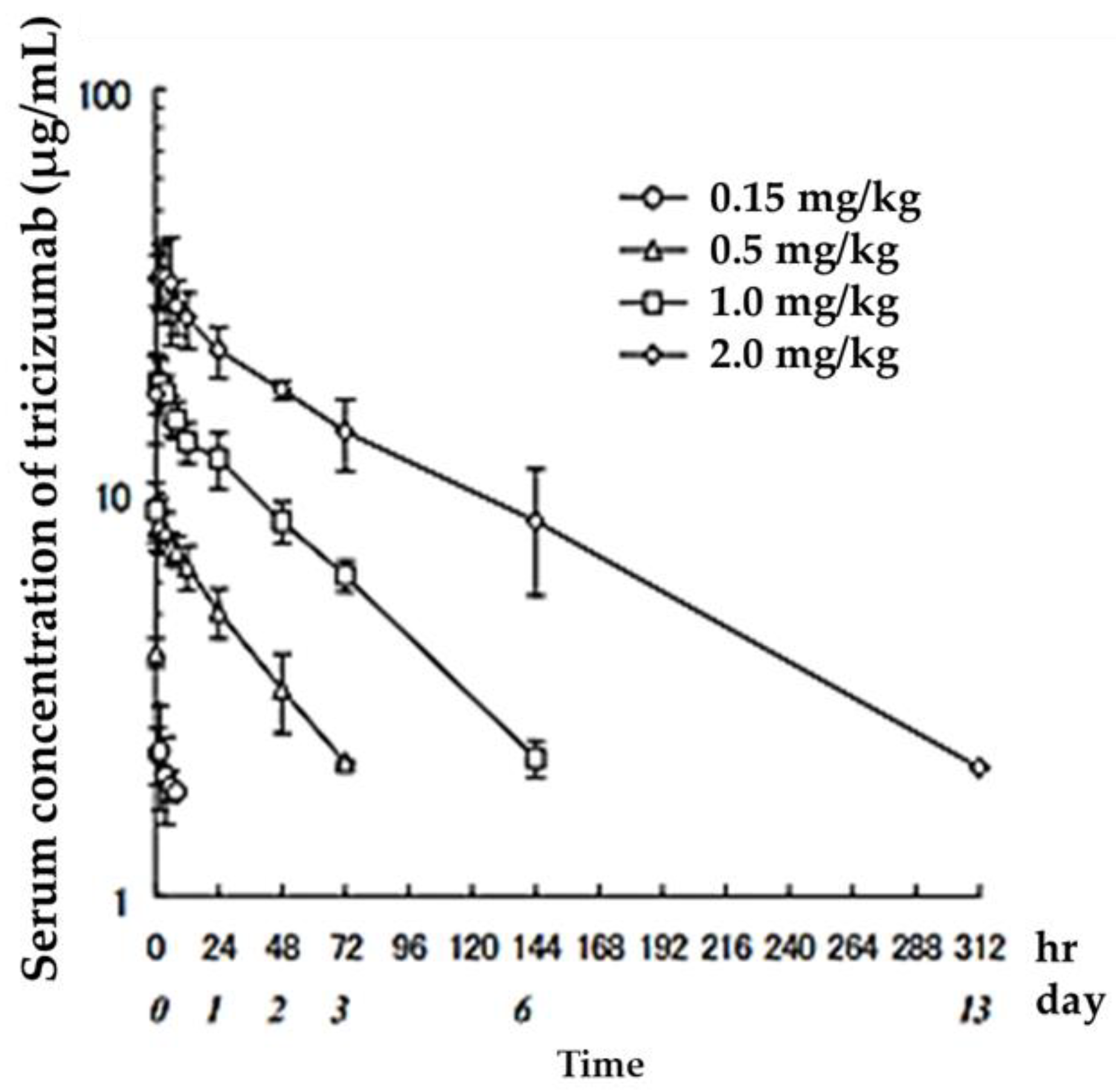
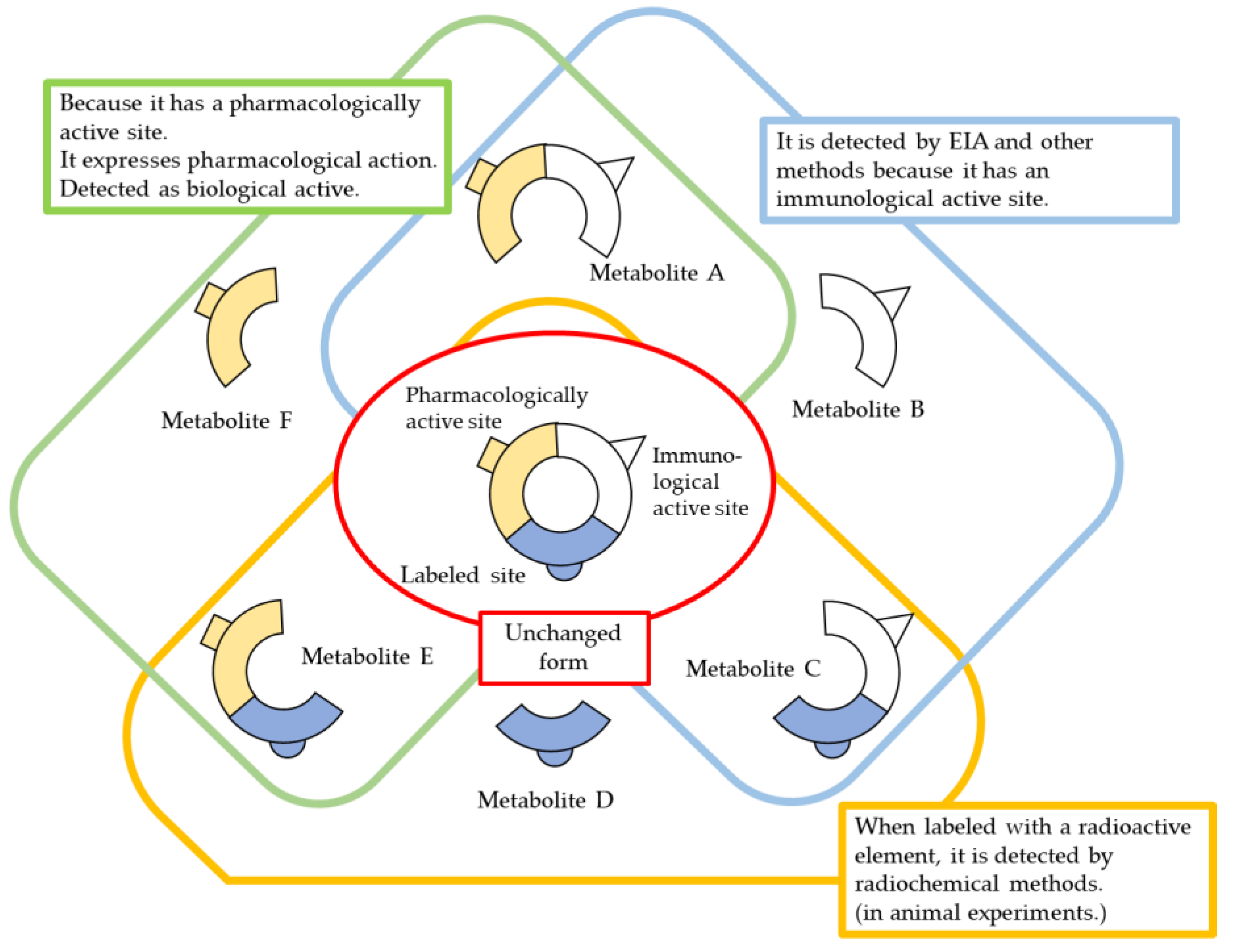
3. Drug Quantification and Evaluation of Blood Concentration
4. Kinetic Innovation of Biopharmaceuticals
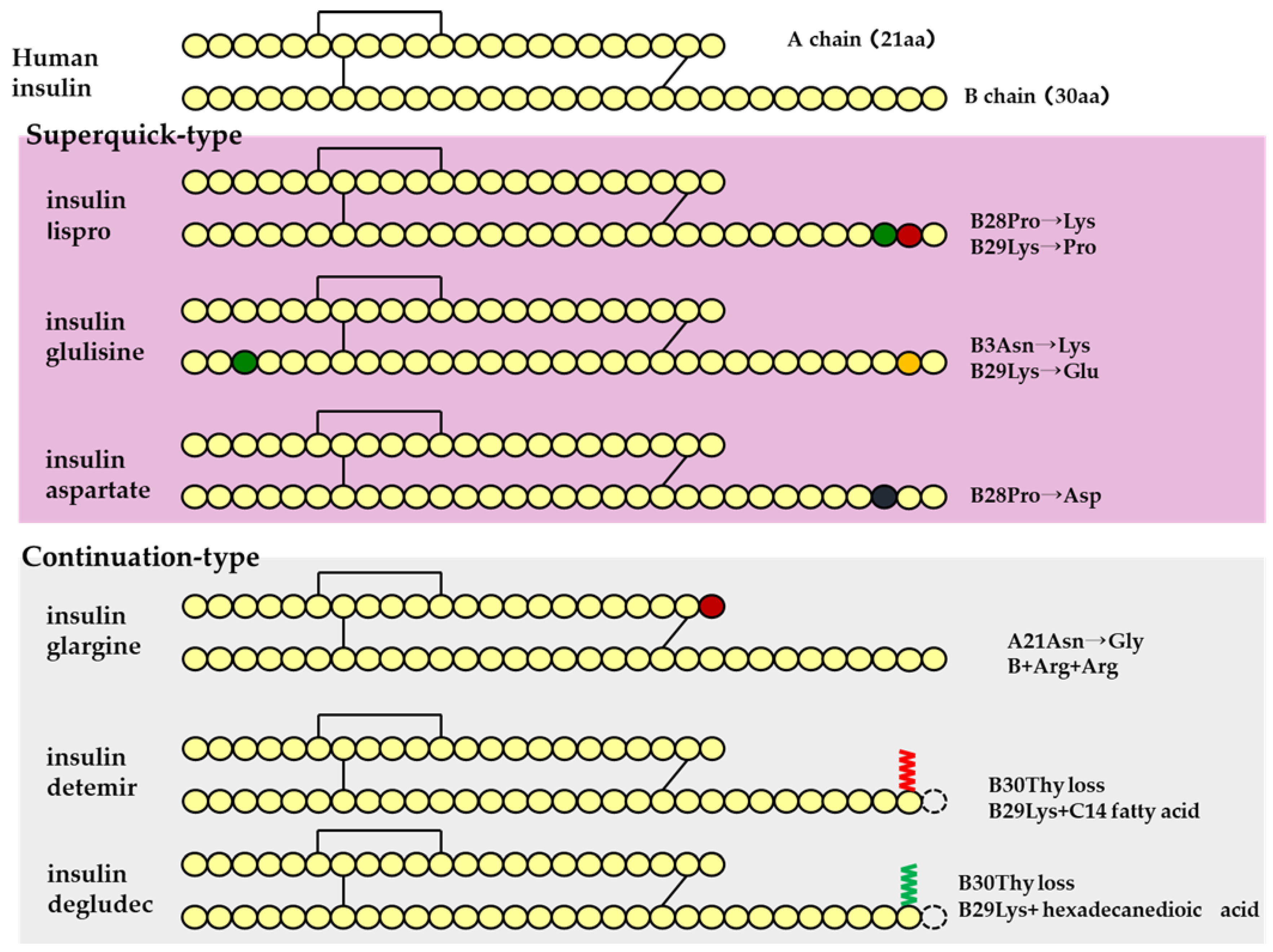
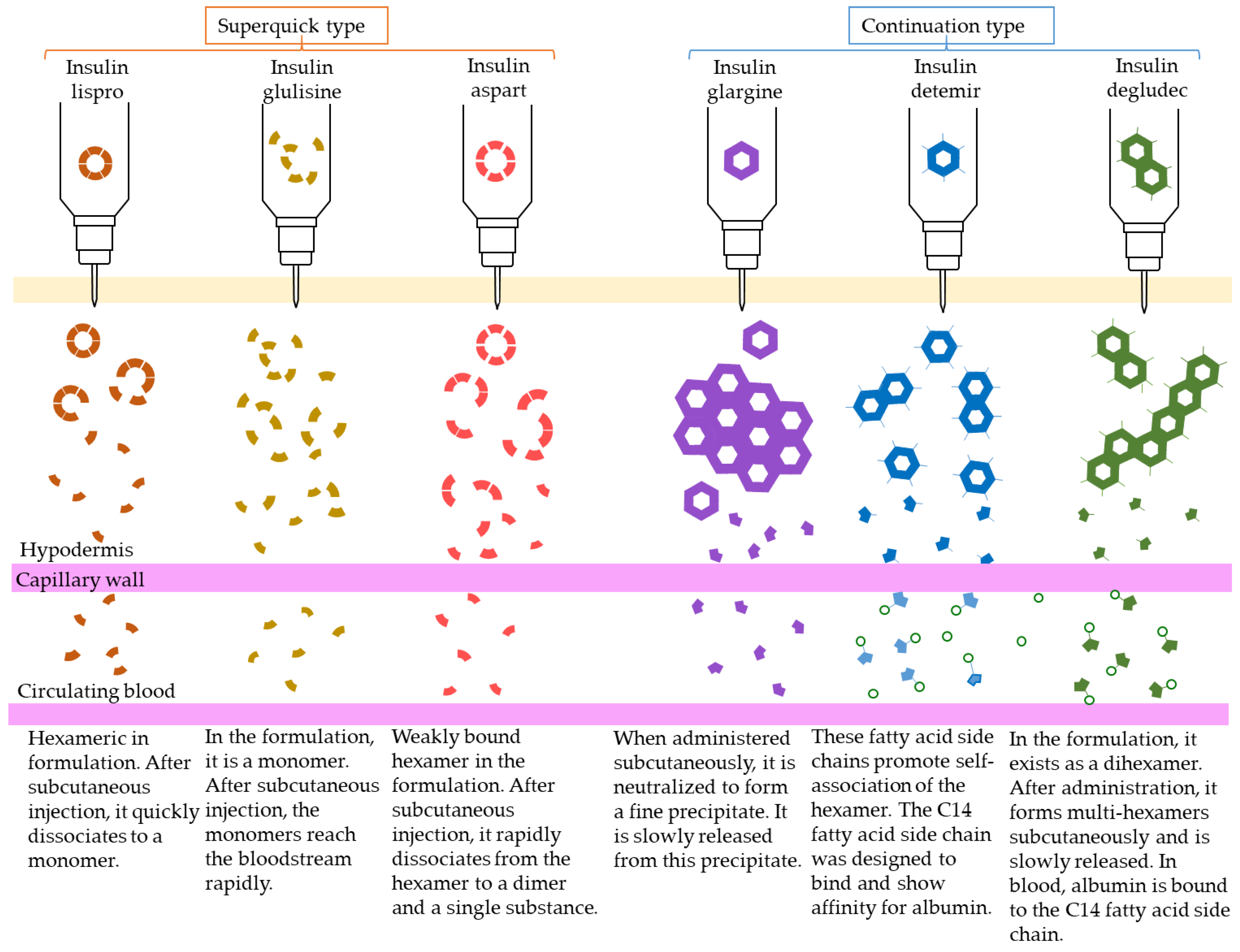
4.1. Insulin
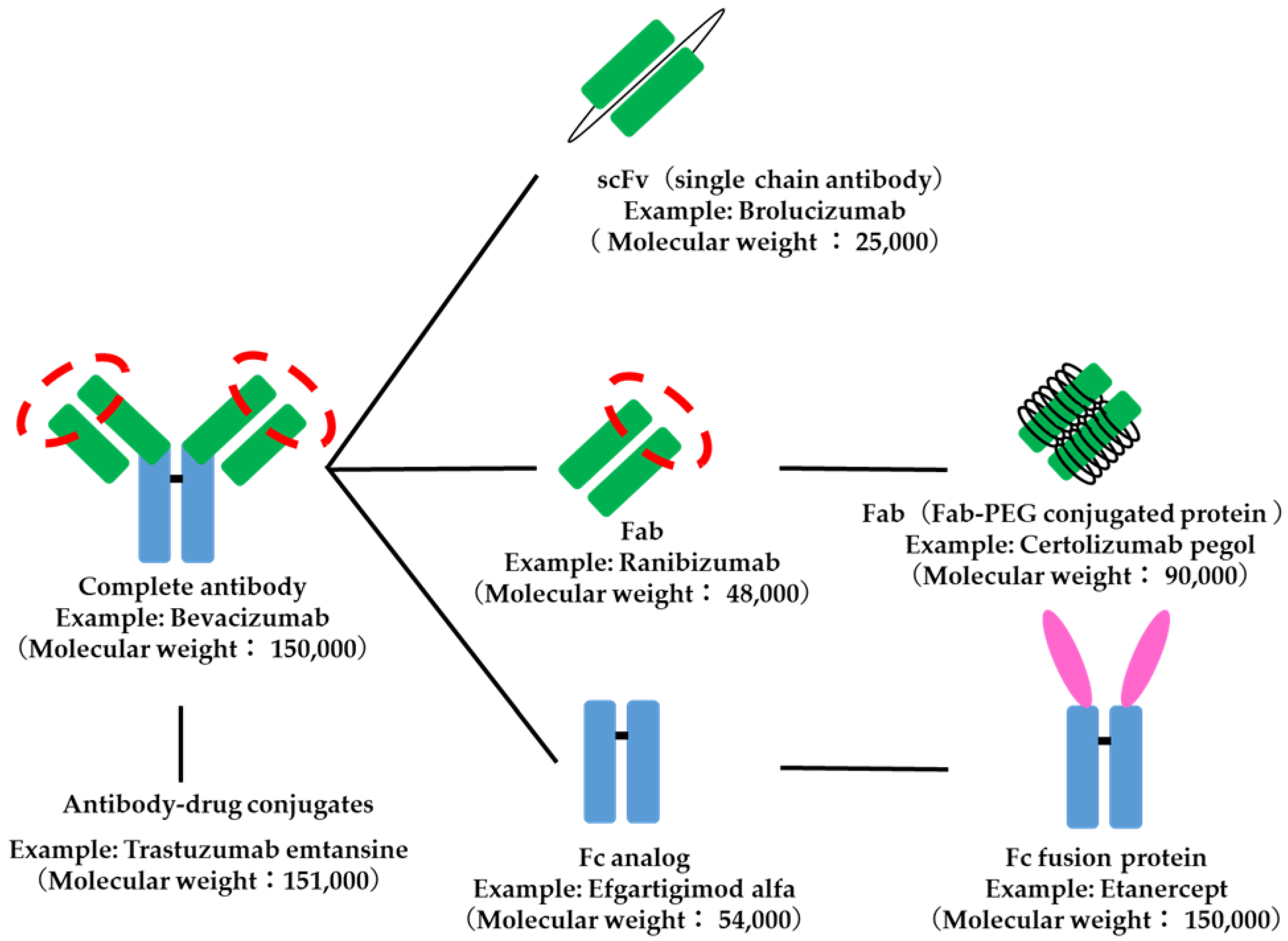
4.2. Antibody-Related Drugs
4.3. Blood Coagulation Factors
4.4. Interferon
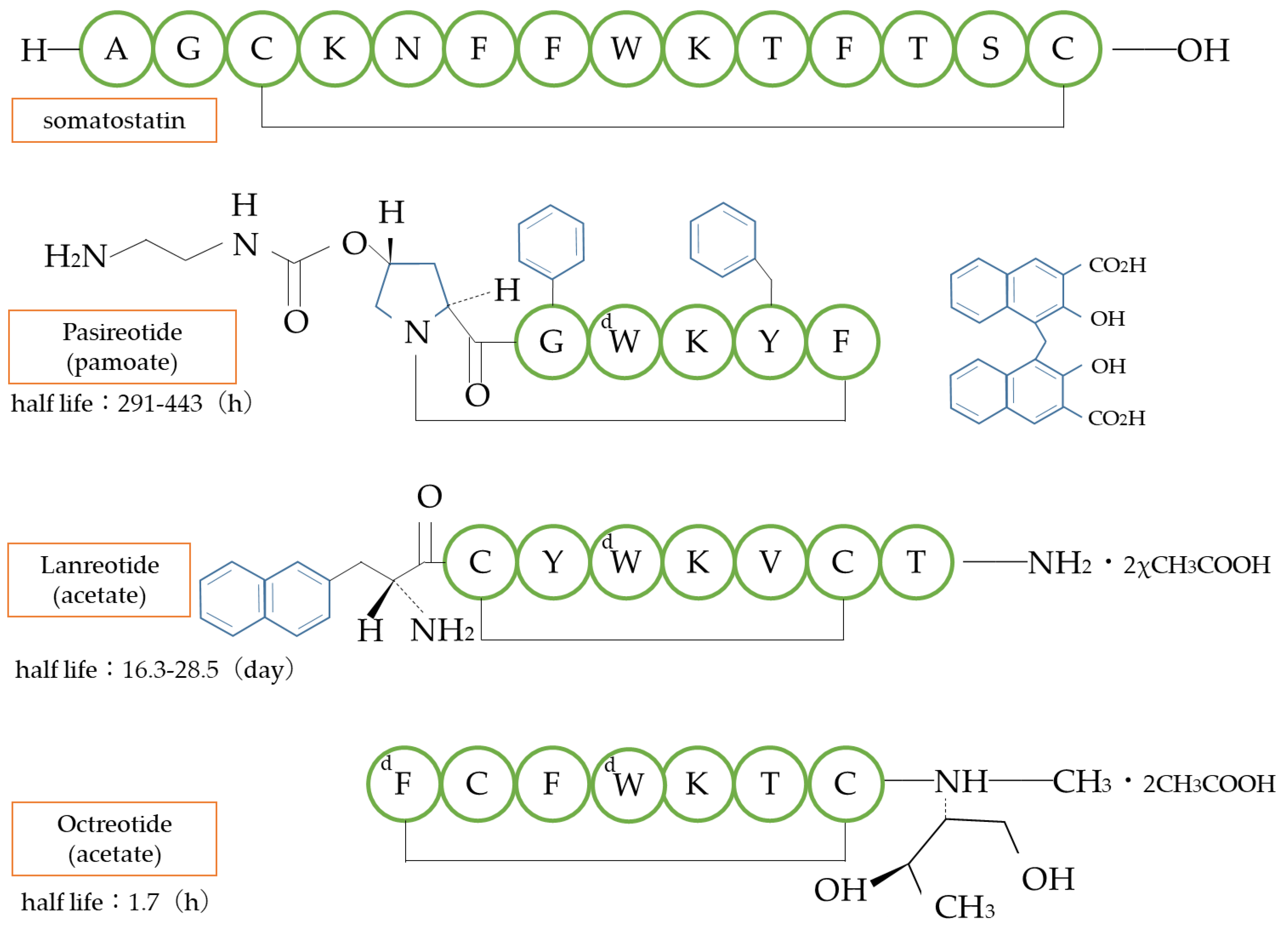
4.5. Other Examples
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chapter 2 Pharmaceuticals Ranking: Global Blockbusters in 2019 Top Global Sales Products (over $1 Billion in Sales). In Monthly Mix, Additional Issue; MIX, Inc.: Tokyo, Japan, 2019; pp. 54–56. Available online: https://www.mixonline.jp/tabid55.html?artid=69890 (accessed on 19 March 2023).
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. MABS 2020, 12, 1703531. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MABS 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Kagan, L.; Gershkovich, P.; Mendelman, A.; Amsili, S.; Ezov, N.; Hoffman, A. The role of the lymphatic system in subcutaneous absorption of macromolecules in the rat model. Eur. J. Pharm. Biopharm. 2007, 67, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, N.; Shen, X.; Cunningham, P.; Fauty, S.; Michel, K.; Wang, B.; Hong, X.; Adreani, C.; Nunes, C.N.; et al. Lymphatic transport and catabolism of therapeutic proteins after subcutaneous administration to rats and dogs. Drug Metab. Dispos. 2012, 40, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Misaizu, T.; Shinkai, H.; Tanaka, H.; Tanimoto, M.; Takahashi, H.; Kikuchi, K.; Takiwa, T. Metabolic fate of KRN8601: Plasma level, distribution, metabolism and excretion of I-KRN8601 after a single intravenous administration to rats. Drug Metab. Pharmacokinet. 1990, 5, 283–305. [Google Scholar] [CrossRef]
- Managuli, R.S.; Raut, S.Y.; Reddy, M.S.; Mutalik, S. Targeting the intestinal lymphatic system: A versatile path for enhanced oral bioavailability of drugs. Expert Opin. Drug Deliv. 2018, 15, 787–804. [Google Scholar] [CrossRef]
- Griffin, C.T.; Gao, S. Building discontinuous liver sinusoidal vessels. J. Clin. Investig. 2017, 127, 790–792. [Google Scholar] [CrossRef]
- Takeuchi, M.; Suzuki, T.; Shiba, T.; Urita, Y.; Murakuni, H. Experience with MR-20 in Pancreatitis and Blood Concentrations in Normal Subjects. Jpn. J. Clin. Exp. Med. 1985, 62, 626–630. [Google Scholar]
- Behr, T.M.; Goldenberg, D.M.; Becker, W. Reducing the renal uptake of radiolabeled antibody fragments and peptides for diagnosis and therapy: Present status, future prospects and limitations. Eur. J. Nucl. Med. 1998, 25, 201–212. [Google Scholar] [CrossRef]
- Ueno, T.; Takeuchi, S.; Takahashi, M.; Arai, H.; Kobayashi, N.; Maekawa, T.; Tsukada, H. Studies on Metabolism and Mechanism of Action of Urokinase (Report 2). J. Med. Enzymol. 1975, 1, 540–544. [Google Scholar]
- Ueno, T.; Takeuchi, S.; Takahashi, M.; Arai, H.; Kobayashi, N.; Maekawa, T.; Tsukada, H. Studies on Metabolism and Mechanism of Action of Urokinase (Report 3). J. Med. Enzymol. 1975, 1, 703–707. [Google Scholar]
- Tocilizumab Interview Form; Items related to pharmacokinetics; 2019.
- Ohzawa, N.; Takahashi, Y.; Tsuchiya, T.; Ogihara, T.; Nakai, Y.; Kohda, K.; Tanaka, S.; Ishiguro, J. Studies on the Metabolic Fate of Ulinastatin (1): Pharmacokinetics in Rats, Mice and Rabbits following a Single Intravenous Dose. Drug Metab. Pharmacokinet. 1990, 5, 739–753. [Google Scholar] [CrossRef]
- Simpson, D.; McCormack, P.L.; Keating, G.M.; Lyseng-Williamson, K.A. Insulin lispro: A review of its use in the management of diabetes mellitus. Drugs 2007, 67, 407–434. [Google Scholar] [CrossRef]
- Miikkulainen, K.; Caruso, A.; Mast, O.; Zhang, R.; Borisenko, O. Systematic literature review of use of blood glucose monitoring in phase III clinical studies of insulin analogs. BMC Endocr. Disord. 2016, 16, 21. [Google Scholar] [CrossRef]
- Becker, R.H.; Frick, A.D. Clinical pharmacokinetics and pharmacodynamics of insulin glulisine. Clin. Pharmacokinet. 2008, 47, 7–20. [Google Scholar] [CrossRef]
- Haahr, H.; Heise, T. Fast-Acting Insulin Aspart: A Review of its Pharmacokinetic and Pharmacodynamic Properties and the Clinical Consequences. Clin. Pharmacokinet. 2020, 59, 155–172. [Google Scholar] [CrossRef]
- Rasmussen, C.H.; Røge, R.M.; Ma, Z.; Thomsen, M.; Thorisdottir, R.L.; Chen, J.W.; Mosekilde, E.; Colding-Jørgensen, M. Insulin aspart pharmacokinetics: An assessment of its variability and underlying mechanisms. Eur. J. Pharm. Sci. 2014, 62, 65–75. [Google Scholar] [CrossRef]
- Hoy, S.M. MYL1501D Insulin Glargine: A Review in Diabetes Mellitus. BioDrugs 2020, 34, 245–251. [Google Scholar] [CrossRef]
- Kurtzhals, P.; Havelund, S.; Jonassen, I.; Kiehr, B.; Larsen, U.D.; Ribel, U.; Markussen, J. Albumin binding of insulins acylated with fatty acids: Characterization of the ligand-protein interaction and correlation between binding affinity and timing of the insulin effect in vivo. Biochem. J. 1995, 312, 725–731. [Google Scholar] [CrossRef]
- Whittingham, J.L.; Havelund, S.; Jonassen, I. Crystal structure of a prolonged-acting insulin with albumin-binding properties. Biochemistry 1997, 36, 2826–2831. [Google Scholar] [CrossRef]
- Heinemann, L.; Sinha, K.; Weyer, C.; Loftager, M.; Hirschberger, S.; Heise, T. Time-action profile of the soluble, fatty acid acylated, long-acting insulin analogue NN304. Diabet. Med. 1999, 16, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, I.; Havelund, S.; Hoeg-Jensen, T.; Steensgaard, D.B.; Wahlund, P.O.; Ribel, U. Design of the novel protraction mechanism of insulin degludec, an ultra-long-acting basal insulin. Pharm. Res. 2012, 29, 2104–2114. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Moreau, A.C.; Del Tedesco, E.; Rinaudo, M.; Phelip, J.M.; Genin, C.; Peyrin-Biroulet, L.; Roblin, X. Pharmacokinetics of adalimumab in inflammatory bowel diseases: A systematic review and meta-analysis. Inflamm. Bowel. Dis. 2014, 20, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Homšek, A.; Radosavljević, D.; Miletić, N.; Spasić, J.; Jovanović, M.; Miljković, B.; Stanojković, T.; Vučićević, K. Review of the Clinical Pharmacokinetics, Efficacy and Safety of Pembrolizumab. Curr. Drug Metab. 2022, 23, 460–472. [Google Scholar]
- Wong, A.C.; Ma, B. An update on the pharmacodynamics, pharmacokinetics, safety and clinical efficacy of nivolumab in the treatment of solid cancers. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1255–1261. [Google Scholar] [CrossRef]
- Liu, X.; Lu, Y.; Qin, S. Atezolizumab and bevacizumab for hepatocellular carcinoma: Mechanism, pharmacokinetics and future treatment strategies. Future Oncol. 2021, 17, 2243–2256. [Google Scholar] [CrossRef]
- Chisari, C.G.; Sgarlata, E.; Arena, S.; Toscano, S.; Luca, M.; Patti, F. Rituximab for the treatment of multiple sclerosis: A review. J. Neurol. 2022, 269, 159–183. [Google Scholar] [CrossRef]
- Restellini, S.; Afif, W. Update on TDM (Therapeutic Drug Monitoring) with Ustekinumab, Vedolizumab and Tofacitinib in Inflammatory Bowel Disease. J. Clin. Med. 2021, 10, 1242. [Google Scholar] [CrossRef]
- Maximiano, S.; Magalhães, P.; Guerreiro, M.P.; Morgado, M. Trastuzumab in the Treatment of Breast Cancer. BioDrugs 2016, 30, 75–86. [Google Scholar] [CrossRef]
- Hemperly, A.; Vande Casteele, N. Clinical Pharmacokinetics and Pharmacodynamics of Infliximab in the Treatment of Inflammatory Bowel Disease. Clin. Pharmacokinet. 2018, 57, 929–942. [Google Scholar] [CrossRef]
- Rocca, A.; Andreis, D.; Fedeli, A.; Maltoni, R.; Sarti, S.; Cecconetto, L.; Pietri, E.; Schirone, A.; Bravaccini, S.; Serra, P.; et al. Pharmacokinetics, pharmacodynamics and clinical efficacy of pertuzumab in breast cancer therapy. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1647–1663. [Google Scholar] [CrossRef]
- Rosario, M.; Dirks, N.L.; Milch, C.; Parikh, A.; Bargfrede, M.; Wyant, T.; Fedyk, E.; Fox, I. A Review of the Clinical Pharmacokinetics, Pharmacodynamics, and Immunogenicity of Vedolizumab. Clin. Pharmacokinet. 2017, 56, 1287–1301. [Google Scholar] [CrossRef]
- Paul, B.; Hamadeh, I.; Atrash, S.; Bhutani, M.; Voorhees, P.; Usmani, S.Z. Daratumumab subcutaneous formulation for the treatment of multiple myeloma. Expert Opin. Biol. Ther. 2020, 20, 1253–1259. [Google Scholar] [CrossRef]
- Lee, A.; Scott, L.J. Certolizumab Pegol: A Review in Moderate to Severe Plaque Psoriasis. BioDrugs 2020, 34, 235–244. [Google Scholar] [CrossRef]
- Mazzarella, L.; Guida, A.; Curigliano, G. Cetuximab for treating non-small cell lung cancer. Expert Opin. Biol. Ther. 2018, 18, 483–493. [Google Scholar] [CrossRef]
- Le Louedec, F.; Alix-Panabières, C.; Lafont, T.; Allal, B.C.; Garrel, R.; Digue, L.; Guigay, J.; Cupissol, D.; Delord, J.P.; Lallemant, B.; et al. Cetuximab pharmacokinetic/pharmacodynamics relationships in advanced head and neck carcinoma patients. Br. J. Clin. Pharmacol. 2019, 85, 1357–1366. [Google Scholar] [CrossRef]
- Coiffier, B.; Losic, N.; Rønn, B.B.; Lepretre, S.; Pedersen, L.M.; Gadeberg, O.; Frederiksen, H.; van Oers, M.H.; Wooldridge, J.; Kloczko, J.; et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic associations of ofatumumab, a human monoclonal CD20 antibody, in patients with relapsed or refractory chronic lymphocytic leukaemia: A phase 1-2 study. Br. J. Haematol. 2010, 150, 58–71. [Google Scholar] [CrossRef]
- Elter, T.; Molnar, I.; Kuhlmann, J.; Hallek, M.; Wendtner, C. Pharmacokinetics of alemtuzumab and the relevance in clinical practice. Leuk. Lymphoma 2008, 49, 2256–2262. [Google Scholar] [CrossRef]
- Moore, D.C.; Elmes, J.B.; Shibu, P.A.; Larck, C.; Park, S.I. Mogamulizumab: An Anti-CC Chemokine Receptor 4 Antibody for T-Cell Lymphomas. Ann. Pharmacother. 2020, 54, 371–379. [Google Scholar] [CrossRef]
- Kast, J.; Dutta, S.; Upreti, V.V. Panitumumab: A Review of Clinical Pharmacokinetic and Pharmacology properties after over a decade of experience in patients with solid tumors. Adv. Ther. 2021, 38, 3712–3723. [Google Scholar] [CrossRef]
- Gibiansky, L.; Sutjandra, L.; Doshi, S.; Zheng, J.; Sohn, W.; Peterson, M.C.; Jang, G.R.; Chow, A.T.; Pérez-Ruixo, J.J. Population pharmacokinetic analysis of denosumab in patients with bone metastases from solid tumours. Clin. Pharmacokinet. 2012, 51, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Sebba, A. Tocilizumab: The first interleukin-6-receptor inhibitor. Am. J. Health Syst. Pharm. 2008, 65, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Radin, A.; Li, M.; Hamilton, J.D.; Kajiwara, M.; Davis, J.D.; Takahashi, Y.; Hasegawa, S.; Ming, J.E.; DiCioccio, A.T.; et al. Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Dupilumab in Healthy Adult Subjects. Clin. Pharmacol. Drug Dev. 2020, 9, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Broadwell, A.; Fan, Y.; Hu, C.; Adedokun, O.J.; Chakravarty, S.D.; Zhou, H.; Xu, Z.; Leu, J.H. Population Pharmacokinetic and Exposure-Response Model Simulations: Predicted Exposure and Efficacy for Maintenance Doses of Intravenous Golimumab Every 6 or 8 Weeks in Patients with Moderately to Severely Active Rheumatoid Arthritis. Clin. Ther. 2022, 44, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Avery, R.L.; Castellarin, A.A.; Steinle, N.C.; Dhoot, D.S.; Pieramici, D.J.; See, R.; Couvillion, S.; Nasir, M.A.; Rabena, M.D.; Maia, M.; et al. Systemic pharmacokinetics and pharmacodynamics of intravitreal aflibercept bevacizumab, and ranibizumab. Retina 2017, 37, 1847–1858. [Google Scholar] [CrossRef]
- Guo, G.; You, X.; Wu, W.; Chen, J.; Ke, M.; Lin, R.; Huang, P.; Lin, C. Physiologically-Based Pharmacokinetic Modeling of Omalizumab to Predict the Pharmacokinetics and Pharmacodynamics in Pediatric Patients. Clin. Pharmacol. Ther. 2023, 113, 724–734. [Google Scholar] [CrossRef]
- Sweet, B.V. Natalizumab update. Am. J. Health Syst. Pharm. 2007, 64, 705–716. [Google Scholar] [CrossRef]
- Patel, R.; Bock, M.; Polotti, C.F.; Elsamra, S. Pharmacokinetic drug evaluation of atezolizumab for the treatment of locally advanced or metastatic urothelial carcinoma. Expert Opin. Drug Metab. Toxicol. 2017, 13, 225–232. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Nathan, P.; Sacco, J.J.; Orloff, M.; Hernandez-Aya, L.F.; Yang, J.; Luke, J.J.; Butler, M.O.; Stanhope, S.; Collins, L.; et al. Phase I study of safety, tolerability, and efficacy of tebentafusp using a step-up dosing regimen and expansion in patients with metastatic uveal melanoma. J. Clin. Oncol. 2022, 40, 1939–1948. [Google Scholar] [CrossRef]
- Mahlangu, J.; Iorio, A.; Kenet, G. Emicizumab state-of-the-art update. Haemophilia 2022, 28 (Suppl. 4), 103–110. [Google Scholar] [CrossRef]
- Jackson, K.; Chua, L.; Velez de Mendizabal, N.; Pitou, C.; Rodriguez Capriles, C.; Paller, A.S.; Lansang, P.; Seyger, M.M.B.; Papp, K. Population pharmacokinetic and exposure-efficacy analysis of ixekizumab in paediatric patients with moderate-to-severe plaque psoriasis (IXORA-PEDS). Br. J. Clin. Pharmacol. 2022, 88, 1074–1086. [Google Scholar] [CrossRef]
- Hedrich, W.D.; Fandy, T.E.; Ashour, H.M.; Wang, H.; Hassan, H.E. Antibody-Drug Conjugates: Pharmacokinetic/Pharmacodynamic Modeling, Preclinical Characterization, Clinical Studies, and Lessons Learned. Clin. Pharmacokinet. 2018, 57, 687–703. [Google Scholar] [CrossRef]
- Diéras, V.; Bachelot, T. The success story of trastuzumab emtansine, a targeted therapy in HER2-positive breast cancer. Target Oncol. 2014, 9, 111–122. [Google Scholar] [CrossRef]
- Lee, J.; Park, Y.H. Trastuzumab deruxtecan for HER2+ advanced breast cancer. Future Oncol. 2022, 18, 7–19. [Google Scholar] [CrossRef]
- Sjögreen-Gleisner, K.; Dewaraja, Y.K.; Chiesa, C.; Tennvall, J.; Lindén, O.; Strand, S.E.; Ljungberg, M. Dosimetry in patients with B-cell lymphoma treated with [(90)Y]ibritumomab tiuxetan or [(131)I]tositumomab. Q. J. Nucl. Med. Mol. Imaging 2011, 55, 126–154. [Google Scholar]
- Chinn, P.C.; Leonard, J.E.; Rosenberg, J.; Hanna, N.; Anderson, D.R. Preclinical evaluation of 90Y-labeled anti-CD20 monoclonal antibody for treatment of non-Hodgkin’s lymphoma. Int. J. Oncol. 1999, 15, 1017–1025. [Google Scholar] [CrossRef]
- Spies, S.M. Imaging and dosing in radioimmunotherapy with yttrium 90 ibritumomab tiuxetan (Zevalin). Semin. Nucl. Med. 2004, 34, 10–13. [Google Scholar] [CrossRef]
- Chen, R.; Chen, B. Brentuximab vedotin for relapsed or refractory Hodgkin’s lymphoma. Drug Des. Devel. Ther. 2015, 9, 1729–1733. [Google Scholar] [CrossRef]
- Bradley, A.M.; Devine, M.; DeRemer, D. Brentuximab vedotin: An anti-CD30 antibody-drug conjugate. Am. J. Health Syst. Pharm. 2013, 70, 589–597. [Google Scholar] [CrossRef]
- Keating, G.M. BAY 81-8973 (Octocog Alfa; Kovaltry®): A Review in Haemophilia A. BioDrugs 2016, 30, 453–459. [Google Scholar] [CrossRef]
- Witarto, B.S.; Visuddho, V.; Witarto, A.P.; Sutanto, H.; Wiratama, B.S.; Wungu, C.D.K. Efficacy, safety, and immunogenicity of rurioctocog alfa pegol for prophylactic treatment in previously treated patients with severe hemophilia A: A systematic review and meta-analysis of clinical trials. F1000Research 2021, 10, 1049. [Google Scholar] [CrossRef]
- Ezban, M.; Hansen, M.; Kjalke, M. An overview of turoctocog alfa pegol (N8-GP; ESPEROCT®) assay performance: Implications for postadministration monitoring. Haemophilia 2020, 26, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.; Deeks, E.D. Damoctocog Alfa Pegol: A Review in Haemophilia A. Drugs 2019, 79, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- George, L.A.; Camire, R.M. Profile of efraloctocog alfa and its potential in the treatment of hemophilia A. J. Blood Med. 2015, 6, 131–141. [Google Scholar] [PubMed]
- Syed, Y.Y. Correction to: Nonacog Beta Pegol: A Review in Haemophilia B. Drugs 2018, 78, 1169. [Google Scholar] [CrossRef]
- Hoy, S.M. Eftrenonacog Alfa: A Review in Haemophilia B. Drugs 2017, 77, 1235–1246. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Coagulation Factor IX (Recombinant), Albumin Fusion Protein (Albutrepenonacog Alfa; Idelvion®): A Review of Its Use in Haemophilia B. Drugs 2017, 77, 97–106. [Google Scholar] [CrossRef]
- Yeh, M.L.; Huang, J.F.; Dai, C.Y.; Yu, M.L.; Chuang, W.L. Pharmacokinetics and pharmacodynamics of pegylated interferon for the treatment of hepatitis B. Expert Opin. Drug Metab. Toxicol. 2019, 15, 779–785. [Google Scholar] [CrossRef]
- Perry, C.M.; Jarvis, B. Peginterferon-alpha-2a (40 kD): A review of its use in the management of chronic hepatitis C. Drugs 2001, 61, 2263–2288. [Google Scholar] [CrossRef]
- Takeuchi, T.; Miyasaka, N.; Kawai, S.; Sugiyama, N.; Yuasa, H.; Yamashita, N.; Sugiyama, N.; Wagerle, L.C.; Vlahos, B.; Wajdula, J. Pharmacokinetics, efficacy and safety profiles of etanercept monotherapy in Japanese patients with rheumatoid arthritis: Review of seven clinical trials. Mod. Rheumatol. 2015, 25, 173–186. [Google Scholar] [CrossRef]
- Bruce, S.P.; Boyce, E.G. Update on abatacept: A selective costimulation modulator for rheumatoid arthritis. Ann. Pharmacother. 2007, 41, 1153–1162. [Google Scholar] [CrossRef]
- Tournis, S.; Yavropoulou, M.P.; Polyzos, S.A.; Doulgeraki, A. Hypophosphatasia. J. Clin. Med. 2021, 10, 5676. [Google Scholar] [CrossRef]
- Dorsey, M.J.; Rubinstein, A.; Lehman, H.; Fausnight, T.; Wiley, J.M.; Haddad, E. PEGylated Recombinant Adenosine Deaminase Maintains Detoxification and Lymphocyte Counts in Patients with ADA-SCID. J. Clin. Immunol. 2023. [Google Scholar] [CrossRef]
- Bryson, H.M.; Sorkin, E.M. Dornase alfa. A review of its pharmacological properties and therapeutic potential in cystic fibrosis. Drugs 1994, 48, 894–906. [Google Scholar] [CrossRef]
- Deicher, R.; Hörl, W.H. Differentiating factors between erythropoiesis-stimulating agents: A guide to selection for anaemia of chronic kidney disease. Drugs 2004, 64, 499–509. [Google Scholar] [CrossRef]
- Overbay, D.K.; Manley, H.J. Darbepoetin-alpha: A review of the literature. Pharmacotherapy 2002, 22, 889–897. [Google Scholar] [CrossRef]
- Fishbane, S.; Pannier, A.; Liogier, X.; Jordan, P.; Dougherty, F.C.; Reigner, B. Pharmacokinetic and pharmacodynamic properties of methoxy polyethylene glycol-epoetin beta are unaffected by the site of subcutaneous administration. J. Clin. Pharmacol. 2007, 47, 1390–1397. [Google Scholar] [CrossRef]
- Yang, B.B.; Kido, A. Pharmacokinetics and pharmacodynamics of pegfilgrastim. Clin. Pharmacokinet. 2011, 50, 295–306. [Google Scholar] [CrossRef]
- Jacobsen, L.V.; Flint, A.; Olsen, A.K.; Ingwersen, S.H. Liraglutide in Type 2 Diabetes Mellitus: Clinical Pharmacokinetics and Pharmacodynamics. Clin. Pharmacokinet. 2016, 55, 657–672. [Google Scholar] [CrossRef]
- Granhall, C.; Søndergaard, F.L.; Thomsen, M.; Anderson, T.W. Pharmacokinetics, Safety and Tolerability of Oral Semaglutide in Subjects with Renal Impairment. Clin. Pharmacokinet. 2018, 57, 1571–1580. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Li, Y.; Zhao, X.; Zhou, W.; Loghin, C.; Tham, L.S.; Cui, X.; Cui, Y.; Wang, W. Pharmacokinetics, Pharmacodynamics, and Safety of Dulaglutide After Single or Multiple Doses in Chinese Healthy Subjects and Patients with T2DM: A Randomized, Placebo-Controlled, Phase I Study. Adv. Ther. 2022, 39, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Fares, F.A.; Suganuma, N.; Nishimori, K.; LaPolt, P.S.; Hsueh, A.J.; Boime, I. Design of a long-acting follitropin agonist by fusing the C-terminal sequence of the chorionic gonadotropin beta subunit to the follitropin beta subunit. Proc. Natl. Acad. Sci. USA 1992, 89, 4304–4308. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Jobanputra, A.; Saxena, B.; Nivsarkar, M. Development and Characterization of Saturated Fatty Acid-Engineered, Silica-Coated Lipid Vesicular System for Effective Oral Delivery of Alfa-Choriogonadotropin. AAPS PharmSciTech 2021, 22, 118. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.S. What do we do now that the long-acting growth hormone is here? Front. Endocrinol. 2022, 13, 980979. [Google Scholar] [CrossRef]
- Nedelman, J.; Fisch, R.; Hu, K.; Paule, I.; Zhou, J. Population Pharmacokinetics of Subcutaneous Pasireotide in Healthy Volunteers and Cushing’s Disease Patients. Clin. Pharmacokinet. 2018, 57, 855–866. [Google Scholar] [CrossRef]
- Hu, M.; Tomlinson, B. Pharmacokinetic evaluation of lanreotide. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1301–1312. [Google Scholar] [CrossRef]
- Chanson, P.; Timsit, J.; Harris, A.G. Clinical pharmacokinetics of octreotide. Therapeutic applications in patients with pituitary tumours. Clin. Pharmacokinet. 1993, 25, 375–391. [Google Scholar] [CrossRef]
- Ogihara, T. (Ed.) Tsuji’s Episodic Pharmacokinetics, 2nd ed.; Kyoto-Hirokawa Publishing Inc.: Kyoto, Japan, 2023; Chapter 11; pp. 331–354. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Origin | Main Indications | t1/2 | |
|---|---|---|---|---|
| Human immunoglobulin | Plasma | No, or hypogammaglobulinemia | 21–48 | day |
| Human serum albumin | Plasma | Hypoalbuminemia, hemorrhagic shock | 15–20 | day |
| Human alpha 1-proteinase inhibitor | Plasma | Severe alpha 1-antitrypsin deficiency | 150.4 | h |
| Human fibrinogen | Plasma | Bleeding tendency in congenital hypofibrinogenemia | 3.3–4.2 | day |
| Human antithrombin III | Plasma | Tendency to thrombus formation based on congenital antithrombin III deficiency | 65 | h |
| Human haptoglobin | Plasma | Hemoglobinemia, hemoglobinuria | 20 | h |
| Human blood coagulation factor | Plasma | Bleeding tendency in patients with blood coagulation factor deficiency | 11.6–25.6 | h |
| Human protrobin complex | Plasma | Bleeding tendency during surgery and procedures requiring urgent care | 4.2–59.7 | h |
| Human activated protein C | Plasma | Deep vein thrombosis, acute pulmonary thromboembolism | 71.5 | min |
| Urinastatin | Urine | Acute exacerbation phase of acute pancreatitis and chronic recurrent pancreatitis | 40 | min |
| Urokinase | Urine | Cerebral thrombosis | 17–33 | min |
| Dosage (mg/kg) | Cmax (μg/mL) | AUClast (μg·h/mL) | t1/2 (h) | CLtotal (mL/h/kg) | MRT (h) | Vd,ss (mL/kg) |
|---|---|---|---|---|---|---|
| 0.15 | 2.4 ± 0.6 | 11 ± 6 | 17 ± 16 | 3.8 ± 2.3 | 25 ± 22 | 63.4 ± 16.6 |
| 0.5 | 8.5 ± 1.2 | 285 ± 73 | 33 ± 4 | 1.3 ± 0.2 | 47 ± 5 | 58.4 ± 7.1 |
| 1.0 | 19.5 ± 2.7 | 1009 ± 222 | 49 ± 5 | 0.8 ± 0.1 | 69 ± 8 | 57.3 ± 10.9 |
| 2.0 | 37.6 ± 8.8 | 2532 ± 569 | 74 ± 9 | 0.6 ± 0.2 | 107 ± 16 | 65.9 ± 8.3 |
| Example | Note | ||||
|---|---|---|---|---|---|
| Control of half life | |||||
| Amino acid modifier | Insulin analog | Insulin lispro | Several amino acids were modified to prolong or shorten the half-life | ||
| Insulin glargine | |||||
| PEG modifier | Cytokine | Pegfilgrastim |  | Improved stability against proteolytic enzymes Prolonged half-life due to inhibition of glomerular filtration Improved water solubility as a formulation advantage Reduced antigenicity as a safety benefit | |
| Fatty acid modifier | GLP-1 analog | Liraglutide | |||
| Albumin fusion protein | Blood coagulation factor | Albutrepenonacog alfa | |||
| Fc fusion protein | Enzyme | Veraglucerase alfa | Proteins that fuse functional proteins such as receptors, peptides, and enzymes with the Fc domains of antibodies | ||
| Fc analog | Efgartigimod alfa | Internal stability | |||
| Targeting | |||||
| Fab Analog | Ranibizumab | Targeting to the tissues on a kinetic basis | |||
| Receptor analogue | TNF-alpha antagonist | Etanercept | Fc fusion protein in which the extracellular sequence (domain) of the TNF receptor is bound to the Fc region of human IgG. | ||
| Control of half life/Targeting | |||||
| Monoclonal antibody | Adalimumab | A single antibody that binds specifically to disease-related molecules. Takes advantage of the antibody’s unique targeting and long half-life | |||
| Antibody-drug conjugates | Trastuzumab emtansine | Complexes of monoclonal antibodies with specific drugs | |||
| Drug Name | Daily Dose Number of Times | Expression of Action Pattern (h) | Onset of Action | Maximum Action Expression (h) | Duration of Action(h) | ||
|---|---|---|---|---|---|---|---|
| human insulin preparation | fast-acting | insulin | 0 3 (Before every meal) |  | 0.5 (h) * | 1–3 | 8 * |
| intermediate type | Insulin (protamine preparation 30% mixture) | 1-2 |  | 0.5 (h) * | 2–8 | 24 * | |
| insulin (protamine preparation) | 1 (Before breakfast) | 1.5 (h) * | 4–12 | 24 * | |||
| insulin analog | insulin lispro | 3 (Before every meal) |  | <0.25 (h) | 0.5–1.5 | 3–5 | |
| superquick type | insulin glulisine | 3 (Before every meal) | <0.25 (h) | 0.5–1.5 | 3–5 | ||
| insulin asparte | 3 (Before every meal) | 10–20 (m) | 1–3 | 3–5 | |||
| mixture type (intermediate type) | insulin asparte (protamine preparation 30% mixture) | 1 (Before breakfast) |  | 10–20 (m) | 1–4 | 24 * | |
| insulin asparte (protamine preparation 50% mixture) | 1 (Before breakfast) |  | 10–20 (m) | 1–4 | 24 * | ||
| insulin asparte (protamine preparation 70% mixture) | 1 (Before breakfast) | 0.5–1 (h) | 1–4 | 24 * | |||
| insulin glargine | 1 (Before breakfast) |  | 1.11 (h) | – | 24 * | ||
| continuation type | insulin detemir | 1 (Before dinner) | 1.0 (h)* | 3–14 | 24 * | ||
| insulin degludec | 1 | stationary | – | >42 |
| Drug Name | Indications | Dose | Dosing Interval (Week) | Route *1 | Parameters *2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| t1/2 | CLtot | Vd(mL/kg) | BA(%) | Reference | |||||||||
| Adalimumab | rheumatism | 40 | mg/body | 2 | sc | 12.4 | h | 18.0 | mL/h | [25] | |||
| Pembrolizumab | cancer | 200 | mg/body | 3 | iv | 18.4 | day | 2.46 | mL/day/kg | 65.3 | mL/kg | [26] | |
| Nivolumab | cancer | 2 | mg/kg | 3 | iv | 13.3 | h | 5.04 | mL/day/kg | 69.7 | mL/kg | [27] | |
| Bevacizumab | cancer | 5 | mg/kg | 2< | iv | 13.40 | day | 3.94 | mL/day/kg | 73.47 | mL/kg | [28] | |
| Rituximab | cancer | 375 | mg/m2 | 1 | iv | 16.2 | h | - | - | - | [29] | ||
| Ustekinumab | psoriasis | 390 | mg/body | 8 | iv | 24.7 | day | - | - | - | [30] | ||
| Trastuzumab | cancer | 4 | mg/kg | 1 | iv | 5.9 | day | 7.4 | mL/day/kg | 63 | mL/kg | [31] | |
| Infliximab | rheumatism | 3 | mg | 2–6 | iv | 9.5 | day | - | - | 3 | L | [32] | |
| Pertuzumab | cancer | 840 | mg/body | 3 | iv | 16.8 | day | 4.25 | mL/day/kg | 94.1 | mL/kg | [33] | |
| Vedolizumab | ulcerative colitis | 300 | mg/body | 1–8 | iv | 9.46 | day | 0.258 | L/day | 3.50 | L | [34] | |
| Daratumumab | cancer | 16 | mg/kg | 1–4 | iv | 17.0 | h | 3.15 | mL/day/kg | 72 | mL/kg | [35] | |
| Certolizumab pegol | rheumatism | 400 | mg | 1–4 | sc | 10.7 | day | - | - | - | - | - | [36,37] |
| Cetuximab | rectumcancer | 400 | mg/m2 | 1 | iv | 101 | h | 0.016 | L/h/m2 | 2.14 | L/m2 | [38] | |
| Ofatumumab | chronic lymphocytic leukemia | 300 | mg | 1 | iv | 10 | h | 199.2 | mL/h | [39] | |||
| Alemtuzumab | chronic lymphocytic leukemia | 3 | mg | 7 | iv | 24.06 | h | 37.7 | mL/h/kg | [40] | |||
| Mogamulizumab | T-cell leukemia-lymphoma | 1 | mg/kg | 1 | iv | 422 | h | [41] | |||||
| Panitumumab | cancer | 6 | mg/kg | 2 | iv | 6.72 | day | 8.49 | mL/day/kg | [42] | |||
| Denosumab | osteoporosis | 60 | mg | 6 | sc | 62 | [43] | ||||||
| Tocilizumab | rheumatism | 8 | mg/kg | 4 | iv | 160 | h | 0.6 | mL/h/kg | 137 | mL/kg | [44] | |
| Dupilumab | atopic dermatitis | 600 | mg | 2 | sc | 8.77 | day | 61–64 | [45] | ||||
| Golimumab | rheumatism | 50 | mg | 4 | sc | 11.92 | day | 51 | [46] | ||||
| Ranibizumab | macular degeneration | 0.5 | mg | 1 | the vitreous | [47] | |||||||
| Omalizumab | asthma | 75–600 | mg | 2–4 | sc | 21 | day | 62–71 | [48] | ||||
| Natalizumab | multiple sclerosis | 300 | mg | 4 | iv | 365 | h | 7.28 | mL/h | 3.51 | L | [49] | |
| Atezolizumab | cancer | 1200 | mg | 3 | iv | 13 | day | 0.213 | L/day | 3.82 | L | [50] | |
| Durvalumab | cancer | 10 | mg/kg | 2 | iv | [51] | |||||||
| Emicizumab | haemophilia | 3 | mg/kg | 1 | sc | 28.3–29.0 | day | 80.4–93.1 | [52] | ||||
| Ixekizumab | psoriasis | 160 | mg | 2 | sc | 11.4–12.2 | day | 54–90 | [53] | ||||
| Drug Name *1 | Linker *2 | Site of Action | Indications | t1/2(day) *3 | Reference |
|---|---|---|---|---|---|
| Trastuzumab emtansine | MCClinker (thioether) | DM1 (Maytansine Derivatives) | HER2-positive inoperable or recurrent breast cancer | 2.39–3.74 | [55] |
| Emtansine | |||||
| Trastuzumab deruxtecan | Camptothecin Derivatives | HER2-positive inoperable or recurrent breast cancer | 5.50 | [56] | |
| Deruxtecan | 5.77 | ||||
| Yttrium (90Y) ibritumomab thiuxetan | Tiuxetan | Yttrium (90Y) | CD20-positive non-Hodgkin’s lymphoma, etc. | 34.7–39.3 | [57,58,59] |
| Indium (111In) ibritumamob thiuxetan | Tiuxetan | Indium (111In) | Ibritsumamobuchiukisetan Identification of accumulation sites | 38.6 | [59] |
| Brentuximab vedotin | MMAE (Monomethyl auristatin E) | CD30-positive non-Hodgkin’s lymphoma, etc. | 3.75–4.54 | [60,61] | |
| Vedotin | |||||
| Gemtuzumab ozogamicin | Ozogamicin (caricamycin) | CD33-positive acute myelogenous leukemia | 51–59 | [54] | |
| Classification (Stem) | Drug | Dosing Interval | Parameter * | |||||
|---|---|---|---|---|---|---|---|---|
| t1/2 | Vd | CLtot | BA | Reference | ||||
| (h) | (mL/kg) | (mL/hr/kg) | (%) | |||||
| Factor VIII’ (–octocog) | Plasmaderivative | dried concentrated human blood coagulation factor VIII | 15.1 | |||||
| Octocog alpha | 13.96 | 45.8–98.4 | [62] | |||||
| Octocog beta | 2–3 times a week | 12.8 | 52.7 | |||||
| Rurioctocog alpha | 13.00 | [63] | ||||||
| Lonoctocog alpha | 2–3 times a week | 14.2 | 56.7 | 3.00 | ||||
| Turoctocog alpha | Every other day/3 times a week | 12.61 | [64] | |||||
| PEG-modified | Lurioctocog alpha Pegol | Twice a week | 14.3 | 50 | 2.8 | |||
| Turoctogog alpha pegol | 1–2 times a week | 19.9 | 37.7 | 1.4 | ||||
| Damoktokog alpha pegol | Every 8–48h | 16.3 | 42.4 | 1.83 | [65] | |||
| Fc fusion protein | Efraloctocog alpha | Every 3–5 days | 19.0 | 49.1 | 1.95 | [66] | ||
| Factor IX (–nonacog) | Plasmaderivative | dried concentrated human blood coagulation factor IX | 8.2/20.3(α/β) | |||||
| Nonacog alpha | 20.2–24.5 | |||||||
| Nonakog gamma | Twice a week | 24.5–27.9 | ||||||
| PEG–modified | Nonakog beta pegol | 83 | 47 | [67] | ||||
| Fc fusion protein | Eftrenoacog alpha | Once a week | 5.03/82.12 | 314.8 | 3.19 | [68] | ||
| Albumin fusion protein | Albutrepenonacog alpha | Once a week | 104.2 | 100 | [69] | |||
| Drug Name | Dosing Interval | Route *1 | Parameter *2 | |||
|---|---|---|---|---|---|---|
| t1/2(h) | Tmax(h) | BA(%) | ||||
| Interferon | alpha (natural type) | Once every 1–2 days | sc | 9.6 | 6.0 | |
| alpha 2b | 3 times a week | im | 5.2 | 5.7–6 | ||
| beta | Once a day | iv | - | - | - | |
| beta-1a | Once a week | im | - | 13.0 | ||
| beta 1b | every other day | sc | - | - | - | |
| beta gamma 1a | Once a day | iv | 12.82 | 8.3 | ||
| Peginterferon alpha-2a | Once a week | sc | 32.5–42.8 | 70.9–73.0 | 84 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogihara, T.; Mizoi, K.; Ishii-Watabe, A. Pharmacokinetics of Biopharmaceuticals: Their Critical Role in Molecular Design. Biomedicines 2023, 11, 1456. https://doi.org/10.3390/biomedicines11051456
Ogihara T, Mizoi K, Ishii-Watabe A. Pharmacokinetics of Biopharmaceuticals: Their Critical Role in Molecular Design. Biomedicines. 2023; 11(5):1456. https://doi.org/10.3390/biomedicines11051456
Chicago/Turabian StyleOgihara, Takuo, Kenta Mizoi, and Akiko Ishii-Watabe. 2023. "Pharmacokinetics of Biopharmaceuticals: Their Critical Role in Molecular Design" Biomedicines 11, no. 5: 1456. https://doi.org/10.3390/biomedicines11051456
APA StyleOgihara, T., Mizoi, K., & Ishii-Watabe, A. (2023). Pharmacokinetics of Biopharmaceuticals: Their Critical Role in Molecular Design. Biomedicines, 11(5), 1456. https://doi.org/10.3390/biomedicines11051456