
Scandium-44 Radiolabeled Peptide and Peptidomimetic Conjugates Targeting Neuropilin-1 Co-Receptor as Potential Tools for Cancer Diagnosis and Anti-Angiogenic Therapy

 , , , ,
, , , ,  and
and
Abstract
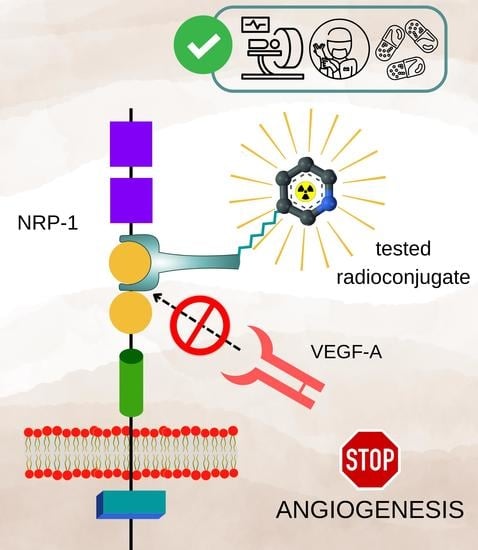
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Analytical Methods
2.2.2. Syntheses
Preparation of [44Sc]Sc-DOTA-Ahx-A7R (44Sc-1), [44Sc]Lys(DOTA-Sc)-A7R (44Sc-1bis), [44Sc]Sc-DOTA-Ahx-K4R (44Sc-2) and [44Sc]DR7A-DLys(DOTA-Sc) (44Sc-3)
Preparation of [68Ga]Lys(DOTA-Ga)-A7R (68Ga-1bis) and [68Ga]DR7A-DLys(DOTA-Ga) (68Ga-3)
Preparation of [177Lu]Lys(DOTA-Lu)-A7R (177Lu-1bis) and [177Lu]DR7A-DLys(DOTA-Lu) (177Lu-3)
Preparation of Cold Reference Compounds Sc-DOTA-Ahx-A7R (Sc-1), Lys(DOTA-Sc)-A7R (Sc-1bis), Lys(hArg)-Dab(Ahx-DOTA-Sc)-Pro-Arg (Sc-2), DR7A-DLys(DOTA-Sc) (Sc-3) and DR7A-DLys(DOTA-Ga) (Ga-3) for ELISA Tests
2.2.3. Physicochemical Properties Study of the Radioconjugates
Radioconjugate Stability Tests
Lipophilicity Test
2.2.4. Competitive NRP-1-Binding Inhibition Assay
3. Results
3.1. Syntheses of Conjugates
3.2. Syntheses of Radioconjugates
3.3. Radioconjugates Stability Studies in PBS Buffer, Cysteine, Histidine and Human Serum
3.4. Lipophilicity Study
3.5. Syntheses of Cold Reference Compounds
3.6. Biological Activity Study with ELISA Competitive Test
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A7R | Ala-Thr-Trp-Leu-Pro-Pro-Arg peptide |
| AAT | Anti-Angiogenic Therapy |
| ADME | administration, distribution, metabolism, excretion |
| Ahx | 6-aminohexanoic acid |
| Alloc | Allyloxycarbonyl group |
| Boc | Tert-butyloxycarbonyl group |
| BSA | Bovine Serum Albumin |
| CendR | C-End-Rule |
| Cys | Cysteine |
| DR7A | DArg-DPro-DPro-DLeu-DTrp-DThr-DAla peptide |
| Dab | 2,4-Diaminobutyric Acid |
| DCM | Dichloromethane |
| DIC | N,N’-Di(propan-2-yl)methanediimine |
| DMF | N,N-Dimethylformamide |
| DOTA | 2,2′,2″,2″-(1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetrayl) tetraacetic acid |
| DOTA-tris(tBu)-NHS | tri-tert-butyl 2,2′,2″-(10-(2-((2,5-dioxopyrrolidin-1-yl)oxy)-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate |
| ECL | Enhanced chemiluminescence |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| ESI-MS | Electrospray ionization mass spectrometry analyses |
| Et3N | Triethylamine |
| Fmoc | 9-fluorenylmethoxycarbonyl group |
| hArg | Homoarginine |
| HCl | Hydrogen chloride |
| His | Histidine |
| HOBt | 1H-1,2,3-Benzotriazol-1-ol |
| HS | Human serum |
| IC50 | Half maximal inhibitory concentration |
| K4R | Lys(hArg)-Dab-Pro-Arg peptidomimetic |
| logP | Decimal logarithm of partition coefficient |
| m/z | Mass-to-charge ratio |
| NRP-1 | Neuropilin-1 |
| NS | Negative control |
| P | Partition coefficient |
| P | Positive control |
| Pbf | Pentamethyl-2,3-dihydrobenzofuran-5-sulfonyl group |
| PBS | Phosphate-buffered saline |
| PET | Positron Emission Tomography |
| PhOH | Phenol |
| PTFE | Polytetrafluoroethylene membrane microfilter |
| R/KXXR/K | Amino acid sequence containing: R-Arginine, K-Lysine, X- any amino acid |
| RP-HPLC | Reverse-Phase High Pressure Liquid Chromatography |
| RT | Retention time |
| S | Measured signal |
| SD | Standard Deviation |
| SPPS | Solid Phase Peptide Synthesis |
| t1/2 | Radionuclide half-life time |
| tBu | Tert-butyl group |
| TFA | Trifluoroacetic acid |
| TIPS | Tri(propan-2-yl)silane |
| TRNT | Targeted Radionuclide Therapy |
| VEGF | Vascular Endothelial Growth Factor |
| VEGF-A165 | Vascular Endothelial Growth Factor-A165 |
| VEGFR-2 | Vascular Endothelial Growth Factor Receptor 2 |
| β+ Emax | Beta emitter with a maximum energy |
| (bt)-VEGF-A165 | Biotinylated VEGF-A165 |
| (p,n) | Proton-neutron nuclear reaction |
References
- Folkman, J. History of angiogenesis. In Angiogenesis: An Integrative Approach from Science to Medicine; Figg, W.D., Folkman, J., Eds.; Springer: New York, NY, USA, 2008; pp. 1–14. [Google Scholar] [CrossRef]
- Hall, A.P. The role of angiogenesis in cancer. Comp. Clin. Path. 2005, 13, 95–99. [Google Scholar] [CrossRef]
- Ribatti, D.; Crivellato, E. “Sprouting angiogenesis”, a reappraisal. Dev. Biol. 2012, 372, 157–165. [Google Scholar] [CrossRef]
- Ribatti, D. Judah Folkman, a pioneer in the study of angiogenesis. Angiogenesis 2008, 11, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: From bench to bedside. In Tumour Angiogenesis: Basic Mechanisms and Cancer Therapy; Marmé, D., Fusenig, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 3–28. [Google Scholar] [CrossRef]
- Murukesh, N.; Dive, C.; Jayson, G.C. Biomarkers of angiogenesis and their role in the development of VEGF inhibitors. Br. J. Cancer 2010, 102, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Backer, M.V.; Backer, J.M. Imaging key biomarkers of tumor angiogenesis. Theranostics 2012, 2, 502. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Chen, X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr. Drug Targets 2010, 11, 1000–1017. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, F.S.; Prota, A.E.; Giese, A.; Ballmer-Hofer, K. Structure–function analysis of VEGF receptor activation and the role of coreceptors in angiogenic signaling. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2010, 1804, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, S.; Driscoll, P.C. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov. 2013, 18, 447–455. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Niland, S.; Eble, J.A. Neuropilin: Handyman and Power Broker in the Tumor Microenvironment. In Tumor Microenvironment. Advances in Experimental Medicine and Biology; Birbrair, A., Ed.; Springer: Cham, Switzerland, 2020; Volume 1223, pp. 31–67. [Google Scholar] [CrossRef]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular pharmacology of VEGF-A isoforms: Binding and signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [PubMed]
- Grandclement, C.; Borg, C. Neuropilins: A new target for cancer therapy. Cancers 2011, 3, 1899–1928. [Google Scholar] [CrossRef]
- Ellis, L.M. The role of neuropilins in cancer. Mol. Cancer Ther. 2006, 5, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Bai, Y.; Zhu, Q.; Hu, B.; Xu, Y. Targeting VEGF–neuropilin interactions: A promising antitumor strategy. Drug Discov. 2019, 24, 656–664. [Google Scholar] [CrossRef]
- Kiselyov, A.; Balakin, K.V.; Tkachenko, S.E. VEGF/VEGFR signalling as a target for inhibiting angiogenesis. Expert Opin. Investig. Drugs 2007, 16, 83–107. [Google Scholar] [CrossRef]
- Masłowska, K.; Halik, P.K.; Tymecka, D.; Misicka, A.; Gniazdowska, E. The Role of VEGF receptors as molecular target in nuclear medicine for cancer diagnosis and combination therapy. Cancers 2021, 13, 1072. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Chen, X. Multimodality molecular imaging of tumor angiogenesis. J. Nucl. Med. 2008, 49 (Suppl. S2), 113S–128S. [Google Scholar] [CrossRef]
- Lu, L.; Chen, H.; Hao, D.; Zhang, X.; Wang, F. The functions and applications of A7R in anti-angiogenic therapy, imaging and drug delivery systems. Asian J. Pharm. Sci. 2019, 14, 595–608. [Google Scholar] [CrossRef]
- Majkowska-Pilip, A.; Bilewicz, A. Macrocyclic complexes of scandium radionuclides as precursors for diagnostic and therapeutic radiopharmaceuticals. J. Inorg. Biochem. 2011, 105, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Domnanich, K.A.; Umbricht, C.A.; van der Meulen, N.P. Scandium and terbium radionuclides for radiotheranostics: Current state of development towards clinical application. Br. J. Radiol. 2018, 91, 20180074. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, S.; Cydzik, I.; Abbas, K.; Bulgheroni, A.; Simonelli, F.; Holzwarth, U.; Bilewicz, A. Cyclotron production of 44Sc for clinical application. Radiochim. Acta 2013, 101, 333–338. [Google Scholar] [CrossRef]
- Roesch, F. Scandium-44: Benefits of a long-lived PET radionuclide available from the 44Ti/44Sc generator system. Curr. Radiopharm. 2012, 5, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Walczak, R.; Krajewski, S.; Szkliniarz, K.; Sitarz, M.; Abbas, K.; Choiński, J.; Jakubowski, A.; Jastrzębski, J.; Majkowska, A.; Simonelli, F.; et al. Cyclotron production of 43Sc for PET imaging. EJNMMI Phys. 2015, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Xu, P.; Li, X.; Vander Kooi, C.W. Structural basis for selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1. J. Biol. Chem. 2012, 287, 11082–11089. [Google Scholar] [CrossRef]
- Vander Kooi, C.W.; Jusino, M.A.; Perman, B.; Neau, D.B.; Bellamy, H.D.; Leahy, D.J. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. USA 2007, 104, 6152–6157. [Google Scholar] [CrossRef]
- Soker, S.; Miao, H.Q.; Nomi, M.; Takashima, S.; Klagsbrun, M. VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J. Cell. Biochem. 2002, 85, 357–368. [Google Scholar] [CrossRef]
- Guo, H.F.; Vander Kooi, C.W. Neuropilin functions as an essential cell surface receptor. J. Biol. Chem. 2015, 290, 29120–29126. [Google Scholar] [CrossRef]
- Zanuy, D.; Kotla, R.; Nussinov, R.; Teesalu, T.; Sugahara, K.N.; Alemán, C.; Haspel, N. Sequence dependence of C-end rule peptides in binding and activation of neuropilin-1 receptor. J. Struct. Biol. 2013, 182, 78–86. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef]
- Tymecka, D.; Lipiński, P.F.; Fedorczyk, B.; Puszko, A.; Wileńska, B.; Perret, G.Y.; Misicka, A. Structure-activity relationship study of tetrapeptide inhibitors of the Vascular Endothelial Growth Factor A binding to Neuropilin-1. Peptides 2017, 94, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Starzec, A.; Vassy, R.; Martin, A.; Lecouvey, M.; Di Benedetto, M.; Crépin, M.; Perret, G.Y. Anti-angiogenic and antitumor activities of peptide inhibiting the vascular endothelial growth factor binding to neuropilin-1. Life Sci. 2006, 79, 2370–2381. [Google Scholar] [CrossRef] [PubMed]
- Starzec, A.; Ladam, P.; Vassy, R.; Badache, S.; Bouchemal, N.; Navaza, A.; Hervé du Penhoat, C.; Perret, G.Y. Structure–function analysis of the antiangiogenic ATWLPPR peptide inhibiting VEGF165 binding to neuropilin-1 and molecular dynamics simulations of the ATWLPPR/neuropilin-1 complex. Peptides 2007, 28, 2397–2402. [Google Scholar] [CrossRef]
- Binetruy-Tournaire, R.; Demangel, C.; Malavaud, B.; Vassy, R.; Rouyre, S.; Kraemer, M.; Plouët, J.; Derbin, C.; Perret, G.Y.; Mazie, J.C. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J. 2000, 19, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Tymecka, D.; Puszko, A.K.; Lipiński, P.F.; Fedorczyk, B.; Wilenska, B.; Sura, K.; Perret, G.Y.; Misicka, A. Branched pentapeptides as potent inhibitors of the vascular endothelial growth factor 165 binding to Neuropilin-1: Design, synthesis and biological activity. Eur. J. Med. Chem. 2018, 158, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Shen, Q.; Liu, Y.; Yan, Z.; Wei, X.; Zhan, C.; Gao, J.; Xie, C.; Yao, B.; Lu, W. Stabilized heptapeptide A7R for enhanced multifunctional liposome-based tumor-targeted drug delivery. ACS Appl. Mater. Interfaces 2016, 8, 13232–13241. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Dong, Q.; Zhai, W.; Zhao, W.; Shi, P.; Wu, Y.; Zhou, X.; Gao, Y. A PD-L1 and VEGFR2 dual targeted peptide and its combination with irradiation for cancer immunotherapy. Pharmacol. Res. 2022, 182, 106343. [Google Scholar] [CrossRef]
- Masłowska, K.; Witkowska, E.; Tymecka, D.; Halik, P.K.; Misicka, A.; Gniazdowska, E. Synthesis, Physicochemical and Biological Study of Gallium-68-and Lutetium-177-Labeled VEGF-A165/NRP-1 Complex Inhibitors Based on Peptide A7R and Branched Peptidomimetic. Pharmaceutics 2022, 14, 100. [Google Scholar] [CrossRef]
- Minegishi, K.; Nagatsu, K.; Fukada, M.; Suzuki, H.; Ohya, T.; Zhang, M.R. Production of scandium-43 and -47 from a powdery calcium oxide target via the nat/44Ca(α,x)-channel. Appl. Radiat. Isot. 2016, 116, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Pruszyński, M.; Loktionova, N.S.; Filosofov, D.V.; Rösch, F. Post-elution processing of 44Ti/44Sc generator-derived 44Sc for clinical application. Appl. Radiat. Isot. 2010, 68, 1636–1641. [Google Scholar] [CrossRef] [PubMed]
- Puszko, A.K.; Sosnowski, P.; Tymecka, D.; Raynaud, F.; Hermine, O.; Lepelletier, Y.; Misicka, A. Neuropilin-1 peptide-like ligands with proline mimetics, tested using the improved chemiluminescence affinity detection method. MedChemComm 2019, 10, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Q.; Borriello, L.; Allain, B.; Pavoni, S.; Lopez, N.; Hermine, O.; Garbay, C.; Raynaud, F.; Lepelletier, Y.; Demange, L. New Peptides Structurally Related to VEGF-A165 Exon-7 and-8 Encoded Domains Antagonize Its Binding to NRP-1 and VEGF-R1. Int. J. Pept. Res. Ther. 2015, 21, 117–124. [Google Scholar] [CrossRef]
- Su, H.; Zhao, L.; Yu, B.; Zeng, H.; Yang, J.; Zhu, M.; Zhao, J. Preparation and bioevaluation of [99mTc]Tc-labeled A7R and DA7R for SPECT imaging of triple-negative breast cancer. New J. Chem. 2022, 46, 21401–21408. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (Radio)compound | RP-HPLC System | RT [min] | logP ± SD |
|---|---|---|---|
| 44Sc-1 | 3 | 11.00 | −3.44 ± 0.02 |
| 44Sc-1bis | 3 | 10.68 | −3.56 ± 0.06 |
| 44Sc-2 | 4 | 10.43 | −2.57 ± 0.02 |
| 44Sc-3 | 3 | 10.87 | −3.61 ± 0.03 |
| 68Ga-3 | 3 | 10.72 | −4.11 ± 0.06 |
| 177Lu-3 | 3 | 10.28 | −4.48 ± 0.05 |
| 68Ga-1bis | 3 | 10.32 | −4.18 ± 0.07 |
| 177Lu-1bis | 3 | 10.38 | −4.45 ± 0.03 |
| 68Ga-1 * | 3 | 11.27 | −3.92 ± 0.03 |
| 177Lu-1 * | 3 | 11.08 | −3.40 ± 0.14 |
| 68Ga-2 * | 4 | 10.63 | −4.57 ± 0.05 |
| 177Lu-2 * | 4 | 10.78 | −3.75 ± 0.08 |
| Sc-1 | 3 | 10.71 | ----- |
| Sc-1bis | 3 | 10.26 | ----- |
| Sc-2 | 4 | 10.46 | ----- |
| Sc-3 | 3 | 10.60 | ----- |
| Ga-3 | 3 | 10.45 | ----- |
| Biomolecule | Sequence | IC50 ± SD | logIC50 ± SD |
|---|---|---|---|
| or Reference Compound | [µM] | ||
| A7R | Ala-Thr-Trp-Leu-Pro-Pro-Arg | 17.47 ± 1.49 | −4.74 ± 0.02 |
| Sc-1 | Sc-DOTA-Ahx-A7R | 22.48 ± 6.56 | −4.65 ± 0.12 |
| Sc-1bis | Lys(DOTA-Sc)-A7R | 18.08 ± 0.46 | −4.74 ± 0.01 |
| K4R | Lys(hArg)-Dab-Pro-Arg | 4.83 ± 0.25 | −5.32 ± 0.12 |
| Sc-2 | Sc-DOTA-Ahx-K4R | 17.93 ± 1.80 | −4.73 ± 0.03 |
| DR7A | dArg-dPro-dPro-dLeu-dTrp-dThr-dAla | >100 | n/a |
| Ga-3 | DR7A-DLys(DOTA-Ga) | >100 | n/a |
| Sc-3 | DR7A-DLys(DOTA-Sc) | >100 | n/a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masłowska, K.; Redkiewicz, P.; Halik, P.K.; Witkowska, E.; Tymecka, D.; Walczak, R.; Choiński, J.; Misicka, A.; Gniazdowska, E. Scandium-44 Radiolabeled Peptide and Peptidomimetic Conjugates Targeting Neuropilin-1 Co-Receptor as Potential Tools for Cancer Diagnosis and Anti-Angiogenic Therapy. Biomedicines 2023, 11, 564. https://doi.org/10.3390/biomedicines11020564
Masłowska K, Redkiewicz P, Halik PK, Witkowska E, Tymecka D, Walczak R, Choiński J, Misicka A, Gniazdowska E. Scandium-44 Radiolabeled Peptide and Peptidomimetic Conjugates Targeting Neuropilin-1 Co-Receptor as Potential Tools for Cancer Diagnosis and Anti-Angiogenic Therapy. Biomedicines. 2023; 11(2):564. https://doi.org/10.3390/biomedicines11020564
Chicago/Turabian StyleMasłowska, Katarzyna, Patrycja Redkiewicz, Paweł Krzysztof Halik, Ewa Witkowska, Dagmara Tymecka, Rafał Walczak, Jarosław Choiński, Aleksandra Misicka, and Ewa Gniazdowska. 2023. "Scandium-44 Radiolabeled Peptide and Peptidomimetic Conjugates Targeting Neuropilin-1 Co-Receptor as Potential Tools for Cancer Diagnosis and Anti-Angiogenic Therapy" Biomedicines 11, no. 2: 564. https://doi.org/10.3390/biomedicines11020564
APA StyleMasłowska, K., Redkiewicz, P., Halik, P. K., Witkowska, E., Tymecka, D., Walczak, R., Choiński, J., Misicka, A., & Gniazdowska, E. (2023). Scandium-44 Radiolabeled Peptide and Peptidomimetic Conjugates Targeting Neuropilin-1 Co-Receptor as Potential Tools for Cancer Diagnosis and Anti-Angiogenic Therapy. Biomedicines, 11(2), 564. https://doi.org/10.3390/biomedicines11020564