
Characteristic Analysis of Featured Genes Associated with Cholangiocarcinoma Progression

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Processing of Gene Expression Data
2.2. Gene Co-Expression Network Construction
2.3. Construction of CCA Modules with Clinical Relationships
2.4. Functional Annotations for DEGs
2.5. Hub Gene Validation
2.6. Identification of Hub Gene Functional Annotations
2.7. Human Samples
2.8. Immunohistochemical Staining
2.9. Construction of PPI Network
2.10. Statistical Analysis
3. Results
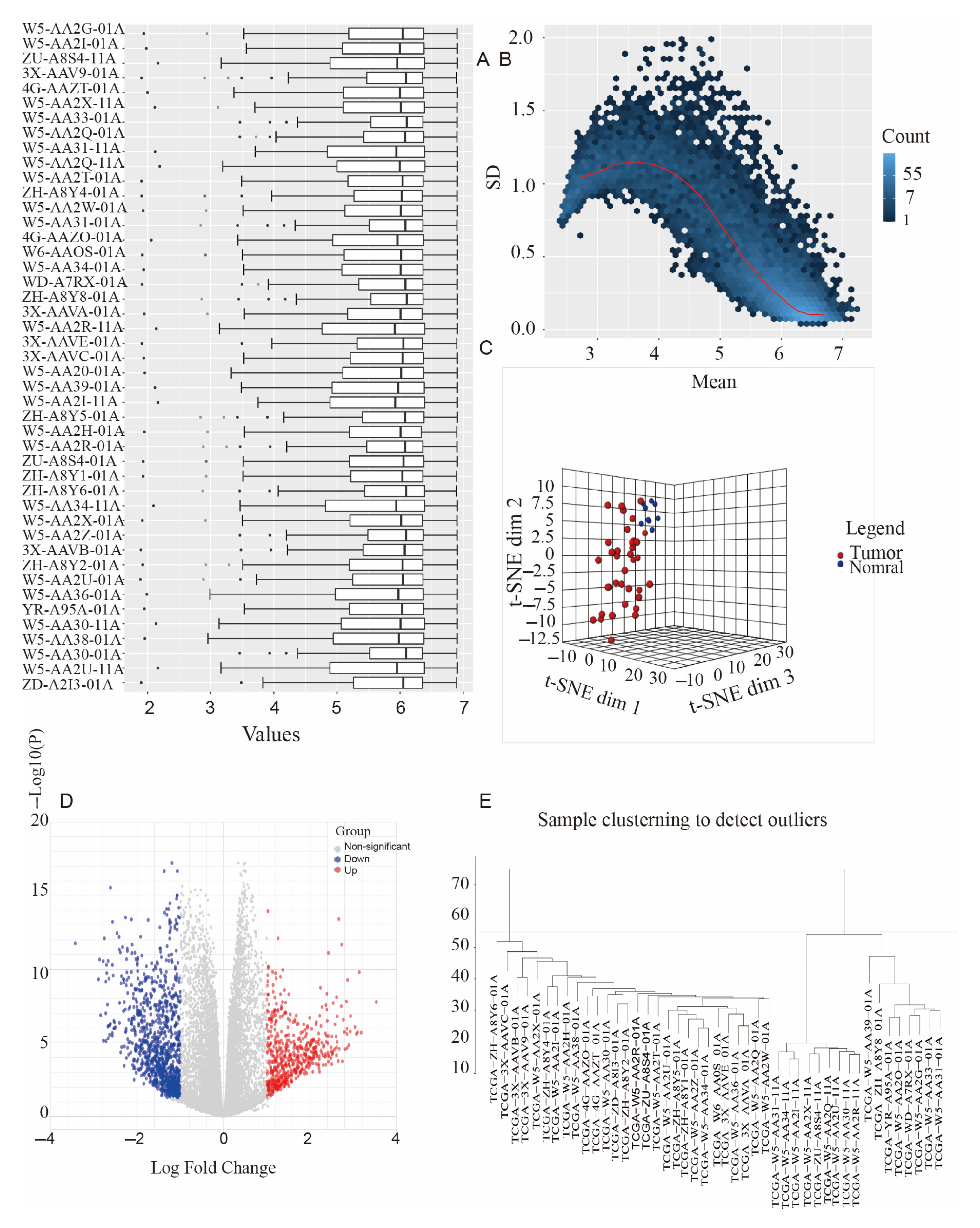
3.1. Data Collection and Processing
3.2. Functional Annotations of Hub Genes
3.3. Weighting Coefficient β Selection
3.4. Co-Expression Network Construction
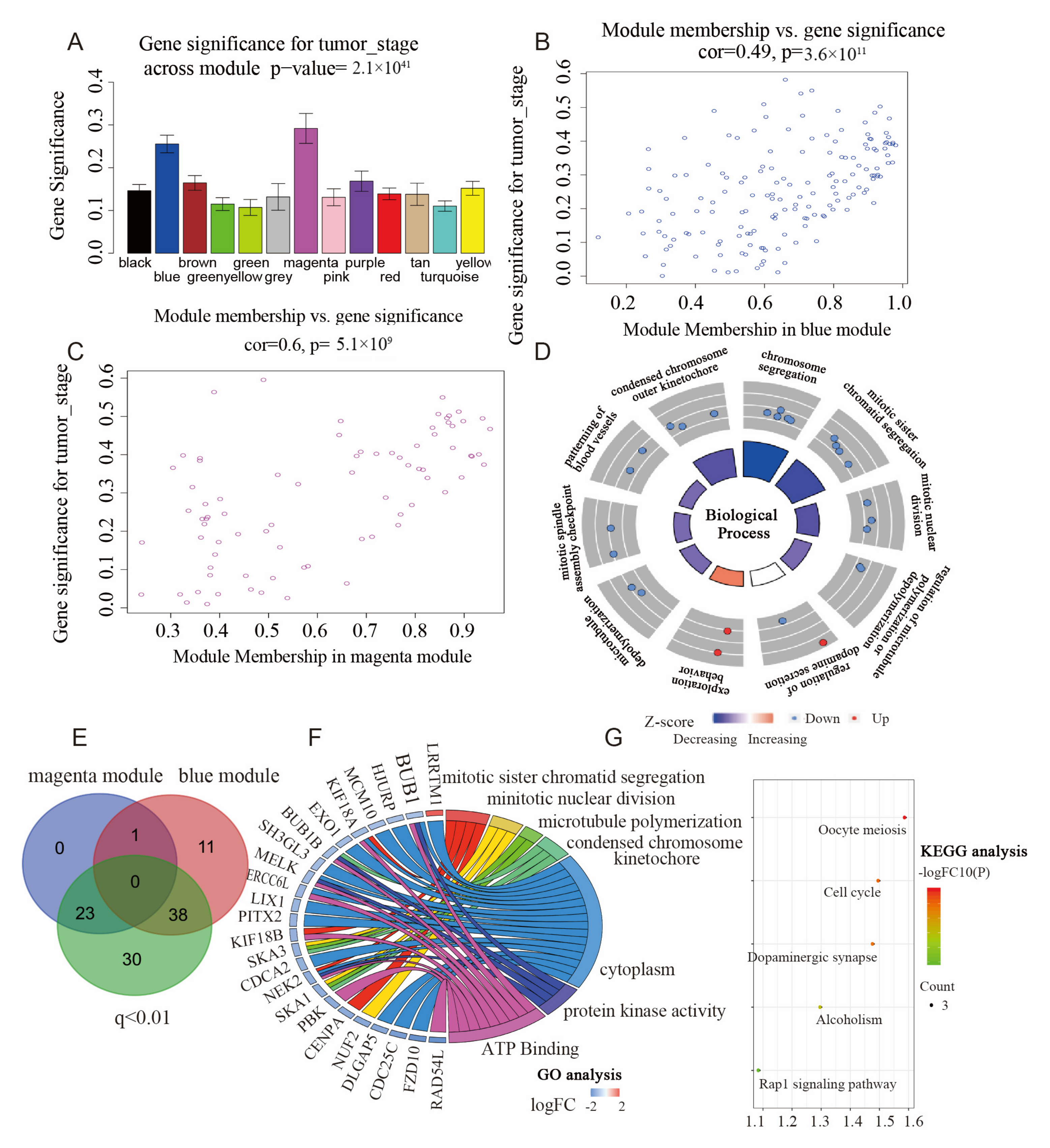
3.5. Identification of Clinical Modules
3.6. Identification of Hub Genes
3.7. Functional Enrichment Analyses of the Hub Genes in Hub Modules
3.8. Construction of PPI Network and Verification of Hub Genes
3.9. Survival Analysis
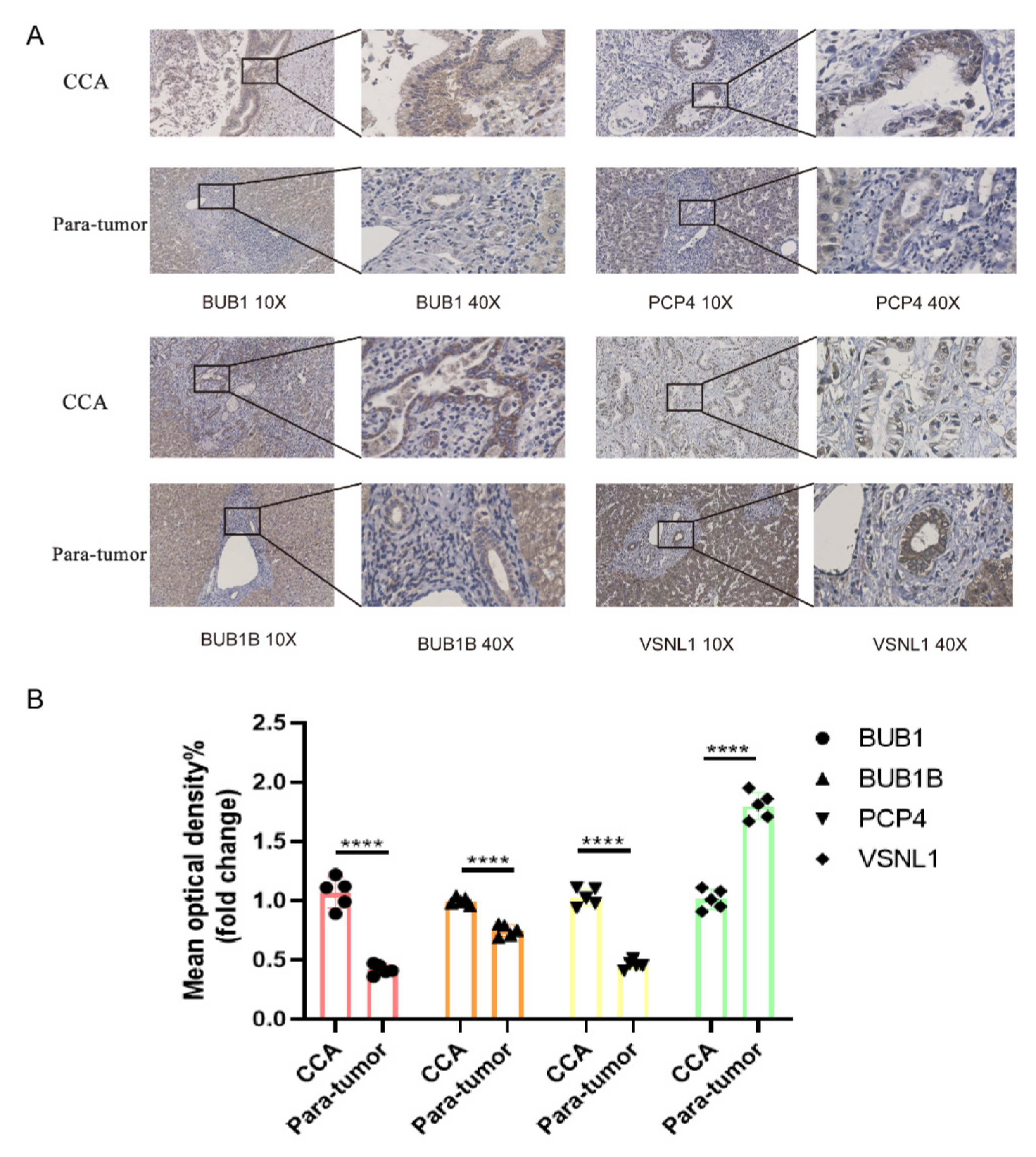
3.10. Hub Gene Analysis and Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rizvi, S.; Gores, G. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, V.; Bragazzi, M.C.; Carpino, G.; Torrice, A.; Fraveto, A.; Gentile, R.; Pasqualino, V.; Melandro, F.; Aliberti, C.; Bastianelli, C.; et al. Cholangiocarcinoma: Increasing burden of classifications. HepatoBiliary Surg. Nutr. 2013, 2, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Khan, S.; Hallemeier, C.; Kelley, R.; Gores, G. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2017, 15, 95–111. [Google Scholar] [CrossRef]
- Sapisochin, G.; De Lope, C.R.; Gastaca, M.; De Urbina, J.O.; Suarez, M.A.; Santoyo, J.; Castroagudín, J.F.; Varo, E.; López-Andujar, R.; Palacios, F.; et al. “Very Early” Intrahepatic Cholangiocarcinoma in Cirrhotic Patients: Should Liver Transplantation Be Reconsidered in These Patients? Am. J. Transplant. 2014, 14, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.; Li, T.; Huang, H.-Y.; Lin, J.-H.; Chen, W.-S.; Shyu, W.-C.; Wu, H.-C.; Peng, C.-Y.; Su, I.-J.; Jeng, L.-B. Next-Generation Sequencing-Based Quantitative Detection of Hepatitis B Virus Pre-S Mutants in Plasma Predicts Hepatocellular Carcinoma Recurrence. Viruses 2020, 12, 796. [Google Scholar] [CrossRef]
- Dobie, R.; Wilson-Kanamori, J.R.; Henderson, B.E.; Smith, J.R.; Matchett, K.P.; Portman, J.R.; Wallenborg, K.; Picelli, S.; Zagorska, A.; Pendem, S.V.; et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep. 2019, 29, 1832–1847. [Google Scholar] [CrossRef]
- Gasparello, J.; Papi, C.; Allegretti, M.; Giordani, E.; Carboni, F.; Zazza, S.; Pescarmona, E.; Romania, P.; Giacomini, P.; Scapoli, C.; et al. A Distinctive microRNA (miRNA) Signature in the Blood of Colorectal Cancer (CRC) Patients at Surgery. Cancers 2020, 12, 2410. [Google Scholar] [CrossRef]
- Li, B.; Pu, K.; Wu, X. Identifying novel biomarkers in hepatocellular carcinoma by weighted gene co-expression network analysis. J. Cell. Biochem. 2019, 120, 11418–11431. [Google Scholar] [CrossRef]
- Peng, W.; Bai, F.; Shao, K.; Shen, L.; Li, H.; Huang, S. The key genes underlying pathophysiology association between the type 2-diabetic and colorectal cancer. J. Cell. Physiol. 2018, 233, 8551–8557. [Google Scholar] [CrossRef]
- Banales, J.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, S.; Chen, J.-M.; Wang, G.-P.; Wang, Z.-F.; Zhou, B.; Jin, C.-H.; Yang, Y.-T.; Feng, X.-S. Serum CA242, CA199, CA125, CEA, and TSGF are Biomarkers for the Efficacy and Prognosis of Cryoablation in Pancreatic Cancer Patients. Cell Biochem. Biophys. 2014, 71, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Feng, F.; Zhao, X. Combined detection tumor markers for diagnosis and prognosis of gallbladder cancer. World J. Gastroenterol. 2014, 20, 4085–4092. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, X.; Huang, X.; Yong, H.; Shen, J.; Tang, Q.; Zhu, J.; Ni, J.; Feng, Z. Nicotinamide N-methyltransferase: A potential biomarker for worse prognosis in gastric carcinoma. Am. J. Cancer Res. 2016, 6, 649–663. [Google Scholar]
- Li, X.; Pasche, B.; Zhang, W.; Chen, K. Association of MUC16 Mutation with Tumor Mutation Load and Outcomes in Patients with Gastric Cancer. JAMA Oncol. 2018, 4, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Lycke, M.; Kristjansdottir, B.; Sundfeldt, K. A multicenter clinical trial validating the performance of HE4, CA125, risk of ovarian malignancy algorithm and risk of malignancy index. Gynecol. Oncol. 2018, 151, 159–165. [Google Scholar] [CrossRef]
- Tian, A.; Pu, K.; Li, B.; Li, M.; Liu, X.; Gao, L.; Mao, X. Weighted gene coexpression network analysis reveals hub genes involved in cholangiocarcinoma progression and prognosis. Hepatol. Res. 2019, 49, 1195–1206. [Google Scholar] [CrossRef]
- Javle, M.; Churi, C.; Kang, H.; Shroff, R.; Janku, F.; Surapaneni, R.; Zuo, M.; Barrera, C.; Alshamsi, H.; Krishnan, S.; et al. HER2/neu-directed therapy for biliary tract cancer. J. Hematol. Oncol. 2015, 8, 1–9. [Google Scholar] [CrossRef]
- Sia, D.; Villanueva, A.; Friedman, S.; Llovet, J. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Huang, S.; Bai, Y.; Mao, J.; Wang, A.; Lin, Y.; Yang, X.; Wang, D.; Lin, J.; Bian, J.; et al. Transcriptional landscape of cholangiocarcinoma revealed by weighted gene coexpression network analysis. Briefings Bioinform. 2020, 22, bbaa224. [Google Scholar] [CrossRef]
- Høgdall, D.; Lewinska, M.; Andersen, J. Desmoplastic Tumor Microenvironment and Immunotherapy in Cholangiocarcinoma. Trends Cancer 2018, 4, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Baiocchi, L.; Sato, K.; Ekser, B.; Kennedy, L.; Francis, H.; Ceci, L.; Lenci, I.; Alvaro, D.; Franchitto, A.; Onori, P.; et al. Cholangiocarcinoma: Bridging the translational gap from preclinical to clinical development and implications for future therapy. Expert Opin. Investig. Drugs 2020, 30, 1–11. [Google Scholar] [CrossRef]
- Nault, J.; Villanueva, A. Biomarkers for Hepatobiliary Cancers. Hepatology 2020, 73, 115–127. [Google Scholar] [CrossRef]
- Louis, C.; Edeline, J.; Coulouarn, C. Targeting the tumor microenvironment in cholangiocarcinoma: Implications for therapy. Expert Opin. Ther. Targets 2021, 25, 153–162. [Google Scholar] [CrossRef]
- Sarkar, S.; Sahoo, P.; Mahata, S.; Pal, R.; Ghosh, D.; Mistry, T.; Ghosh, S.; Bera, T.; Nasare, V.D. Mitotic checkpoint defects: En route to cancer and drug resistance. Chromosom. Res. 2021, 29, 131–144. [Google Scholar] [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Pollok, J.; Tinguely, P.; Berenguer, M.; Niemann, C.U.; Raptis, D.A.; Spiro, M.; Mayr, A.; Dominguez, B.; Muller, E.; Rando, K.; et al. Enhanced recovery for liver transplantation: Recommendations from the 2022 International Liver Transplantation Society consensus conference. Lancet Gastroenterol. Hepatol. 2023, 8, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Cui, D.; Wang, J.; Qin, J.; Wang, S.; Wang, Z.; Zhai, X.; Ma, H.; Ma, D.; Liu, Y.; et al. Overexpression of HMGA1 confers radioresistance by transactivating RAD51 in cholangiocarcinoma. Cell Death Discov. 2021, 7, 322. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.-H.; Huang, W.-C.; Kuo, K.-T.; Yuan, R.-H.; Chen, Y.-L.; Jeng, Y.-M. S100P immunostaining identifies a subset of peripheral-type intrahepatic cholangiocarcinomas with morphological and molecular features similar to those of perihilar and extrahepatic cholangiocarcinomas. Histopathology 2012, 61, 1106–1116. [Google Scholar] [CrossRef]
- Sutphen, C.L.; McCue, L.; Herries, E.M.; Xiong, C.; Ladenson, J.H.; Holtzman, D.M.; Fagan, A.M.; Adni, O.B.O. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 869–879. [Google Scholar] [CrossRef]
- Luo, X.; Hou, L.; Shi, H.; Zhong, X.; Zhang, Y.; Zheng, D.; Tan, Y.; Hu, G.; Mu, N.; Chan, J.; et al. CSF levels of the neuronal injury biomarker visinin-like protein-1 in Alzheimer’s disease and dementia with Lewy bodies. J. Neurochem. 2013, 127, 681–690. [Google Scholar] [CrossRef]
- Korshunov, A.; Okonechnikov, K.; Sahm, F.; Ryzhova, M.; Stichel, D.; Schrimpf, D.; Ghasemi, D.R.; Pajtler, K.W.; Antonelli, M.; Donofrio, V.; et al. Transcriptional profiling of medulloblastoma with extensive nodularity (MBEN) reveals two clinically relevant tumor subsets with VSNL1 as potent prognostic marker. Acta Neuropathol. 2019, 139, 583–596. [Google Scholar] [CrossRef]
- Dai, Q.; Wang, Y.; Jiang, Y.; Li, L.; Wang, H. VSNL1 Promotes Gastric Cancer Cell Proliferation and Migration by Regulating P2X3/P2Y2 Receptors and Is a Clinical Indicator of Poor Prognosis in Gastric Cancer Patients. Gastroenterol. Res. Pract. 2020, 2020, 1–9. [Google Scholar] [CrossRef]
- Akagi, T.; Hijiya, N.; Inomata, M.; Shiraishi, N.; Moriyama, M.; Kitano, S. Visinin-like protein-1 overexpression is an indicator of lymph node metastasis and poor prognosis in colorectal cancer patients. Int. J. Cancer 2011, 131, 1307–1317. [Google Scholar] [CrossRef]
- Harashima, S.; Wang, Y.; Horiuchi, T.; Seino, Y.; Inagaki, N. Purkinje cell protein 4 positively regulates neurite outgrowth and neurotransmitter release. J. Neurosci. Res. 2011, 89, 1519–1530. [Google Scholar] [CrossRef]
- Kobuke, K.; Oki, K.; Gomez-Sanchez, C.; Ohno, H.; Itcho, K.; Yoshii, Y.; Yoneda, M.; Hattori, N. Purkinje Cell Protein 4 Expression Is Associated with DNA Methylation Status in Aldosterone-Producing Adenoma. J. Clin. Endocrinol. Metab. 2017, 103, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Kim, H.; Ji, Z.; Zhang, T.; Chen, B.; Ge, Y.; Hu, Y.; Feng, X.; Han, X.; Xu, H.; et al. The BUB3-BUB1 Complex Promotes Telomere DNA Replication. Mol. Cell 2018, 70, 395–407. [Google Scholar] [CrossRef]
- Ding, Y.; Hubert, C.; Herman, J.; Corrin, P.; Toledo, C.M.; Skutt-Kakaria, K.; Vazquez, J.; Basom, R.; Zhang, B.; Risler, J.K.; et al. Cancer-Specific Requirement for BUB1B/BUBR1 in Human Brain Tumor Isolates and Genetically Transformed Cells. Cancer Discov. 2013, 3, 198–211. [Google Scholar] [CrossRef]
- Hadders, M.; Hindriksen, S.; Truong, M.A.; Mhaskar, A.N.; Wopken, J.P.; Vromans, M.J.; Lens, S.M. Untangling the contribution of Haspin and Bub1 to Aurora B function during mitosis. J. Cell Biol. 2020, 219, e201907087. [Google Scholar] [CrossRef]
- Izumi, H.; Matsumoto, Y.; Ikeuchi, T.; Saya, H.; Kajii, T.; Matsuura, S. BubR1 localizes to centrosomes and suppresses centrosome amplification via regulating Plk1 activity in interphase cells. Oncogene 2009, 28, 2806–2820. [Google Scholar] [CrossRef]
- Piao, J.; Zhu, L.; Sun, J.; Li, N.; Dong, B.; Yang, Y.; Chen, L. High expression of CDK1 and BUB1 predicts poor prognosis of pancreatic ductal adenocarcinoma. Gene 2019, 701, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Hu, C.; Ning, Z.; Huang, J.; Zhu, Z. Identification of key genes controlling cancer stem cell characteristics in gastric cancer. World J. Gastrointest. Surg. 2020, 12, 442–459. [Google Scholar] [CrossRef] [PubMed]
- Fujibayashi, Y.; Isa, R.; Nishiyama, D.; Sakamoto-Inada, N.; Kawasumi, N.; Yamaguchi, J.; Kuwahara-Ota, S.; Matsumura-Kimoto, Y.; Tsukamoto, T.; Chinen, Y.; et al. Aberrant BUB1 Overexpression Promotes Mitotic Segregation Errors and Chromosomal Instability in Multiple Myeloma. Cancers 2020, 12, 2206. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, X.; Liu, Z.; Wang, W.; Li, F.; Fu, W. Characteristic Analysis of Featured Genes Associated with Stemness Indices in Colorectal Cancer. Front. Mol. Biosci. 2020, 7, 563922. [Google Scholar] [CrossRef]
- Jiao, C.; Feng, Q.; Li, C.X.; Wang, D.; Han, S.; Zhang, Y.D.; Jiang, W.J.; Chang, J.; Wang, X.; Li, X.C. BUB1B promotes extrahepatic cholangiocarcinoma progression via JNK/c-Jun pathways. Cell Death Dis. 2021, 12, 63. [Google Scholar] [CrossRef]
- Weng, Y.; Liang, W.; Ji, Y.; Li, Z.; Jia, R.; Liang, Y.; Ning, P.; Xu, Y. Key Genes and Prognostic Analysis in HER2+ Breast Cancer. Technol. Cancer Res. Treat. 2021, 20. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhang, S.; Wang, P.; Wang, H.; Sha, B.; Peng, H.; Ju, Z.; Rao, J.; Lu, L. BUB1B promotes hepatocellular carcinoma progression via activation of the mTORC1 signaling pathway. Cancer Med. 2020, 9, 8159–8172. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Q.; Chen, W.; Gao, F.; Wu, Y.; Zhou, L.; Xu, H.; Yu, J.; Zhu, X.; Wang, L.; Li, L.; et al. Characteristic Analysis of Featured Genes Associated with Cholangiocarcinoma Progression. Biomedicines 2023, 11, 847. https://doi.org/10.3390/biomedicines11030847
Yao Q, Chen W, Gao F, Wu Y, Zhou L, Xu H, Yu J, Zhu X, Wang L, Li L, et al. Characteristic Analysis of Featured Genes Associated with Cholangiocarcinoma Progression. Biomedicines. 2023; 11(3):847. https://doi.org/10.3390/biomedicines11030847
Chicago/Turabian StyleYao, Qigu, Wenyi Chen, Feiqiong Gao, Yuchen Wu, Lingling Zhou, Haoying Xu, Jong Yu, Xinli Zhu, Lan Wang, Lanjuan Li, and et al. 2023. "Characteristic Analysis of Featured Genes Associated with Cholangiocarcinoma Progression" Biomedicines 11, no. 3: 847. https://doi.org/10.3390/biomedicines11030847
APA StyleYao, Q., Chen, W., Gao, F., Wu, Y., Zhou, L., Xu, H., Yu, J., Zhu, X., Wang, L., Li, L., & Cao, H. (2023). Characteristic Analysis of Featured Genes Associated with Cholangiocarcinoma Progression. Biomedicines, 11(3), 847. https://doi.org/10.3390/biomedicines11030847