Abstract
Carnosic acid (CA) and carnosol (CAR) are two major diterpenes of the rosemary plant (Rosmarinus officinalis). They possess a phenolic structural moiety and are endowed with the power to remove cellular reactive oxygen species (ROS) either through direct scavenging reaction or indirectly through upregulation of antioxidant defences. Hand in hand with these activities are their multiple biological effects and therapeutic potential orchestrated through modulating various signalling pathways of inflammation, including the NF-κB, MAPK, Nrf2, SIRT1, STAT3 and NLRP3 inflammasomes, among others. Consequently, they ameliorate the expression of pro-inflammatory cytokines (e.g., TNF-α, IL-1 and IL-6), adhesion molecules, chemokines and prostaglandins. These anti-inflammatory mechanisms of action as a therapeutic link to various effects of these compounds, as in many other natural products, are scrutinised.
Keywords:
rosemary; diterpenes; carnosic acid; carnosol; inflammation; MAPK; NLRP3 inflammasome; Nrf2; NF-κB; SIRT1; STAT3 1. Introduction
Any immune response to tissue insult resulting from infection or physical/chemical injury involves an initial stage of pro-inflammatory activity followed by a period of resolution of inflammation called the anti-inflammatory phase. The regulated process of inflammation allows the body to clear pathogens, dead cells and debris, by which means a normal tissue structure and homeostasis are sustained. Acute inflammation is largely a function of resident immune cells (dendritic cells, macrophages, mast cells, etc.) and is characterised by a temporal and limited upregulation of inflammatory activity followed by prompt resolution once the insult stimulus is removed. The classic example of an acute inflammatory response is that initiated through infection by pathogens. The process involves the interaction of pathogen-associated molecular patterns (PAMPs) expressed on pathogens with host immune cells’ receptors called pattern recognition receptors (PRRs) or danger-associated molecular pattern (DAMP) receptors. The PPRs include the Toll-like receptors (TLRs) as well as nucleotide-binding oligomerisation domain (NOD)-like receptors (NLR) (and mannose-binding lectin (MBL)) [1,2,3], which are all activated by various extracellular bacterial (e.g., endotoxins) and viral products [4] as well as cellular molecules (e.g., uric acid, ATP and DNA) released from damaged cells [5]. We also have non-TLRs as PRRs that activate the innate immune complexes, the inflammasome system [6,7]. Any physical, chemical or inflammatory signal of metabolic origin can also initiate an acute inflammatory response due to the release of DAMPs [8]. Meanwhile, chronic inflammatory conditions constitute long-term inflammatory activity that involves continuous recruitment and infiltration of leucocytes at the site of injury. This results in major structural and physiological alterations in the tissues and organs involved, resulting in diseases such as arthritis, asthma, type 2 diabetes (T2D) and autoimmune, cardiovascular and neurodegenerative (e.g., Alzheimer’s and Parkinson’s) diseases. The most prominent mediators of chronic inflammation are pro-inflammatory cytokines such as interleukin (IL)-1α, IL-1α, IL-1β, IL-6 and tumour necrosis factor-α (TNF-α). Not surprisingly, the most promising current therapeutic strategies for treating chronic inflammatory conditions are based on antibodies targeting pro-inflammatory cytokines (e.g., [9,10]) such as TNF-α (infliximab, adalimumab, etanercept, golimumab and certolizumab), IL-6 (targeting its receptor with tocilizumab or sarilumab or targeting the cytokine with siltuximab) and IL-1 (targeting its receptor with anakinra or rilonacept by binding with IL-1α and IL-1β, and canakinumab as an anti-IL-1β antibody). Other mediators include lipids of cyclooxygenase-2 (COX-2) products such as prostaglandin-E2 (PGE2), chemokines (proteins, lipids and other origins) and nitric oxide (NO) as a product of the inducible NO synthase (iNOS), among others. Antagonists of lipid mediators among the old-generation aspirin-like drugs (nonselective COX inhibitors) and the latest selective inhibitors (COX-2 inhibitors), as well as lipoxygenase inhibitors, are common anti-inflammatory agents on the market, though they are mostly of limited use for chronic inflammatory diseases. Following the release of inflammatory mediators, adhesion molecules both on leucocytes and endothelial cell surfaces are activated/upregulated, resulting in extravascular immigration of leucocytes, which also involves leucocytes’ interaction with extracellular matrix (ECM) proteins. Given the crucial role of leucocyte adhesion in the pathology of chronic inflammatory diseases, therapeutic strategies using antibodies are also common (e.g., natalizumab against human α4 integrin; see also the review by Slack et al. [11]). Linking the expression of inflammatory mediators to activation of PRRs are drug targets of the signal transduction pathways such as the mitogen-activated protein kinase (MAPK) [12] and transcription factors [13], including the nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells (NF-κB). All these pro-inflammatory pathways, as well as those of inflammation resolution, such as that by IL-10 production [14], are subject to modulation by anti-inflammatory agents including natural products. In the following sections, the therapeutic potential of the two most abundant rosemary diterpenes, carnosic acid (CA) and carnosol (CAR), orchestrated through multiple anti-inflammatory mechanisms is discussed.
2. Phytochemical Overview
The chemistry and biosynthesis route of carnosic acid and carnosol (Figure 1) have been outlined previously [15]. They belong to the labdane type of diterpenes of the 20-carbon skeleton. What is unique about these compounds is that they possess a phenolic structural moiety which is common in flavonoids and other aromatic compounds of the shikimic acid and acetate metabolic pathways in plants. Hence, they are commonly known for their antioxidant properties as they can directly interact with reactive oxygen species (ROS). These two compounds are the major constituents of the aerial parts of rosemary, which also contain other related diterpenes including glycoside forms at minor concentrations [15]. They have a very limited distribution in plants and have been isolated from few genera of the Lamiaceae family (including Salvia (sage such as S. officinalis), Rosmarinus, Lepechinia, Oreganum and Thymus) [16]. The conversion of CA to carnosol under various stressful conditions has been noted [17], and exposure to high temperatures such as under cooking conditions can also lead to the production of a range of other structurally related phenols and quinones [18]. The two major diterpenes of rosemary (CA and CAR) have diverse pharmacological effects, but only their molecular pharmacology related to anti-inflammatory mechanisms of action is discussed in this communication.
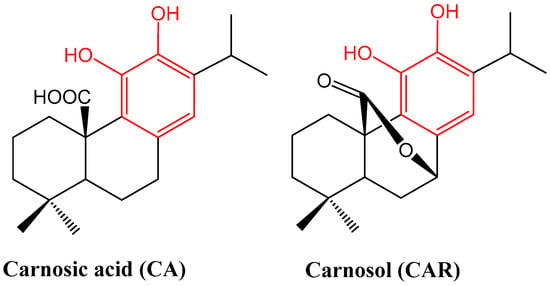
Figure 1.
Structures of carnosic acid and carnosol. One common structural moiety for the two compounds is the orthodihydroxybenzene (catechol) group shown in red. This makes them similar to polyphenolic compounds such as flavonoids, which are known for their general antioxidant properties. They are also made from the 20-carbon diterpene skeleton. The concentration of CA can be up to 5-fold higher than CAR, but oxidation under various conditions can transform it into other products including CAR [17].
3. Methodology
Rosemary as well as its most abundant diterpenes CA and CAR are among the most investigated natural products. A literature search in PUBMED using the keyword “carnosic acid” returned 986 hits, and for “carnosol”, 393. The hit rate on ScienceDirect is higher at 2212 and 1706, respectively. For this review, the search hits were filtered by using additional keywords “inflammation”, “anti-inflammatory” and “anti-inflammatory” and included entries up to 10 January 2023. The findings were grouped under sections arranged by inflammatory mechanisms such as leucocyte activation, as leucocytes are among the major targets for anti-inflammatory drugs, and disease types such as lung, skin, cardiac, renal, hepatic, neuronal, endothelial, diabetes- and obesity-associated diseases and gut inflammation. The key findings associated with the outlined mechanisms of action are summarised in two tables as in vitro (Table 1) and in vivo (Table 2) outcomes.

Table 1.
Overview of anti-inflammatory mechanisms of CA and CA based on in vitro studies.
Table 2.
Overview of anti-inflammatory mechanisms of CA and CA based on in vivo studies.
4. Mechanisms Related to Macrophages’ and Other Immune Cells’ Activation
Macrophages play a key role in the immune response and can be induced to polarise into a pro-inflammatory phenotype by stimulation with a variety of agents such as bacterial lipopolysaccharide (LPS) and IFN-γ. This is a feature where they release pro-inflammatory cytokines such as IL-1, IL-6 and TNF-α, which are markers of chronic inflammatory diseases. They also release anti-inflammatory cytokines such as IL-10, which are relevant to the restoration stage of wound healing or the late stage of inflammation. The signalling cascades of these pro- and anti-inflammatory mechanisms and modulation by therapeutic agents are routinely assessed using macrophages and other immune cell cultures in vitro. These assessments are presented in this section along with some in vivo studies confirming the therapeutic potential of CA and CAR.
Several studies employed the transformed cell line RAW 264.7 cells as a model of macrophage activation where the potential anti-inflammatory effect of rosemary diterpenes was evaluated. In the study by Oh et al. [19], CA (5–20 μg/mL) was shown to inhibit the release of nitric oxide (NO), TNF-α and prostaglandin E₂ (PGE₂) from stimulated RAW 264.7 cells. As a stimulus, they used Toll-like receptor (TLR)-2 ligands, Gram-positive-bacteria-derived peptidoglycan (PGN), pam3CSK (TLR2/TLR1 ligand) and the TLR4 ligand and Gram-negative-bacteria-derived lipopolysaccharide (LPS). CA blocked the nuclear translocation of NF-κB and its upstream signalling including Syk/Src, phosphoinositide 3-kinase (PI3K), Akt (protein kinase B), inhibitor of κBα (IκBα) kinase (IKK) and IκBα for NF-κB activation. Kinase assays revealed that Syk could be a direct enzymatic target for CA in its anti-inflammatory action. Meanwhile, a direct effect of CA on bacteria should not be ruled out as the compound arrested the growth of dermatitis-inducing Gram-positive and Gram-negative microorganisms such Propionibacterium acnes, Pseudomonas aeruginosa and Staphylococcus aureus. In the same concentration range (up to 10 μM), both CA and CAR were also shown to inhibit the secretion of matrix metallopeptidase 9 (MMP-9), and monocyte chemoattractant protein-1 (MCP-1) from LPS stimulated RAW 264.7 cells’ as well as TNF-α-stimulated rat vascular smooth muscle cells’ (VSMCs’) activity in a cell migration assay in vitro [20]. The production of NO from LPS-stimulated RAW 264.7 cells in vitro was also shown to be suppressed both by CA and CAR within the concentration range of 12.5–50 μg/mL [21]. Wang et al. [22] also investigated the signalling pathway associated with the anti-inflammatory activity of CA (2.5–20 μM) in LPS-stimulated RAW 264.7 cells. In addition to a reduction in the levels of NO and TNF-α, the compound also downregulated cyclooxygenase-2 (COX-2) protein expression, as well as the transcriptional level of inflammatory genes including Nos2, Tnfα, Cox2 and Mcp1. Moreover, the mitogen-activated protein kinases (MAPKs) including extracellular signal-regulated kinase 1/2 (ERK), Jun N-terminal kinase (JNK) and p38, along with NF-κB, and FoxO signalling pathways, were suppressed, as evidenced by the inactivation of IKKβ/IκB-α/NF-κB, MAPKs and FoxO1/3. A similar study for CAR was conducted by Lo et al. [23] using the LPS-stimulated RAW 264.7 macrophage. Inhibition of NO production (IC50 of 9.4 μM) coupled with iNOS mRNA and protein expression, a reduction in NF-κB subunits’ translocation, and DNA binding activity of NF-κB were also observed. The downregulation of IKK activity by CAR (5 μM) was further shown to be associated with inhibition of LPS-induced phosphorylation as well as degradation of IκBα. As with the above-mentioned effect of CA, the LPS-induced p38 and p44/42 MAPK activation was also inhibited by CAR (20 μM). Other similar studies by Lee et al. [24] demonstrated the inhibitory effect of CAR (1, 2 and 5 μM) on LPS-induced NO and expression of iNOS and COX-2 in RAW 264.7 cells. In this case, inhibition of the phosphorylation of signal transducer and activator of transcription 3 (STAT3) and DNA binding activity in RAW 264.7 cells coupled with a docking model showing potential direct binding activity to the DNA binding domain of STAT3 was revealed. In addition, both CA and CAR within the concentration range of 5–15 μM reduced NO and prostaglandin E₂ (PGE₂) production in LPS-stimulated RAW 264.7. This activity was linked to inhibition of gene expression of iNOS, cytokines/interleukins (IL-1α, IL-6) and chemokines including CCL5/RANTES and CXCL10/IP-10, coupled with suppression of nuclear translocation of NF-κBp65, as evidenced in IL-1β-stimulated cells [25].
To study the anti-inflammatory properties of CAR in vitro, Shi et al. [26] employed primary mouse bone-marrow-derived macrophages (BMDMs), THP-1 cells and human peripheral blood mononuclear cells (hPBMCs). Their major finding was the inhibition (2.5–10 µM in a concentration-dependent manner) of NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation by directly targeting heat-shock protein 90 (HSP90). This was coupled with inhibition of pro-inflammatory cytokine expression (pro-IL-1β, TNF-α and IL-6) by pre-treatment of hPBMCs with CAR prior to LPS induction. They also performed experiments in vivo where administration of CAR (20 or 40 mg/kg, i.p.) in mice could ameliorate the endotoxemia or IL-1β and TNF-α production induced by LPS, a process inhibited by an NLRP3 inflammasome inhibitor, MCC950. The ethanolic rosemary extract (as well as CA and CAR) were also shown to suppress the secretion and mRNA expression of IL-8, IL-1β and TNF-α in Propionibacterium acnes-stimulated monocytic THP-1 cells [76].
Studies both in human whole-blood stimulated with LPS, and a cell-free system also showed that CA and CAR did not affect the activity of COX-1 and COX-2 directly but modulated the activity of microsomal PGE2 synthase (mPGES)-1, an enzyme upstream of COX [27,28]. The differential effect of the two compounds in the cell system, however, need further clarification. As a model of allergic inflammatory reaction, Crozier et al. [29] employed a mast cell culture in vitro using anti-TNP IgE as a sensitising agent. They showed that CA (1.59–100 µM) could ameliorate the allergen-induced ROS generation, Ca2+ mobilisation and degranulation. Hand in hand with these effects were suppression of the release of pro-inflammatory cytokines (IL-6, IL-13 and TNF) and chemokines (CCL2, CCL3 and CCL9), based on their protein and gene expression levels, though the effects were demonstrated at moderate concentrations (50 and 100 μM). As a mechanism of action, CA (15 and 50 μM) reduced the phosphorylation of both IKK and IκBα while it also decreased NF-κB2 mRNA, a relevant gene for transcribing the p52/100 subunit of NF-κB. Other allergen-specific genes suppressed by the compound were c-jun, Egr1 and Egr2. CA (15 and 50 μM) further inhibited the phosphorylation levels of Syk (Tyr352 and 525/526) and Akt, which are known to be involved in NF-κB-pro-inflammatory signalling. Of the MAPKs affected was the upstream TAK1 (Ser412), which is known to be relevant to allergic reactions, but unlike activities reported from macrophages, the phosphorylation of MAPKs ERK, JNK and p38 in primary cultures of mast cells was not affected. Hence, the major signalling affected by CA in allergic inflammation induced in mast cells appears to be inhibition of Syk activity and phosphorylation, TAK1 (Ser412), Akt (Ser473) and upstream NF-κB signalling [29]. The studies outlined in this section overall revealed the dual antioxidant and anti-inflammatory properties of CA and CAR. These effects are in line with our knowledge of rosemary extract’s role in ameliorating allergen-mediated mast cell activation [77].
The macrophage equivalent cell system in the CNS, operated by microglia, also appears to be modulated by rosemary diterpenes. The study by Foresti et al. [30] using BV2 mouse microglial cells treated with CA or CAR (5 μM) revealed induction of HO-1 coupled with inhibition of the LPS and INF-γ-induced NO and TNF-α, and PGE2 production. In BV2 cells subjected to oxygen-glucose deprivation (OGD), CAR decreased the levels of malondialdehyde (MDA), lipid peroxidation (LPO), TNF-α, IL-1β and IL-6, and increased the levels of the reduced form of glutathione (GSH), superoxide dismutase (SOD), IL-4 and IL-10 [30,78]. Readers should bear in mind that this effect is mainly an activation of a survival mechanism by CAR under stress conditions via the PI3K/Akt/mTOR signalling pathway as selective inhibitors antagonise the positive effect of CAR in these cells. Furthermore, these data are in line with our understanding of this pathway as a survival mechanism in cancer cells and a means of their resistance to chemotherapy [79].
5. Arthritis
Hosokawa et al. [31] employed IL-1β- or TNF-α-stimulated human periodontal ligament cells to study the anti-inflammatory effect of CA. They showed that the compounds could effectively suppress the release of IL-6 and production of CXC chemokine ligand (CXCL)10, CC chemokine ligand (CCL)2 and CCL20. As demonstrated in the macrophage system, the compounds could also suppress the JNK, NF-κB and STAT3 pathways of activation induced by IL-1β or TNF-α. These data are consistent with the authors’ previous report using human oral epithelial cell line (TR146) cells stimulated by IL-27, where CA (3.125–50 µM) showed a suppressive effect on chemokine (CXCL9, CXCL10 and CXCL11) production along with significant inhibition of the phosphorylation of STAT1, STAT3 and Akt [32]. In an adjuvant arthritis model in rats, administration of methotrexate (0.3 mg/kg) in combination with CA (100 mg/kg, p.o.) for up to 28 days was shown to suppress hind paw swelling, the levels of IL-17A, MMP-9 and MCP-1 in plasma, and gamma-glutamyltransferase (GGT) activity in joint homogenates [52]. As a further mechanism of action, the mRNA expression levels of HO-1 and catalase (CAT) were increased, while IL-1β was reduced in the liver by the drug combination but not individual components alone.
Liu et al. [33] used an in vitro (osteoclasts and fibroblast-like synoviocytes) and collagen-induced arthritis experimental model in rats to study the anti-inflammatory potential of CA. The pro-inflammatory proteins suppressed by the compound included TNF-ɑ, IL-1β, IL-6, IL-8, IL-17, MMP-3 and receptor activator for nuclear factor-κB ligand (RANKL). Along with the observed inhibition of osteoclastogenesis and joint destruction, the RANKL-induced activation of NF-κB and MAPKs (JNK and p38) leading to the downregulation of NFATc1 (plays a role in the inducible expression of cytokine genes) was also ameliorated by the compound. In a collagen-induced arthritis db/db mice model of rheumatoid arthritis, Xia et al. [53] demonstrated improvement in bone loss by CA (30 and 60 mg/kg, i.p. daily for 4 weeks) along with modulation of the fasting blood glucose and glucose levels in an oral glucose tolerance test (OGTT) and insulin tolerance test (ITT). In vitro (bone marrow cells and osteoblasts), CA (10 or 20 μM) suppressed (RANKL)- and macrophage colony-stimulating factor (M-CSF)-induced osteoclastogenesis. Among the oxidative stress and inflammation markers suppressed both in vitro and in vivo were ROS (while upregulating SOD and glutathione peroxidase (GPx) activity) the RANKL- and M-CSF-induced p38 MAPK, NF-κB phosphorylation and cytokine (TNF-α, IL-1β and IL-18) and COX2 expression [53].
Li et al. [54] used a type II collagen-induced arthritis DBA/1 model in mice to study the potential effects of rosmanol (40 mg/kg/d, p.o.) and CAR (40 mg/kg/d, p.o.) alone. The compounds could alleviate rheumatoid arthritis symptoms (swelling, redness and synovitis; decreased the arthritis index score) along with the serum level of pro-inflammatory cytokine (IL-6, MCP-1 and TNF-α). Other inflammation markers blocked by the compounds were the TLR4/ NF-κB/JNK and p38 MAPK pathways in synovial tissue. In a drug combination study, a 20 mg/kg dose of each resulted in higher activity than individual compounds, suggesting a possible additive/synergistic anti-inflammatory effect.
In the chondrosarcoma cell line SW1353 and in primary human chondrocytes, CA and CAR, at a concentration range of 5–15 µM, inhibited IL-1β-induced catabolic genes such MMP-13 and ADAMTS-4. These downregulated genes contributed to cartilage erosion, while the expression of anabolic genes including Col2A1 and aggrecan was shifted by the compounds toward the pre-pathophysiological homeostasis state. The induced nuclear translocation of NF-κBp65 was also inhibited [25]. Overall, CA and CAR appear to suppress the arthritis inflammatory score and markers including NF-κB, MAPK, STATs, pro-inflammatory cytokines, chemokines and matrix degradation by lowering the MMP level.
6. Lung Inflammation
Direct evidence for the therapeutic potential of CA in alleviating lung inflammation came from the study by Tsai et al. [34] both in vitro and in vivo. Human neutrophils primed for respiratory burst (superoxide anion and ROS release) with N-formyl-L-methionyl-L-leucyl-L-phenylalanine (fMLF) (FPR1 agonist), MMK1 (FPR2 agonist) and PMA (protein kinase C activator) showed a reduced level of respiratory burst when treated by CA (1–10 μM). As a measure of inflammation, the fMLF-stimulated expression of integrin adhesion molecules (CD11b) and neutrophil adhesion to the surface of endothelial cells (bEND 3 cells) were suppressed at the same concentration range through a mechanism associated with inhibition of phosphorylation of MAPKs (ERK, JNK and p38). The in vivo experimental model employed in the study was acute respiratory distress syndrome (ARDS) in mice induced by LPS spray into the trachea [34]. In this case, administration of CA (5 or 10 mg/kg, i.v.) alleviated the symptoms as assessed by qualitative (histology) and quantitative (MPO activities, immunohistochemistry and immunofluorescence staining) assays. The oxidative state and neutrophil infiltration level, as assessed using anti-Ly6G (targeting the component of the myeloid differentiation antigen) and anti-4-HNE (targeting the lipid peroxidation product, 4-hydroxy-2-nonenal) antibodies, suggested the anti-inflammatory properties of CA [34]. An LPS-induced acute lung injury (ALI) experimental model in mice was also used to study the effect of CA (10, 20 and 40 mg/kg doses, i.p.). A protective effect was evidenced by histologic results and a reduction in the wet-to-dry ratio of lung tissues, while an anti-inflammatory effect was evident from the suppression of neutrophil apoptosis, and production (mRNA and protein) of IL-1β, IL-6, TNF-α, TLR4 and NF-κB expression, as well as of NF-κB phosphorylation in lung tissues [55]. When bleomycin was used to induce lung damage in rats, CAR (at doses of 10, 20 and 40 mg/kg, p.o.) was shown to reduce the levels of oxidative markers (MDA, NO, protein carbonyl) and pro-inflammatory cytokines (TNF-α and IL-6 levels) and the myeloperoxidase (MPO) activity in the lungs. Meanwhile, antioxidant markers (GSH content, catalase, GPx and SOD activities) were increased in line with the improvement of lung fibrosis and histopathological changes [56]. Lee and Im [57] used an ovalbumin-induced allergic asthma experimental model in mice to reveal the effect of CAR (5 mg/kg, i.p.). In addition to suppressing the increase in the number of eosinophils in the bronchoalveolar lavage fluids, cytokine production, including IL-4 and IL-13, was also suppressed in both bronchoalveolar lavage fluids (BALF) and the lungs. These findings are consistent with the known suppressive effect of rosemary extract on allergic airway inflammation [80].
In human lung NCI-H1975 cells, CAR (3 μM) upregulated the HO-1 level and protected cells from H2O2-induced cell death. It also protected lung tissues in an excised-lung organ culture ischemia model [35]. This ex vivo culture was sourced from mice treated with carnosol-enriched Callicarpa longissima extract (30 mg/kg, p.o.), which was associated with upregulation of HO-1 expression.
7. Skin Inflammation
Mengoni et al. [21] used a phorbol 12-myristate 13-acetate (PMA)-induced ear inflammation model in mice where CA and CAR reduced oedema with EC50 values, respectively, of 10.20 μg/cm2 and 10.70 μg/cm2. The inhibition of leucocyte infiltration and epidermal ulceration induced by PMA were also coupled with reduction in the skin tissue expression levels of IL-1β, TNF-α and COX-2 (not COX-1), and to a less extent, fibronectin, and ICAM-1 expression. The carrageenan-induced mouse hyperalgesia model in the hind paw is one of the most commonly used anti-inflammatory assays in vivo. In one study, in a mouse model of pleurisy induced by carrageenan, both the crude rosemary plant extract as well as CAR and rosmarinic acid decreased the pro-inflammatory markers (MPO, adenosine-deaminase, NO and IL-17A) while increasing the anti-inflammatory cytokine, IL-10, level [58]. In an in vivo mouse model of P. acnes-induced ear swelling and granulomatous inflammation, the crude extract of rosemary was shown to alleviate inflammation [76].
In a mouse atopic dermatitis experimental model induced by 5% phthalic anhydride, CAR (0.05 µg/cm2) was shown to suppress skin inflammation along with inhibition of the expression of iNOS and COX-2 in skin tissue [24]. This activity was coupled with inhibition of the activation of STAT3 in skin tissue, while in the blood serum, the levels of TNF-α, IL-1β and immunoglobulin-E were also suppressed. The experimental model employed the administration of CAR (0.05 μg/cm2) together with an inflammatory inducer, followed 3 h later by 100 μL (20 μL/cm2) of 10 μM CAR. Skin inflammation can also be induced by exposure to UVB, in which case CAR also demonstrated efficacy, as reported by Yeo et al. [59]. Topical application of CAR (0.05 µg/cm2) on UVB (540 mJ/cm2, for 3 successive days)-induced skin inflammation in HR1 mice was shown to reduce erythema, epidermal thickness and inflammatory responses such as reduction in the levels of immunoglobulin-E and IL-1β in blood serum. The suppressed inflammatory markers included iNOS and COX-2 in the back skin, coupled with decreased activation of STAT3 and its upstream signal, Janus kinase (JAK). By using the carrageenan-induced oedema model, Maione et al. [28] demonstrated that CAR and CA (30 or 100 µg per paw) displayed a dose-dependent anti-inflammatory effect (oedema) and suppressed microsomal prostaglandin E synthase-1 (mPGES-1)- and 5-LO-derived products.
Oh et al. [19] studied an in vitro inflammatory skin model and showed that CA (5– 20 μg/mL) could suppress the production of IL-6, IL-8 and MCP-1 in keratinocyte HaCaT cells stimulated with sodium lauryl sulphate (SLS) and retinoic acid (RA).
8. Neuroinflammation
The direct anti-inflammatory effect of test compounds can be assessed in neuronal cultures in vitro. By using the SH-SY5Y cells exposed to paraquat, de Oliveira et al. [36] showed the neuroprotective effect of CA (1 μM) through anti-inflammatory mechanisms such as reduction in the production of IL-1β, TNF-α and COX-2. They also showed that the Nrf2 and HO-1 signalling pathway of cytoprotection was activated while the activation of the NF-κB transcription factor was suppressed by CA. In the neuronal cell line of PC12 cells subjected to serum starvation, CAR (10 µM) was shown to increase the induction of HO-1 (protein level) and increased Nrf2 expression [37]. This survival mechanism induced by CAR, unlike that described for LPS (Section 4) and other pro-inflammatory agents’ stimulation, involves activation or phosphorylation of the MAPKs (ERK, p38, JNK, Akt and its downstream effector PI3K). Large volume of literature is available in demonstrating the neuroprotective effect of rosemary diterpenes through antioxidant mechanisms. For example, CA as a potential therapeutic agent for treating Parkinson’s disease was shown by [38] to ameliorate 6-hydroxydopamine (6-OHDA)-induced neuronal death both in vitro (in SH-SY5Y cells, 1 µM) and in vivo in rats (20 mg/kg, p.o.). Beyond reversing the behavioural changes, LPO was reduced while GSH level and SOD activity were enhanced. Moreover, cell apoptosis induced by 6-OHDA via activation of the MAPKs was inhibited, as evidenced by suppression of the phosphorylation of JNK and p38. This was also effectively demonstrated in PC12 cells subjected to hypoxia-induced neuronal damage where CA ameliorated inflammation, oxidative stress and cell death [39].
When neuroinflammation was induced in mice by exposure to organophosphate pesticide (chlorpyrifos), daily administration of CA (30 and 60 mg/kg p.o. for 14 days) was shown to ameliorate the biochemical changes in a dose-dependent manner. This included reversal of the increased serum concentrations of pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α), a reduced level of acetylcholinesterase (AChE) and antioxidant markers (GSH, GPx, SOD and CAT) and an increased level of prooxidant (MDA and NO) markers in cerebral and ocular tissues [60].
Maynard et al. [61] employed repetitive mild traumatic brain injury (TBI) in mice, where CA at a small dose (1 mg/kg, i.p.) was shown to alleviate neuronal damage. By activating the Nrf2 pathway of the antioxidant/anti-inflammatory mechanism, the compound further suppressed the transcription factor, NF-κB. Another similar neuroprotective study was that by Wang et al. [62], which demonstrated that CAR protects against spinal cord injury through Nrf2 upregulation. The oxidative stress markers of increased ROS generation, total oxidant levels, LPO content, protein carbonyl and sulfhydryl levels were also suppressed, along with the expression of NF-κB and COX-2, while the disease-associated or -altered phosphorylated Akt and Nrf2 levels were reversed by the compound. The neuroprotective effect of CA (0.3, 1.0 or 3.0 mg/kg, i.p.) in traumatic brain injury in mice was also shown to be mediated through activation of the Nrf2–ARE pathways [63]. Hence, oxidative markers were reduced while mitochondrial respiratory dysfunction and neuronal damage, as shown by cytoskeletal damage and biochemical markers’ (4-HNE (lipid peroxidation) levels in the hippocampus and cortex and 3-NT (protein nitration) in the cortex, were reduced by the compound.
Teng et al. [64] used the subarachnoid haemorrhage (SAH) of early brain injury model, where CA displayed a protective effect. Among the improved parameters were decreasing ROS levels, brain oedema and blood–brain barrier permeability, neuronal cell death and neuronal function improvement. Furthermore, CA was shown to increase the SIRT1, MnSOD and Bcl-2 expression while apoptosis markers were suppressed. More studies in this line showing the anti-inflammatory effect of the compound are needed. In the APP/PS1 mouse model of Alzheimer’s disease, administration of CA was shown to reduce β-amyloid (Aβ) deposition and ameliorate cognitive impairment and pro-inflammatory cytokine (IL-1β, TNFα and IL-6) production [65]. By blocking the interaction of CEBPβ with NF-κB p65, the transcription of the NF-κB target genes TNF-α and IL-6, as well as Aβ secretion, were suppressed. A good example of neuroinflammation is that of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS). Li et al. [66] showed that CAR could reduce demyelination, inhibit Th17 cell differentiation and STAT3 phosphorylation and block the translocation of NF-κB. The compound could also switch the inflammatory phenotypes of infiltrated macrophage/microglia in the chronic stage of the disease.
In BV2 microglial cells in vitro, CAR (5–20 μM) was one of the compounds identified to induce HO-1 and Nrf2, thereby inhibiting the production of TNF-α, PGE2 and NO stimulated by interferon-γ (INF-γ) or (LPS) [30]. The anti-inflammatory effect in the neuronal system could also mark an indirect effect via inhibition of amyloid beta (Aβ) production, as shown for CA (30 µM). It ameliorated Aβ (1-40 and 1-42) production by activating α-secretase TACE (tumour necrosis factor-α-converting enzyme or disintegrin and metalloproteinase-17 or ADAM17) in cultured SH-SY5Y human neuroblastoma cells [40]. The selectivity of this enzyme was demonstrated as no effect was observed in β-secretase BACE1. It (50 μM) also suppressed the secretion and release of Aβ peptides (1-40, 1-42 and 1-43) in U373MG human astrocytoma cells by increasing the mRNA expression of an α-secretase (TACE) without affecting other secretases (BACE1 and PS1) [40]. At the same time, increases in the HO-1 mRNA level were also observed in this experiment, suggesting the neuroprotective potential of the compound from inflammation and oxidative stress.
Other neuronal effects of these compounds include the antinociceptive effect of CAR (10 mg/kg, p.o.) to chemical agents (acetic acid, formalin, glutamate, capsaicin and cinnamaldehyde) [81], formalin-induced pain response of CAR [82] and anti-inflammatory and anti-nociceptive effects of CA and CAR [28]. The overall trend thus seems to indicate that neuroprotection and inhibition of neuroinflammation are enacted by CA and CAR in a variety of experimental models.
9. Diabetes-Associated Inflammation
Several studies investigated the antidiabetic potential of rosemary extracts, and indeed, the plant was shown to reduce the glucose level both in healthy and diabetic experimental animals [83,84,85,86,87]. Similarly, rosemary extract and diterpenes including CA and CAR were shown to increase insulin sensitivity, alleviate insulin resistance and protect cells from high-glucose-induced damage both in vitro and in vivo [88,89,90,91]. Hand in hand with this potential antidiabetic effect, the anti-obesity and lipid-lowering potential of rosemary and its phenolic diterpenes were established [71,92,93,94,95,96]. Given both diabetes and obesity are associated with inflammation, the anti-inflammatory mechanism of these compounds in such pathological conditions is worthy of scrutiny. Administration of CAR (1, 5, 10 mg/kg, i.p. for 4 weeks) in STZ-induced diabetic animals was shown to suppress the serum levels of glucose, IL-6, TNF-α, MDA, TG, TC, LDL-C, GST, SOD, CAT and HDL-C in a dose-dependent manner [67]. Hence, the compound could reduce the diabetes-associated increases in blood glucose level, oxidative stress and inflammation. The study by Ou et al. [68] also showed that CA (30 mg/kg) not only reduced the glucose level in STZ-induced diabetic rats but also the inflammation score. The study by Xie et al. [69] using the same diabetic model also corroborated the antidiabetic potential of CA (30 mg/kg, p.o.), which also involves suppression of the NF-κB activation. In 3T3-L1 adipocytes, CA (1–20 µM) reversed the TNF-α-mediated insulin resistance, as shown by insulin-stimulated glucose uptake and the phosphorylation of Tyr(632) insulin receptor substrate-1 (IRS-1), Akt and FoxO1 levels [42]. As an anti-inflammatory compound, it also attenuated the TNF-α-induced mRNA expression of inflammatory genes, including IL-6 and MCP-1. Furthermore, CA attenuated the TNF-α-mediated activation of ERK and JNK, the phosphorylation of inhibitor-κB (IκB) kinase (IKK)α/β, the phosphorylation and degradation of IκBα, the nuclear translocation of p65 and the DNA-binding activity of NF-κB and AP-1 [42].
10. Cardiac Inflammation
Hu et al. [70] employed the ischaemia/reperfusion model in diabetic mice on a high-fat diet under 30 min occlusion of the anterior descending coronary artery followed by reperfusion of the heart for 3 or 24 h. They showed that pre-treatment with CA (50 mg/kg, p.o.) could suppress the overproduction of ROS and pro-inflammatory cytokines (IL-6 and TNF-α).
The cytoprotective effect of CA was demonstrated by the doxorubicin (DOX)-induced cardiotoxicity in rats at the dose of 10 mg/kg (p.o.) as well as in isolated rat cardiomyocyte (2.4–10 µM) cell cultures [43]. In addition to suppression of the ROS and NO levels, the DOX-induced expression of phospho-p38 and phospho-JNK1 proteins as well as NF–κB (p65) were lowered while the DOX-induced downregulation of Nrf2 in the nucleus and HO-1 in the cardiomyocytes were reversed. Similarly, the study by Zhang et al. [44] investigated using a cardiac muscle cell line, H9C2 cells, in vitro (5–20 μM), and the DOX-induced cardiotoxicity mice in vivo (5 mg/kg, p.o.) treated with CA or carvedilol. They showed a protective effect partly by augmenting the expression and activities of the antioxidant enzymes. In addition, the inflammatory response was significantly suppressed by the two compounds in combination, as shown by suppression of the levels of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β and IL-18) and COX-2, which was associated with the inactivation of NF-κB. The effect of CAR (5–20 μM) on the LPS-stimulated cardiomyocyte cell line (H9C2) was also studied [45]. The major finding was the inhibition of the NF-κB, and the NF-κB-dependent inflammatory pathway associated with cytokine (TNF-α, IL-1β, IL-6) and COX-2 (as well as PGE2) expression. Their in silico analyses further suggested potential interaction of the compound with the binding site of the catalytic domain of IKKβ.
11. Hepato- and Renal Inflammation
The experimental models employed to demonstrate the hepatoprotective effect of CA (10 µM) include a chronic alcoholic liver injury model in rats (15 or 30 mg/kg, i.g.) and an in vitro model using HepG2 cells [46]. One major observation to note was the activation of SIRT1 to account for the antioxidant and anti-inflammatory response, as well as pathological markers of liver cell damage. The level of MnSOD was increased, while NF-κB and the serum level of TNF-α were inhibited. In an ischemia/reperfusion model of liver damage, Li et al. [47] demonstrated that nanoparticle preparations of CA (10 and 20 mg/kg, i.p.) could protect from liver injury progression when coupled with antioxidant (normalising the levels of SOD, CAT, GSH and GPx) and NF-κB signalling pathways of pro-inflammatory cytokine (TNF-α and IL-1β) expression. The in vitro experiment in the same study, using LPS-treated hepatic stellate cells, also showed that CA nanoparticles could inactivate phosphorylated IKKα, IκBα and NF-κB, leading to decreased TNF-α, IL-1β and IL-18 expression.
In a high-fat-diet (HFD)-induced non-alcoholic fatty liver disease (NAFLD) model in mice, the pharmacological activity of CA (15 mg/kg, p.o.) was reported to be related to an improvement in lipid metabolism and inflammation markers [71]. Notably, the serum and hepatic tissue levels of IL-1β, IL-18, TNF-α, IL-2, IL-4, IL-6, IL-12 and IFN-γ were suppressed. The link between these activities and myristoylated alanine-rich C-kinase substrate (MARCKS) via the PI3K/Akt, NLRP3/NF-κB and SREBP-1c signalling pathways of inflammation was established using inhibitors and MARCKS-deficient mice. Another interesting finding both for the antioxidant and anti-inflammatory effect of CA came from a study by Xiang et al. [72] of LPS-induced liver injury in rats. Administration of CA (30 or 60 mg/kg, p.o.) alleviated symptoms of liver damage, as assessed by histology and biochemical markers (alanine aminotransferase, aspartate aminotransferase and alkaline phosphatase). In addition to suppression of immigration of inflammatory cells, the serum levels of TNF-α and IL-6 were suppressed. The oxidative markers suppressed were NO and ROS, while antioxidant levels (SOD, GSH and GPx) in the serum and liver were augmented.
Administration of CA (40 mg/kg, i.p.) in mice after LPS injection was shown to ameliorate histological abnormalities and renal dysfunction [73]. Notably, the pro-inflammatory cytokine (IL-1β, IL-6, TNF-α and MCP-1, mRNA and protein level) expression, immune cell (neutrophil) infiltration and NF-κB activation induced by LPS injection were suppressed by CA. Among the oxidative markers improved by the compound were GSH, NOX4 (mRNA and protein), CAT and MnSOD, which were associated with reduced tubular cell apoptosis. Other data also suggest the renal protective effect of CA, such as those demonstrated by Xie et al. [69]. In STZ-induced diabetic db/db mice, a nephroprotective effect of CA (15 or 30 mg/kg, i.g.) associated with activation of Nrf2 and inhibition of NF-κB was demonstrated. On the other hand, the experiment by Zheng et al. [74] suggested that CAR (3 mg/kg) protects against renal ischemia-reperfusion injury in rats when administered intravenously. The inhibition of apoptotic tubular cell death, caspase-3 activation and activation of the p38 pathway was evident. To study the anti-inflammatory properties of CAR (20 or 40 mg/kg, i.p.), Shi et al. [26] employed an in vivo non-alcoholic steatohepatitis model in mice, which was shown to be associated with downregulation of IL-1β and TNF-α. This activity was associated with suppression of NLRP3 inflammasome activity via a direct effect on heat-shock protein 90 (HSP90), as explained in Section 4 for a macrophage culture in vitro.
12. Obesity-Associated Inflammation
In a-high fat-induced mouse obesity and metabolic syndrome model, CA treatment (10 or 20 mg/kg, p.o.) was shown to decrease the serum levels of triglycerides, total cholesterol, insulin and glucose [75]. As an anti-inflammatory compound, CA decreased the protein expression levels of various pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) in serum and brain tissues, along with suppression of the NF-κB signalling pathway. As a cytoprotective agent, it promoted the expression levels of anti-apoptotic Bcl-2, while decreasing the levels of pro-apoptotic Bax and matrix metallopeptidase 9 [75].
In 3T3-L1 adipocytes stimulated by LPS, the TLR4-mediated elevated mRNA expression of TNF-α, IL-6 and MCP-1 was suppressed by CA (up to 20 µM). In addition to the LPS-induced upregulation of TLR4, myeloid differentiation factor 88, TNF receptor-associated factor 6 and ERK were also suppressed by CA [48]. These data were in line with the study by Tsai et al. [42], which showed that 3T3-L1 adipocytes stimulated with TNF-α could be targeted by CA, leading to reduced levels of mRNA expression of inflammatory genes (IL-6 and MCP-1). Among the signal transduction pathways affected by CA in this study were the activation of ERK and JNK; the phosphorylation of IκB, (IKK)α/β; the phosphorylation and degradation of IκBα, the NF-κB p65 subunit; and the DNA-binding activity of NF-κB and AP-1. The modulation of diabetes, obesity and metabolic syndrome by rosemary extract and diterpenes is also a result of the effect on various other metabolic signalling pathways such as PPAR-γ and AMPK [91,94,96,97].
13. Endothelial Inflammation
The study by D’Agata et al. [49] employed an in vitro culture of human retinal endothelial cells to study high-glucose-induced ROS generation and cell damage and death. By upregulating the expression and activity of Nrf2, HO-1 and ERK1/2, CAR was demonstrated to have a cytoprotective effect in endothelial cells. In addition to increasing the expression of endothelial cadherin (VE-cadherin), thereby improving the integrity of intercellular junctions, CAR (10 µM) was also shown to protect human lung microvascular endothelial cells (HMVEC-L) from tert-butyl hydroperoxide (t-BHP)-induced cell death [50]. The antioxidant mechanism of action was evident from the increased expression of Nrf2 and HO-1, and it also interrupted the Nrf2-Keap1 protein−protein interaction.
14. Gut Inflammation
Xu et al. [51] employed a dextran sulphate sodium (DSS) experimental model of colitis in mice. They showed that induction of an improvement in the clinical symptoms and colonic pathological damage by CAR (50 mg/kg i.p. for 10 days) was associated with reduction in inflammatory cell infiltration and cytokine (TNF-α, IL-1β, IL-6 and IFN-γ) expression. This evidence was further substantiated in vitro (10 µM) using thapsigargin-induced endoplasmic reticulum (ER) stress in HCT-116 cells (an intestinal epithelial cell line) as well as colonic mucosa tissues (2–3 pieces from patients). As with the in vivo data, suppression of pro-inflammatory mediators (TNF-α, IL-6, IFN-γ and CXCL10) was evident in the in vitro assay. Although the main emphasis was on anticancer effect analysis of CA, Li et al. [98] also demonstrated that CA suppressed the inflammatory response in colorectal cancer in mice by reducing the levels of IL-1β, -6 and -17A. Readers should also note that the protective effect of these diterpenes in the gut could be associated with modulation of the microbiota structure and population, as reported under various experimental conditions [68,95,98,99].
15. General Discussion
As a defence mechanism to protect cells and organs from damage induced both from internal and external sources, inflammation is universally involved in disease processes, either at the initiation phase or later stages of pathologies. Anti-inflammatory drugs of the classical steroidal class target phospholipase enzymes, which catalyse the initial step in the synthesis of lipid mediators such as prostaglandins, and leukotrienes. They also have various other mechanisms including suppression of cytokine expression and serve in therapeutics associated with immunosuppression (e.g., organ transplantation). We also have classical examples of small molecules of anti-inflammatory agents that target the active site of enzymes such as COX (selective new generation and non-selective aspirin type) and lipoxygenases. As highlighted in the introduction section, these drugs of high significance to acute inflammatory conditions are, however, of little benefit for chronic inflammatory diseases such as those highlighted in these articles. In this case, antibody approaches targeting pro-inflammatory cytokines, their receptors or effector proteins such as adhesion molecules have been effectively employed in recent years. Given the challenge of using protein-based drugs, the search for a small-molecular-weight antagonist of protein mediators for chronic inflammation therapy is ongoing. Unfortunately, small molecules have little impact on ligand–receptor interaction of the protein–protein type. Meanwhile, the signal transduction pathways of protein signalling molecules (e.g., cytokines) are subject to modulation by small-molecular-weight compounds including natural products. The crosstalk of signalling between inflammation and oxidative stress also creates opportunities to utilise natural products, which are commonly regarded as antioxidants.
In various sections of this article, the antioxidant potential of rosemary diterpenes was shown through experimental evidence where the production of ROS, lipid peroxidation products (e.g., MDA) and NO was reduced while antioxidant defences such as the GSH level and activity of antioxidant enzymes (SOD, CAT, GPx) were augmented. While ROS-generating systems such as NOX4 are inhibited, antioxidant defences including NQO-1 and Nrf2/HO-1 are activated. Hand in hand with these antioxidant effects, the anti-inflammatory activity of CA and CAR was demonstrated through experimental evidence showing suppression of pro-inflammatory markers under several disease models. The common links in these diseases, constituting anti-inflammatory mechanisms at the molecular level of signalling, are summarised in the following sections.
15.1. Rosemary Diterpenes Inhibit Activation of NF-κB
In various sections of this article, the anti-inflammatory properties of CA and CAR have been shown to be associated with inhibition of upregulation of NF-κB under inflammatory conditions. The expression of pro-inflammatory genes leading to cytokines, chemokines, adhesion molecules and enzymes (e.g., COX) for lipid mediators are all under the control of the transcription factor, NF-κB. Inevitably, suppression of inflammation-mediated upregulation of NF-κB inhibits immune cell activation and the inflammatory score under various disease conditions, as highlighted in this article. Readers who would like more insight into the role of NF-κB in various inflammatory diseases should refer to review articles in the field [100,101]. Various physical and chemical pressures, pro-inflammatory cytokines and bacterial and viral products are known to stimulate the activation of NF-κB. As shown for CA and CAR, the common target for inhibiting NF-κB is by modulating a family of inhibitory molecules (IκBs) such as IκBα. The NF-κB proteins (p65/RelA, RelB, c-Rel, p50 and p52) that form dimers are retained in the cytoplasm in their inactive form as they exist in association with the IκB proteins. The NF-κB activation cascade thus involves inactivation of the inhibitory proteins through phosphorylation and their subsequent degradation. The IκB kinase complex (IKK with IKK1/α and IKK2/β subunits and regulatory subunit IKKγ) that does this task is the common target for anti-inflammatory compounds, as shown in this article for CA and CAR (Figure 2). The multiple effects of these compounds in the NF-κB signalling pathways were demonstrated in the study by Oh et al. [19], which showed inhibition of the LPS-mediated and TLR-dependent NF-κB activation by CA was associated with Syk/Src, PI3K and Akt inhibition. The critical role of these pathways in NF-κB activation via IKK was demonstrated through inhibitor studies and various inflammatory disease models [102,103,104]. Many natural products also display anti-inflammatory activities by suppressing NF-κB activation in a similar manner. These include curcumin [105,106,107], gallate derivatives [108], quercetin [109] and resveratrol [110,111], among others.
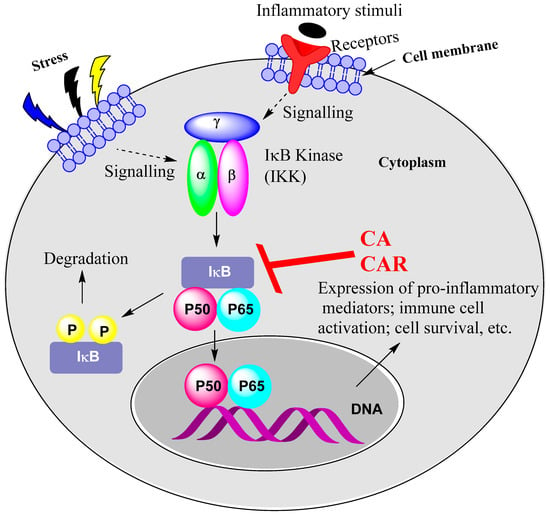
Figure 2.
NF-κB activation as a target for CA and CAR. Stress and inflammatory mediators and growth factors, PAMPs and cytokines such as TNF, IL-1 and IL-6 all activate NF-κB. Their relevant receptors include growth factor receptors, TLRs and cytokine receptors such as TNF receptor. CA and CAR have been shown to suppress the activity of IKK, leading to inhibition of NF-κB activation and/or mobilisation to the nucleus. Upstream signalling pathways could also be a target, as shown for CA, inhibiting the TLR-mediated activation of Syk/Src-PI3K and thereby abolishing the LPS-induced IKK activation.
15.2. Rosemary Diterpenes Modulate the MAPK Pathways
The role of the MAPK pathways in inflammation has been well-established but complexity arises from these pathways also being involved in various other cellular processes such as proliferation, differentiation and cell death (apoptosis). The processes are based on phosphorylation steps in a sequence involving MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK) and MAPK. The MAPKs, in turn, activate via phosphorylation several other enzymes as substrates called MAPK-activated protein kinases (MAPKAPKs). This level of complexity was not demonstrated in studies using CA and CAR, and hence only the relevant MAPKs are described herein. The three most common MAPKs relevant to inflammation are ERK 1/2 (mostly described as ERK, though we also have ERK 3/4, ERK5 and ERK7/8), JNK and p38 MAPK. Various isoforms of these MAPKs are also known, for example, α, β, γ and δ isoforms of p38. As shown in the various sections of this article, these three MAPKs are involved in inflammation induced by a variety of agents (cellular stress, growth factors, PAMPs), pro-inflammatory cytokines and cell survival cases such as neurons subjected to cellular stress from glucose deprivation or direct physical damage. A simplistic presentation of the MAPK pathways is shown in Figure 3, where they all culminate in the activation of transcription factors such as AP-1 and others. The pathways also have a crosstalk with NF-κB activation. For example, activation of MAPKKK (e.g., TAK1) leads to NF-κB activation via IKK phosphorylation. For details of the MAPK signalling pathways, readers are directed to review articles in the field [112,113,114]. The synthesis of pro-inflammatory cytokines involves activation of the MAPKs, and hence the suppressive effect of CA and CAR on this signalling system should be considered as one of their mechanisms of anti-inflammatory effect. Cell survival as neuronal and cardiac cells are damaged under stress also requires activation of these pathways, and hence CA and CAR paradoxically may activate the MAPKs as part of their cytoprotective effect. Meanwhile, the MAPKs are also key targets in cancer of various agents [115] including CA. The toxicity of therapeutic agents and cell death in cardiac cells (e.g., by doxorubicin) was shown to be associated with pro-inflammatory cytokine expression linked to upregulation of the MAPK pathway (e.g., [116]). The association of doxorubicin toxicity in cardiac cells with p38 MAPK overactivity was also established (e.g., [117]). While suppressing MAPKs’ activity has been researched as a key target for therapeutic intervention in cancer and inflammation, their role in antiapoptotic events, once again, is emerging as a complication in adopting such a therapeutic approach. In this line, the study by Martin et al. [37] showing activation of the MAPKs by CAR as a mechanism in neuroprotection is interesting. The pro- and anti-apoptotic roles of the MAPKs have also been reviewed [118]. In addition, it is worth noting that CA displayed antiproliferative and cancer metastasis inhibitory effects by inhibiting the ERK, p-38 and JNK signalling pathways [119]. Hence, further research is needed to establish the link between MAPK signalling and CA/CAR cytoprotective effects through anti-inflammatory mechanisms. Overall, the evidence available so far suggests that CA and CAR display anti-inflammatory effects and promote cell survival through regulation of the MAPK pathways. As shown for TAK1, modulation of the earlier-stage (upstream) MAPK pathways by these compounds could also be possible, so further research in this area is required. As a final point, this effect of rosemary diterpenes is in line with their mechanism of action in their anticancer activity via the inhibition of the MAPK and STAT3 (see Section 15.3) pathways [120].
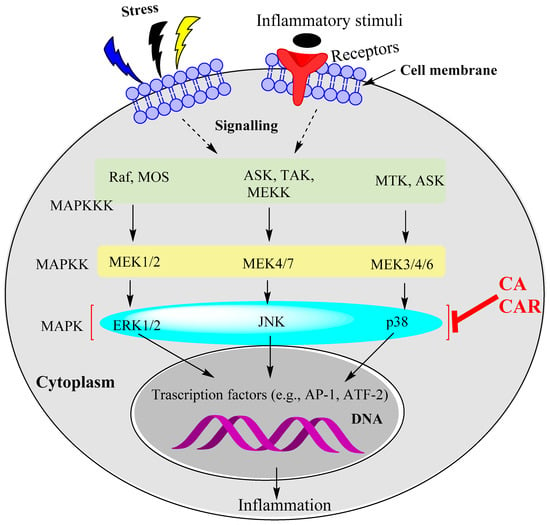
Figure 3.
The MAPK pathways as targets for CA and CAR. The evidence available so far suggests that ERK1/2, JUNK and p38 MAPK phosphorylation are targeted by rosemary diterpenes to induce anti-inflammatory effects. Further research is required to determine whether the other kinases upstream of MAPKs are affected by CA and CAR. At least one study shows that upstream kinases (TAK1) could also be affected.
15.3. Rosemary Diterpenes Modulate the SIRT1/SERT3 Pathways
The STAT signalling pathway relates to the JAK family protein as an upstream regulator. The STATs (including STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6) control various biological functions, and STAT3 is the most characterised in terms of its role in cell proliferation, survival, differentiation and angiogenesis. When cells are induced by a variety of agents such as cytokines and growth factors, activation of STATs via phosphorylation leads to dimerisation and nuclear translocation, DNA binding and target genes’ activation. Overactivition of STAT3 under inflammatory and cancer conditions has led to suggestions that it is a potential therapeutic target. Evidence along this line came from the fact that inhibition of STAT3 inhibits cancer cells’ proliferation and selectively induces apoptosis in cancer cells. Several forms of oncogenic signalling employ STAT3, and chemotherapeutic resistance has also been associated with overactivation of STAT3 [121,122]. Overall, STAT3 is constitutively expressed in cancer cells (only transiently in normal cells) and is involved in the regulation of genes involved in cancer cells’ survival, invasion, angiogenesis and interaction with immune cells. In the latter case, its role in inflammation has been driving research searching for novel drugs for asthma, inflammatory bowel disease and fibrosis, among others [123]. The phosphorylation of both STAT1 and STAT3 has been shown to be inhibited by CA and CAR through the mechanism depicted in Figure 4. In view of the current level of interest in STATs, further research in this field is needed to assess the true potential of CA and CAR as therapeutic leads through this mechanism of action. Meanwhile, SIRT1 opposes STAT3, and its main role is in the expression of genes that ameliorate stress, apoptosis, aging and inflammation [124]. Several isoforms of the SIRTs have been characterised in recent years but the most studied is SIRT1. As a histone deacetylase enzyme, they are mostly found in the nucleus, and their dysregulation appears to be associated with various disease conditions. Although the complexity of the signalling pathway of SIRT1 as a mechanism for the anti-inflammatory activity of rosemary diterpenes is not yet established, activation of the SIRT1 pathways not only suppresses the production of pro-inflammatory cytokines but also the NLRP3 inflammasome signalling pathway (Section 15.5), as established using the most potent SERT1 activator natural product, resveratrol [125,126,127,128,129,130], and the MAPK pathways of inflammation and neuroprotection [131,132]. SIRT1 also protects cells against oxidative stress by various mechanisms including an increase in the expression antioxidant enzymes. Interestingly, many other natural antioxidants such as quercetin [133,134], berberine [135,136] and curcumin [137,138] have been shown to have cytoprotective, antioxidant and anti-inflammatory effects via upregulation of SIRT1 activity.
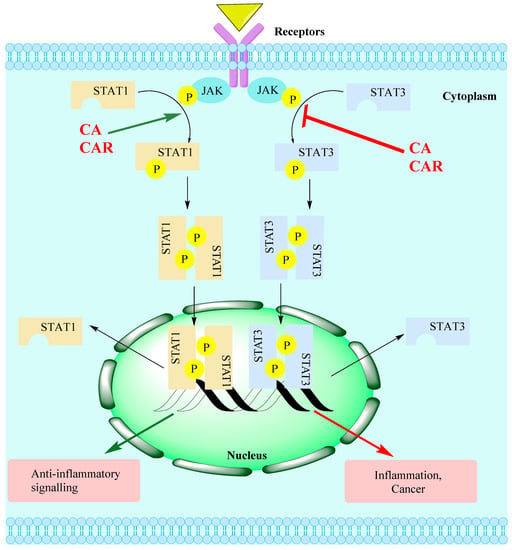
Figure 4.
The JAK-STAT signalling pathway targets for CA/CAR. Several external stimuli such as UV radiation, infection, carcinogens, inflammatory mediators and stress can induce the activation of the STAT pathway. Activation of the JAK/STAT pathway associated with receptors’ activation (e.g., by cytokines) leads to the phosphorylation of JAKs. This, in turn, activates STAT monomers through phosphorylation of tyrosine residues, leading to STATs’ dimerisation. Translocation of activated STAT dimers to the nucleus and subsequent DNA binding activate target genes relevant to various physiological/pathological pathways including inflammation. The level of STAT1/3 activation has been shown to be suppressed by rosemary diterpenes, while SIRT1 has been shown to be activated.
15.4. Rosemary Diterpenes Activate the Nrf2/HO-1 Pathways of Cytoprotection
Previous review articles from our laboratories and others showed the therapeutic potential of natural products via upregulation of the Nrf2/HO-1 pathway [139,140,141]. Cellular stress induced by a variety of agents including ROS activates this pathway of cell survival, as outlined for CA and CAR in Figure 5.
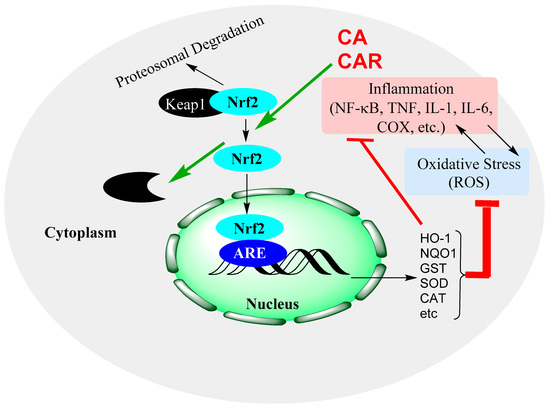
Figure 5.
Induction of the Nrf2 signalling pathway and modulation by CA and CAR. Nrf2 activation leads to the expression of antioxidant genes, which, in turn, increase the levels of antioxidant proteins that resolve oxidative stress in cells. The pathway also inhibits inflammatory genes’ activation and has indirect impacts by removing the oxidative stress component of inflammation, oxidative stress crosstalk.
15.5. Rosemary Diterpenes Suppress the NLRP3 Inflammasome
The inflammasomes are complexes of large-molecular-weight proteins with functional components such as sensors, adaptors (e.g., apoptosis-associated speck-like protein containing a caspase-recruitment domain or ASC) and pro-caspases. The NLRP3 inflammasome is the best example, which is involved both in the innate immune system and inflammatory signalling in health and disease. In the inflammasome complex of NLRP3, the NLRP3 protein binds to ASC, which, in turn, interacts with pro-caspase-1; in this way, the NLRP3–ASC–pro-caspase-1 complex is formed. This recruitment of NLRP3 leads to the cleaving by caspase-1 of pro-cytokines (e.g., pro-IL-1β) into their mature form, thereby activating inflammatory signalling. In immune cells such as macrophages, various triggers for priming and subsequent activation of NLP3 have been identified. Among the various activators of the NLRP3 inflammasome are ion fluxes such as potassium efflux [142], Ca2+ mobilisation from cellular storage sites [143,144], ROS generation and/or mitochondrial dysfunction [145] and lysosomal damage [146]. In terms of the NLRP3 inflammasome’s role in inflammatory diseases, rheumatoid arthritis [147], diabetes [148], cancer [149] and neurodegenerative diseases [150] are just a few to mention. In this emerging role of NLRP3 as a therapeutic target for numerous diseases, CA and CAR have been shown to suppress inflammation activation under various experimental conditions. This is in line with results for other natural products such as apigenin [151], caffeic acid phenethyl ester [152], curcumin [153], resveratrol [154,155] and quercetin [156], among others. The effect could be related to the known effects of lowering the level of ROS and ameliorating mitochondrial dysfunction.
16. Conclusions
Rosemary diterpenes are examples of natural products with phenolic structures that scavenge ROS or remove them indirectly via upregulation of antioxidant defences such as the GSH level and SOD, CAT and GPx activities. They also conduct anti-inflammatory activities by modulating various signalling pathways of inflammation including the NF-κB, MAPK, Nrf2, SIRT and NRLP3 inflammasomes, among others. Through such diverse effects, they downregulate the expression of pro-inflammatory cytokines (e.g., TNF-α, IL-1 and IL-6), adhesion molecules, chemokines and prostaglandins. The therapeutic potential of these diterpenes, as with many natural products, is based on targeting the inflammatory component of the disease through multiple mechanisms.
Funding
This work received no internal of external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Sig. Transduct. Target Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, C.; McKernan, D.P. Anti-Viral Pattern Recognition Receptors as Therapeutic Targets. Cells 2021, 10, 2258. [Google Scholar] [CrossRef]
- Kimura, Y.; Tsukui, D.; Kono, H. Uric Acid in Inflammation and the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 12394. [Google Scholar] [CrossRef]
- Kanneganti, T.D. The inflammasome: Firing up innate immunity. Immunol. Rev. 2015, 265, 1–5. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Kotsovilis, S.; Andreakos, E. Therapeutic human monoclonal antibodies in inflammatory diseases. Methods Mol. Biol. 2014, 1060, 37–59. [Google Scholar]
- Lai, Y.; Dong, C. Therapeutic antibodies that target inflammatory cytokines in autoimmune diseases. Int. Immunol. 2016, 28, 181–188. [Google Scholar] [CrossRef]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef]
- Moens, U.; Kostenko, S.; Sveinbjørnsson, B. The Role of Mitogen-Activated Protein Kinase-Activated Protein Kinases (MAPKAPKs) in Inflammation. Genes 2013, 4, 101–133. [Google Scholar] [CrossRef]
- Kim, M.E.; Kim, D.H.; Lee, J.S. Transcription Factors as Targets of Natural Compounds in Age-Related Diseases and Cancer: Potential Therapeutic Applications. Int. J. Mol. Sci. 2022, 23, 13882. [Google Scholar] [CrossRef]
- Wang, X.; Wong, K.; Ouyang, W.; Rutz, S. Targeting IL-10 Family Cytokines for the Treatment of Human Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a028548. [Google Scholar] [CrossRef]
- Habtemariam, S. The Therapeutic Potential of Rosemary (Rosmarinus officinalis) Diterpenes for Alzheimer’s Disease. Evid. Based Complement. Alternat. Med. 2016, 2016, 2680409. [Google Scholar] [CrossRef]
- Birtić, S. Carnosic acid. Phytochemistry 2015, 115, 9–19. [Google Scholar] [CrossRef]
- Razboršek, M.I.; Ivanović, M. Stability studies and determination of carnosic acid and its oxidative degradation products by gas chromatography–mass spectrometry. Inter. J. Mass Spectr. 2016, 407, 29–39. [Google Scholar] [CrossRef]
- Buchin, Y.; Sakemi, Y.; Hamashima, R.; Morioka, Y.; Yamanaka, D.; Hakuno, F.; Takahashi, S.-I.; Shindo, K. Structures and biological activities of new carnosic acid- and carnosol-related compounds generated by heat treatment of rosemary. Phytochem. Lett. 2019, 30, 43–48. [Google Scholar] [CrossRef]
- Oh, J.; Yu, T.; Choi, S.J.; Yang, Y.; Baek, H.S.; An, S.A.; Kwon, L.K.; Kim, J.; Rho, H.S.; Shin, S.S.; et al. Syk/Src pathway-targeted inhibition of skin inflammatory responses by carnosic acid. Mediators Inflamm. 2012, 2012, 781375. [Google Scholar] [CrossRef]
- Chae, I.G.; Yu, M.H.; Im, N.K.; Jung, Y.T.; Lee, J.; Chun, K.S.; Lee, I.S. Effect of Rosemarinus officinalis L. on MMP-9, MCP-1 levels, and cell migration in RAW 264.7 and smooth muscle cells. J. Med. Food. 2012, 15, 879–886. [Google Scholar] [CrossRef]
- Mengoni, E.S.; Vichera, G.; Rigano, L.A.; Rodriguez-Puebla, M.L.; Galliano, S.R.; Cafferata, E.E.; Pivetta, O.H.; Moreno, S.; Vojnov, A.A. Suppression of COX-2, IL-1β and TNF-α expression and leukocyte infiltration in inflamed skin by bioactive compounds from Rosmarinus officinalis L. Fitoterapia 2011, 82, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Wei, W.H.; Zhang, X.W.; Liu, D.; Zeng, K.W.; Tu, P.F. An Integrated Proteomics and Bioinformatics Approach Reveals the Anti-inflammatory Mechanism of Carnosic Acid. Front. Pharmacol. 2018, 9, 370. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.H.; Liang, Y.C.; Lin-Shiau, S.Y.; Ho, C.T.; Lin, J.K. Carnosol, an antioxidant in rosemary, suppresses inducible nitric oxide synthase through down-regulating nuclear factor-kappaB in mouse macrophages. Carcinogenesis 2002, 23, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Hwang, C.J.; Choi, J.Y.; Park, M.H.; Song, M.J.; Oh, K.W.; Son, D.J.; Lee, S.H.; Han, S.B.; Hong, J.T. Inhibitory Effect of Carnosol on Phthalic Anhydride-Induced Atopic Dermatitis via Inhibition of STAT3. Biomol. Ther. 2017, 25, 535–544. [Google Scholar] [CrossRef]
- Schwager, J.; Richard, N.; Fowler, A.; Seifert, N.; Raederstorff, D. Carnosol and Related Substances Modulate Chemokine and Cytokine Production in Macrophages and Chondrocytes. Molecules 2016, 21, 465. [Google Scholar] [CrossRef]
- Shi, W.; Xu, G.; Zhan, X.; Gao, Y.; Wang, Z.; Fu, S.; Qin, N.; Hou, X.; Ai, Y.; Wang, C.; et al. Carnosol inhibits inflammasome activation by directly targeting HSP90 to treat inflammasome-mediated diseases. Cell Death. Dis. 2020, 11, 252. [Google Scholar] [CrossRef]
- Bauer, J.; Kuehnl, S.; Rollinger, J.M.; Scherer, O.; Northoff, H.; Stuppner, H.; Werz OKoeberle, A. Carnosol and carnosic acids from Salvia officinalis inhibit microsomal prostaglandin E2 synthase-1. J. Pharmacol. Exp. Ther. 2012, 342, 169–176. [Google Scholar] [CrossRef]
- Maione, F.; Cantone, V.; Pace, S.; Chini, M.G.; Bisio, A.; Romussi, G.; Pieretti, S.; Werz, O.; Koeberle, A.; Mascolo, N.; et al. Anti-inflammatory and analgesic activity of carnosol and carnosic acid in vivo and in vitro and in silico analysis of their target interactions. Br. J. Pharmacol. 2017, 174, 1497–1508. [Google Scholar] [CrossRef]
- Crozier, R.W.E.; Yousef, M.; Coish, J.M.; Fajardo, V.A.; Tsiani, E.; MacNeil, A.J. Carnosic acid inhibits secretion of allergic inflammatory mediators in IgE-activated mast cells via direct regulation of Syk activation. J. Biol. Chem. 2023, 102867. [Google Scholar] [CrossRef]
- Foresti, R.; Bains, S.K.; Pitchumony, T.S.; de Castro Brás, L.E.; Drago, F.; Dubois-Randé, J.-L.; Bucolo, C.; Motterlini, R. Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol. Res. 2013, 76, 132–148. [Google Scholar] [CrossRef]
- Hosokawa, I.; Hosokawa, Y.; Ozaki, K.; Matsuo, T. Carnosic acid inhibits inflammatory cytokines production in human periodontal ligament cells. Immunopharmacol. Immunotoxicol. 2020, 42, 373–378. [Google Scholar] [CrossRef]
- Hosokawa, I.; Hosokawa, Y.; Ozaki, K.; Matsuo, T. Carnosic Acid Inhibits CXCR3 Ligands Production in IL-27-Stimulated Human Oral Epithelial Cells. Inflammation 2019, 42, 1311–1316. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, X.; Zhou, L.; Liu, Z.; Yuan, J.; Cheng, J.; Zhao, J.; Wu, L.; Li, H.; Qiu, H.; et al. Carnosic acid inhibits inflammation response and joint destruction on osteoclasts, fibroblast-like synoviocytes, and collagen-induced arthritis rats. J. Cell Physiol. 2018, 233, 6291–6303. [Google Scholar] [CrossRef]
- Tsai, Y.-F.; Yang, S.-C.; Hsu, Y.-H.; Chen, C.-Y.; Chen, P.-J.; Syu, Y.-T.; Lin, C.-H.; Hwang, T.G.-L. Carnosic acid inhibits reactive oxygen species-dependent neutrophil extracellular trap formation and ameliorates acute respiratory distress syndrome. Life Sci. 2022, 121334. [Google Scholar] [CrossRef]
- Kawamura, T.; Momozane, T.; Sanosaka, M.; Sugimura, K.; Iida, O.; Fuchino, H.; Funaki, S.; Shintani, Y.; Inoue, M.; Minami, M.; et al. Carnosol Is a Potent Lung Protective Agent: Experimental Study on Mice. Transplant. Proceed. 2015, 47, 1657–1661. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; de Souza, I.C.C.; Fürstenau, C.R. Carnosic Acid Induces Anti- Inflammatory Effects in Paraquat-Treated SH-SY5Y Cells Through a Mechanism Involving a Crosstalk Between the Nrf2/HO-1 Axis and NF-κB. Mol. Neurobiol. 2018, 55, 890–897. [Google Scholar] [CrossRef]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; de Galarreta, C.M.R.; Cuadrado, A. Regulation of Heme Oxygenase-1 Expression through the Phosphatidylinositol 3-Kinase/Akt Pathway and the Nrf2 Transcription Factor in Response to the Antioxidant Phytochemical Carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef]
- Wu, C.-R.; Tsai, C.-W.; Chang, S.-W.; Lin, C.-Y.; Huang, L.C.; Tsai, C.-W. Carnosic acid protects against 6-hydroxydopamine-induced neurotoxicity in in vivo and in vitro model of Parkinson’s disease: Involvement of antioxidative enzymes induction. Chemico-Biol. Inter. 2015, 225, 40–46. [Google Scholar] [CrossRef]
- Hou, C.-W.; Lin, Y.-T.; Chen, Y.-L.; Wang, Y.H.; Chou, J.L.; Ping, L.Y.; Jeng, K.C. Neuroprotective effects of carnosic acid on neuronal cells under ischemic and hypoxic stress. Nutr. Neurosci. 2012, 15, 257–263. [Google Scholar] [CrossRef]
- Meng, P.; Yoshida, H.; Matsumiya, T.; Imaizumi, T.; Tanji, K.; Xing, F.; Hayakari, R.; Dempoya, J.; Tatsut, T.; Aizawa-Yashiro, T.; et al. Carnosic acid suppresses the production of amyloid-β 1-42 by inducing the metalloprotease gene TACE/ADAM17 in SH-SY5Y human neuroblastoma cells. Neurosci. Res. 2013, 75, 94–102. [Google Scholar] [CrossRef]
- Yoshida, H.; Meng, P.; Matsumiya, T.; Tanji, K.; Hayakari, R.; Xing, F.; Wang, L.; Tsuruga, K.; Tanaka, H.; Mimura, J.; et al. Carnosic acid suppresses the production of amyloid-β 1-42 and 1-43 by inducing an α-secretase TACE/ADAM17 in U373MG human astrocytoma cells. Neurosci. Res. 2014, 79, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.W.; Liu, K.L.; Lin, Y.R.; Kuo, W.C. The mechanisms of carnosic acid attenuates tumor necrosis factor-α-mediated inflammation and insulin resistance in 3T3-L1 adipocytes. Mol. Nutr. Food Res. 2014, 58, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Dewanjee, S.; Joardar, S.; Chakraborty, P.; Bhattacharya, H.; Bhanja, S.; Bhattacharyya, C.; Bhowmik, M.; Bhowmick, S.; Saha, A.; et al. Carnosic acid attenuates doxorubicin-induced cardiotoxicity by decreasing oxidative stress and its concomitant pathological consequences. Food Chem. Toxicol. 2022, 166, 113205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.L.; Yang, J.J.; Zhang, H.S. Carvedilol (CAR) combined with carnosic acid (CAA) attenuates doxorubicin-induced cardiotoxicity by suppressing excessive oxidative stress, inflammation, apoptosis and autophagy. Biomed. Pharmacother. 2019, 109, 71–83. [Google Scholar] [CrossRef]
- Baradaran Rahimi, V.; Momeni-Moghaddam, M.A.; Chini, M.G.; Saviano, A.; Maione, F.; Bifulco, G.; Rahmanian-Devin, P.; Jebalbarezy, A.; Askari, V.R. Carnosol Attenuates LPS- Induced Inflammation of Cardiomyoblasts by Inhibiting NF-κB: A Mechanistic in Vitroand in SilicoStudy. Evid. Based Complement. Alternat. Med. 2022, 2022, 7969422. [Google Scholar] [CrossRef]
- Gao, L.; Shan, W.; Zeng, W.; Hu, Y.; Wang, G.; Tian, X.; Zhang, N.; Shi, X.; Zhao, Y.; Ding, C.; et al. Carnosic acid alleviates chronic alcoholic liver injury by regulating the SIRT1/ChREBP and SIRT1/p66shc pathways in rats. Mol. Nutr. Food Res. 2016, 60, 1902–1911. [Google Scholar] [CrossRef]
- Li, H.; Sun, J.J.; Chen, G.Y.; Wang, W.W.; Xie, Z.T.; Tang, G.F.; Wei, S.D. Carnosic acid nanoparticles suppress liver ischemia/reperfusion injury by inhibition of ROS, Caspases and NF-κB signaling pathway in mice. Biomed. Pharmacother. 2016, 82, 237–246. [Google Scholar] [CrossRef]
- Park, M.Y.; Mun, S.T. Carnosic acid inhibits TLR4-MyD88 signaling pathway in LPS- stimulated 3T3-L1 adipocytes. Nutr. Res. Pract. 2014, 8, 516–620. [Google Scholar] [CrossRef]
- D’Agata, V.; D’Amico, A.G.; Maugeri, G.; Bucolo, C.; Rossi, S.; Giunta, S. Carnosol attenuates high glucose damage in human retinal endothelial cells through regulation of ERK/Nrf2/HO-1 pathway. J. Asian Nat. Prod. Res. 2022; in press. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Q.; Hou, N.; Li, J.; Liu, M.; Peng, S.; Zhang, Y.; Luo, Y.; Zhao, B.; Wang, S.; et al. Carnosol as a Nrf2 Activator Improves Endothelial Barrier Function Through Antioxidative Mechanisms. Int. J. Mol. Sci. 2019, 20, 880. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, G.; Peng, K.; Gao, Y.; Wang, J.; Gao, C.; He, C.; Lu, F. Carnosol Maintains Intestinal Barrier Function and Mucosal Immune Homeostasis in DSS-Induced Colitis. Front. Nutr. 2022, 9, 894307. [Google Scholar] [CrossRef]
- Chrastina, M.; Poništ, S.; Tóth, J.; Czigle, S.; Pašková, Ľ.; Vyletelová, V.; Švík, K.; Bauerová, K. Combination Therapy of Carnosic Acid and Methotrexate Effectively Suppressed the Inflammatory Markers and Oxidative Stress in Experimental Arthritis. Molecules 2022, 27, 7115. [Google Scholar] [CrossRef]
- Xia, G.; Wang, X.; Sun, H.; Qin, Y.; Fu, M. Carnosic acid (CA) attenuates collagen- induced arthritis in db/db mice via inflammation suppression by regulating ROS- dependent p38 pathway. Free Radic. Biol. Med. 2017, 108, 418–432. [Google Scholar] [CrossRef]
- Li, L.; Pan, Z.; Ning, D.; Fu, Y. Rosmanol and Carnosol Synergistically Alleviate Rheumatoid Arthritis through Inhibiting TLR4/NF-κB/MAPK Pathway. Molecules 2022, 27, 78. [Google Scholar] [CrossRef]
- Li, Q.; Liu, L.; Sun, H.; Cao, K. Carnosic acid protects against lipopolysaccharide-induced acute lung injury in mice. Exp. Ther. Med. 2019, 18, 3707–3714. [Google Scholar] [CrossRef]
- Kalantar, H.; Sadeghi, E.; Abolnezhadian, F.; Goudarzi, M.; Hemmati, A.A.; Basir, Z.; Kalantar, M. Carnosol attenuates bleomycin-induced lung damage via suppressing fibrosis, oxidative stress and inflammation in rats. Life Sci. 2021, 287, 120059. [Google Scholar] [CrossRef]
- Lee, J.E.; Im, D.S. Suppressive Effect of Carnosol on Ovalbumin-Induced Allergic Asthma. Biomol. Ther. 2021, 29, 58–63. [Google Scholar] [CrossRef]
- da Rosa, J.S.; Facchin, B.M.; Bastos, J.; Siqueira, M.A.; Micke, G.A.; Dalmarco, E.M.; Pizzolatti, M.G.; Fröde, T.S. Systemic administration of Rosmarinus officinalis attenuates the inflammatory response induced by carrageenan in the mouse model of pleurisy. Planta Med. 2013, 79, 1605–1614. [Google Scholar] [CrossRef]
- Yeo, I.J.; Park, J.H.; Jang, J.S.; Lee, D.Y.; Park, J.E.; Choi, Y.E.; Joo, J.H.; Song, J.K.; Jeon, H.O.; Hong, J.T. Inhibitory effect of Carnosol on UVB-induced inflammation via inhibition of STAT3. Arch. Pharm. Res. 2019, 42, 274–283. [Google Scholar] [CrossRef]
- AlKahtane, A.A.; Ghanem, E.; Bungau, S.G.; Alarifi, S.; Ali, D.; AlBasher, G.; Alkahtani, S.; Aleya, L.; Abdel-Daim, M.M. Carnosic acid alleviates chlorpyrifos-induced oxidative stress and inflammation in mice cerebral and ocular tissues. Environ. Sci. Pollut. Res. Int. 2020, 27, 11663–11670. [Google Scholar] [CrossRef]
- Maynard, M.E.; Underwood, E.L.; Redell, J.B.; Zhao, J.; Kobori, N.; Hood, K.N.; Moore, A.N.; Dash, P.K. Carnosic Acid Improves Outcome after Repetitive Mild Traumatic Brain Injury. J. Neurotrauma 2019, 36, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Xie, Y.X.; Zhang, J.W.; Qiu, X.H.; Cheng, A.B.; Tian, L.; Ma, B.Y.; Hou, Y.B. Carnosol protects against spinal cord injury through Nrf-2 upregulation. J. Recept. Signal Transduct. Res. 2016, 36, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Singh, I.N.; Wang, J.A.; Hall, E.D. Nrf2–ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp. Neurol. 2015, 264, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Fan, L.; Peng, Y.; He, X.; Chen, H.; Duan, H.; Yang, F.; Lin, D.; Lin, Z.; Li, H.; et al. Carnosic Acid Mitigates Early Brain Injury After Subarachnoid Hemorrhage: Possible Involvement of the SIRT1/p66shc Signaling Pathway. Front. Neurosci. 2019, 13, 26. [Google Scholar] [CrossRef]
- Yi-Bin, W.; Xiang, L.; Bing, Y.; Qi, Z.; Fei-Tong, J.; Minghong, W.; Xiangxiang, Z.; Le, K.; Yan, L.; Ping, S.; et al. Inhibition of the CEBPβ-NFκB interaction by nanocarrier-packaged Carnosic acid ameliorates glia-mediated neuroinflammation and improves cognitive function in an Alzheimer’s disease model. Cell Death Dis. 2022, 13, 318. [Google Scholar] [CrossRef]
- Li, X.; Zhao, L.; Han, J.J.; Zhang, F.; Liu, S.; Zhu, L.; Wang, Z.Z.; Zhang, G.X.; Zhang, Y. Carnosol Modulates Th17 Cell Differentiation and Microglial Switch in Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2018, 9, 1807. [Google Scholar] [CrossRef]
- Samarghandian, S.; Borji, A.; Farkhondeh, T. Evaluation of Antidiabetic Activity of Carnosol (Phenolic Diterpene in Rosemary) in Streptozotocin-Induced Diabetic Rats. Cardiovasc. Hematol. Disord. Drug Targets 2017, 17, 11–17. [Google Scholar] [CrossRef]
- Ou, J.; Huang, J.; Zhao, D.; Du, B.; Wang, M. Protective effect of rosmarinic acid and carnosic acid against streptozotocin-induced oxidation, glycation, inflammation and microbiota imbalance in diabetic rats. Food Funct. 2018, 9, 851–860. [Google Scholar] [CrossRef]
- Xie, Z.; Zhong, L.; Wu, Y.; Wan, X.; Yang, H.; Xu, X.; Li, P. Carnosic acid improves diabetic nephropathy by activating Nrf2/ARE and inhibition of NF-κB pathway. Phytomedicine 2018, 47, 161–173. [Google Scholar] [CrossRef]
- Hu, M.; Li, T.; Bo, Z.; Xiang, F. The protective role of carnosic acid in ischemic/reperfusion injury through regulation of autophagy under T2DM. Exp. Biol. Med. 2019, 244, 602–611. [Google Scholar] [CrossRef]
- Song, H.M.; Li, X.; Liu, Y.Y.; Lu, W.P.; Cui, Z.H.; Zhou, L.; Yao, D.; Zhang, H.M. Carnosic acid protects mice from high-fat diet-induced NAFLD by regulating MARCKS. Int. J. Mol. Med. 2018, 42, 193–207. [Google Scholar] [CrossRef]
- Xiang, Q.; Liu, Z.; Wang, Y.; Xiao, H.; Wu, W.; Xiao, C.; Liu, X. Carnosic acid attenuates lipopolysaccharide-induced liver injury in rats via fortifying cellular antioxidant defense system. Food Chem. Toxicol. 2013, 53, 1–9. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hong, H.L.; Kim, G.M.; Leem, J.; Kwon, H.H. Protective Effects of Carnosic Acid on Lipopolysaccharide-Induced Acute Kidney Injury in Mice. Molecules 2021, 26, 7589. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Y.; Zheng, Y.; Zhang, N. Carnosol protects against renal ischemia-reperfusion injury in rats. Exp. Anim. 2018, 67, 545–553. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Hu, M.; Li, Y.H.; Cao, X.H. Carnosic acid alleviates brain injury through NF-κB-regulated inflammation and Caspase-3-associated apoptosis in high fat-induced mouse models. Mol. Med. Rep. 2019, 20, 495–504. [Google Scholar] [CrossRef]
- Tsai, T.H.; Chuang, L.T.; Lien, T.J.; Liing, Y.R.; Chen, W.Y.; Tsai, P.J. Rosmarinus officinalis extract suppresses Propionibacterium acnes-induced inflammatory responses. J. Med. Food. 2013, 16, 324–333. [Google Scholar] [CrossRef]
- Yousef, M.; Crozier, R.W.E.; Hicks, N.J.; Watson, C.J.F.; Boyd, T.; Tsiani, E.; MacNeil, A.J. Attenuation of allergen-mediated mast cell activation by rosemary extract (Rosmarinus officinalis L.). J. Leukoc. Biol. 2020, 107, 843–857. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, Y.; Yang, Y.; Ding, Y.; Sun, X. Carnosol suppresses microglia cell inflammation and apoptosis through PI3K/AKT/mTOR signaling pathway. Immunopharmacol. Immunotoxicol. 2022, 44, 656–662. [Google Scholar] [CrossRef]
- Pungsrinont, T.; Kallenbach, J.; Baniahmad, A. Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 11088. [Google Scholar] [CrossRef]
- Inoue, K.; Takano, H.; Shiga, A.; Fujita, Y.; Makino, H.; Yanagisawa, R.; Ichinose, T.; Kato, Y.; Yamada, T.; Yoshikawa, T. Effects of volatile constituents of a rosemary extract on allergic airway inflammation related to house dust mite allergen in mice. Int. J. Mol. Med. 2005, 16, 315–319. [Google Scholar] [CrossRef]
- Rodrigues, M.R.; Kanazawa, L.K.; das Neves, T.L.; da Silva, C.F.; Horst, H.; Pizzolatti, M.G.; Santos, A.R.; Baggio, C.H.; Werner, M.F. Antinociceptive and anti-inflammatory potential of extract and isolated compounds from the leaves of Salvia officinalis in mice. J. Ethnopharmacol. 2012, 139, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Emami, F.; Ali-Beig, H.; Farahbakhsh, S.; Mojabi, N.; Rastegar-Moghadam, B.; Arbabian, S.; Kazemi, M.; Tekieh, E.; Golmanesh, L.; Ranjbaran, M.; et al. Hydroalcoholic extract of Rosemary (Rosmarinus officinalis L.) and its constituent carnosol inhibit formalin-induced pain and inflammation in mice. Pak. J. Biol. Sci. 2013, 16, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Bakirel, T.; Bakirel, U.; Keleş, O.U.; Ulgen, S.G.; Yardibi, H. In Vivo Assessment of Antidiabetic and Antioxidant Activities of Rosemary (Rosmarinus officinalis) in Alloxan-Diabetic Rabbits. J. Ethnopharmacol. 2008, 116, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Eidi, M.; Eidi, A.; Zamanizadeh, H. Effect of Salvia officinalis L. leaves on serum glucose and insulin in healthy and streptozotocin-induced diabetic rats. J. Ethnopharmacol. 2005, 100, 310–313. [Google Scholar] [CrossRef]
- Lima, C.F.; Azevedo, M.F.; Araujo, R.; Fernandes-Ferreira, M.; Pereira-Wilson, C. Metformin-like effect of Salvia officinalis (common sage): Is it useful in diabetes prevention? Br. J. Nutr. 2006, 96, 326–333. [Google Scholar] [CrossRef]
- Emam, M. Comparative Evaluation of Antidiabetic Activity of Rosmarinus officinalis L. and Chamomile Recutita in Streptozotocin Induced Diabetic Rats. Agric. Biol. J. N. Am. 2012, 3, 247–252. [Google Scholar] [CrossRef]
- Ramadan, K.S.; Khalil, O.A.; Danial, E.N.; Alnahdi, H.S.; Ayaz, N.O. Hypoglycemic and Hepatoprotective Activity of Rosmarinus officinalis Extract in Diabetic Rats. J. Physiol. Biochem. 2013, 69, 779–783. [Google Scholar] [CrossRef]
- Hasei, S.; Yamamotoya, T.; Nakatsu, Y.; Ohata, Y.; Itoga, S.; Nonaka, Y.; Matsunaga, Y.; Sakoda, H.; Fujishiro, M.; Kushiyama, A.; et al. Carnosic Acid and Carnosol Activate AMPK, Suppress Expressions of Gluconeogenic and Lipogenic Genes, and Inhibit Proliferation of HepG2 Cells. Int. J. Mol. Sci. 2021, 22, 4040. [Google Scholar] [CrossRef]
- Vlavcheski, F.; Tsiani, E. Attenuation of Free Fatty Acid-Induced Muscle Insulin Resistance by Rosemary Extract. Nutrients 2018, 10, 1623. [Google Scholar] [CrossRef]
- Naimi, M.; Tsakiridis, T.; Stamatatos, T.C.; Alexandropoulos, D.I.; Tsiani, E. Increased Skeletal Muscle Glucose Uptake by Rosemary Extract through AMPK Activation. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Metab. 2015, 40, 407–413. [Google Scholar] [CrossRef]
- Naimi, M.; Vlavcheski, F.; Murphy, B.; Hudlicky, T.; Tsiani, E. Carnosic Acid as a Component of Rosemary Extract Stimulates Skeletal Muscle Cell Glucose Uptake via AMPK Activation. Clin. Exp. Pharmacol. Physiol. 2017, 44, 94–102. [Google Scholar] [CrossRef]
- Afonso, M.S.; de O Silva, A.M.; Carvalho, E.B.; Rivelli, D.P.; Barros, S.B.; Rogero, M.M.; Lottenberg, A.M.; Torres, R.P.; Mancini-Filho, J. Phenolic Compounds from Rosemary (Rosmarinus officinalis L.) Attenuate Oxidative Stress and Reduce Blood Cholesterol Concentrations in Diet-Induced Hypercholesterolemic Rats. Nutr. Metab. 2013, 10, 19. [Google Scholar] [CrossRef]
- Christensen, K.B.; Jørgensen, M.; Kotowska, D.; Petersen, R.K.; Kristiansen, K.; Christensen, L.P. Activation of the nuclear receptor PPARγ by metabolites isolated from sage (Salvia officinalis L.). J. Ethnopharmacol. 2010, 132, 127–133. [Google Scholar] [CrossRef]
- Park, M.-Y.; Mun, S.T. Dietary Carnosic Acid Suppresses Hepatic Steatosis Formation via Regulation of Hepatic Fatty Acid Metabolism in High-Fat Diet-Fed Mice. Nutr. Res. Pract. 2013, 7, 294–301. [Google Scholar] [CrossRef]
- Romo-Vaquero, M.; Selma, M.V.; Larrosa, M.; Obiol, M.; García-Villalba, R.; González-Barrio, R.; Issaly, N.; Flanagan, J.; Roller, M.; Tomás-Barberán, F.A.; et al. A rosemary extract rich in carnosic acid selectively modulates caecum microbiota and inhibits β-glucosidase activity, altering fiber and short chain fatty acids fecal excretion in lean and obese female rats. PLoS ONE 2014, 9, e94687. [Google Scholar] [CrossRef]
- Zhao, Y.; Sedighi, R.; Wang, P.; Chen, H.; Zhu, Y.; Sang, S. Carnosic Acid as a Major Bioactive Component in Rosemary Extract Ameliorates High-Fat-Diet-Induced Obesity and Metabolic Syndrome in Mice. J. Agric. Food Chem. 2015, 63, 4843–4852. [Google Scholar] [CrossRef]
- Rau, O.; Wurglics, M.; Paulke, A.; Zitzkowski, J.; Meindl, N.; Bock, A.; Dingermann, T.; Abdel-Tawab, M.; Schubert-Zsilavecz, M. Carnosic acid and carnosol, phenolic diterpene compounds of the labiate herbs rosemary and sage, are activators of the human peroxisome proliferator-activated receptor gamma. Planta Med. 2006, 72, 881–887. [Google Scholar] [CrossRef]
- Li, S.; Yang, H.; Li, L.; Wang, W.; Tan, H.Y.; Qu, Y.; Wang, D. The involvement of gut microbiota in the anti-tumor effect of carnosic acid via IL-17 suppression in colorectal cancer. Chem. Biol. Interact. 2022, 365, 110080. [Google Scholar] [CrossRef]
- He, X.; Zhang, M.; Li, S.T.; Li, X.; Huang, Q.; Zhang, K.; Zheng, X.; Xu, X.T.; Zhao, D.G.; Ma, Y.Y. Alteration of gut microbiota in high-fat diet-induced obese mice using carnosic acid from rosemary. Food Sci. Nutr. 2022, 10, 2325–2332. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Bahjat, F.R.; Pine, P.R.; Reitsma, A.; Cassafer, G.; Baluom, M.; Grillo, S.; Chang, B.; Zhao, F.F.; Payan, D.G.; Grossbard, E.B.; et al. An orally bioavailable spleen tyrosine kinase inhibitor delays disease progression and prolongs survival in murine lupus. Arthritis Rheum. 2008, 58, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.K.; Sriskantharajah, S.; Hessel, E.M.; Okkenhaug, K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.S.; Kim, H.G.; Kim, J.H.; Yang, W.S.; Kim, E.; Jeong, D.; Park, J.G.; Aziz, N.; Kim, S.; Parameswaran, N.; et al. Syk-MyD88 Axis Is a Critical Determinant of Inflammatory-Response in Activated Macrophages. Front. Immunol. 2021, 12, 767366. [Google Scholar] [CrossRef]
- Fu, Y.; Gao, R.; Cao, Y.; Guo, M.; Wei, Z.; Zhou, E.; Li, Y.; Yao, M.; Yang, Z.; Zhang, N. Curcumin attenuates inflammatory responses by suppressing TLR4-mediated NF-κB signaling pathway in lipopolysaccharide-induced mastitis in mice. Int. Immunopharmacol. 2014, 20, 54–58. [Google Scholar] [CrossRef]
- Meng, Z.; Yan, C.; Deng, Q.; Gao, D.F.; Niu, X.L. Curcumin inhibits LPS-induced inflammation in rat vascular smooth muscle cells in vitro via ROS-relative TLR4-MAPK/NF-κB pathways. Acta Pharmacol. Sin. 2013, 34, 901–911. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhan, L.; Liao, H.; Chen, L.; Lv, X. Curcumin improves TNBS-induced colitis in rats by inhibiting IL-27 expression via the TLR4/NF-κB signaling pathway. Plant. Med. 2013, 79, 102–109. [Google Scholar] [CrossRef]
- Youn, H.S.; Lee, J.Y.; Saitoh, S.I.; Miyake, K.; Kang, K.W.; Choi, Y.J.; Hwang, D.H. Suppression of MyD88- and TRIF-dependent signaling pathways of Toll-like receptor by (−)-epigallocatechin-3-gallate, a polyphenol component of green tea. Biochem. Pharmacol. 2006, 72, 850–859. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sudhakaran, P.R.; Helen, A. Quercetin attenuates atherosclerotic inflammation and adhesion molecule expression by modulating TLR-NF-κB signaling pathway. Cell Immunol. 2016, 310, 131–140. [Google Scholar] [CrossRef]
- Cianciulli, A.; Calvello, R.; Cavallo, P.; Dragone, T.; Carofiglio, V.; Panaro, M.A. Modulation of NF-κB activation by resveratrol in LPS treated human intestinal cells results in downregulation of PGE2 production and COX-2 expression. Toxicol. In Vitro 2012, 26, 1122–1128. [Google Scholar] [CrossRef]
- Jakus, P.B.; Kalman, N.; Antus, C.; Radnai, B.; Tucsek, Z.; Gallyas, F., Jr.; Sumegi, B.; Veres, B. TRAF6 is functional in inhibition of TLR4-mediated NF-κB activation by resveratrol. J. Nutr. Biochem. 2013, 24, 819–823. [Google Scholar] [CrossRef]
- Machado, T.R.; Machado, T.R.; Pascutti, P.J. The p38 MAPK Inhibitors and Their Role in Inflammatory Diseases. Chem. Eur. 2021, 6, 5729–5742. [Google Scholar] [CrossRef]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell. Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Westenberger, G.; Sellers, J.; Fernando, S.; Junkins, S.; Han, S.M.; Min, K.; Lawan, A. Function of Mitogen-Activated Protein Kinases in Hepatic Inflammation. J. Cell Signal. 2021, 2, 172–180. [Google Scholar]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef]
- Du, L. Doxorubicin-induced Toxicity Through the p38 MAPK Protein Kinase Pathway. Highlights Sci. Eng. Technol. 2022, 19, 9–15. [Google Scholar] [CrossRef]
- Guo, R.M.; Xu, W.M.; Lin, J.C.; Mo, L.Q.; Hua, X.X.; Chen, P.X.; Wu, K.; Zheng, D.D.; Feng, J.Q. Activation of the p38 MAPK/NF-κB pathway contributes to doxorubicin-induced inflammation and cytotoxicity in H9c2 cardiac cells. Mol. Med. Rep. 2013, 8, 603–608. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Jiang, S.; Qiu, Y.; Wang, Z.; Ji, Y.; Zhang, X.; Yan, X.; Zhan, Z. Carnosic Acid Induces Antiproliferation and Anti-Metastatic Property of Esophageal Cancer Cells via MAPK Signaling Pathways. J. Oncol. 2021, 2021, 4451533. [Google Scholar] [CrossRef]
- Wang, X.; Gupta, P.; Jramne, Y.; Danilenko, M.; Liu, D.; Studzinski, G.P. Carnosic acid increases sorafenib-induced inhibition of ERK1/2 and STAT3 signaling which contributes to reduced cell proliferation and survival of hepatocellular carcinoma cells. Oncotarget 2020, 11, 3129–3143. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.-Q.; Sethi, G.; Goh, B.-C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Kasembeli, M.M.; Bharadwaj, U.; Robinson, P.; Tweardy, D.J. Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. Int. J. Mol. Sci. 2018, 19, 2299. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, L.; Chen, X.; Lu, Q.; Yang, Y.; Liu, J.; Ma, X. SIRT1 counteracted the activation of STAT3 and NF-κB to repress the gastric cancer growth. Int. J. Clin. Exp. Med. 2014, 7, 5050–5058. [Google Scholar] [PubMed]
- Li, L.; Sun, Q.; Li, Y.; Yang, Y.; Yang, Y.; Chang, T.; Man, M.; Zheng, L. Overexpression of SIRT1 Induced by Resveratrol and Inhibitor of miR-204 Suppresses Activation and Proliferation of Microglia. J. Mol. Neurosci. 2015, 56, 858–867. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Q.; Zhang, Q.; Lu, Y.; Liu, J.; Li, W.; Lv, S.; Zhou, M.; Zhang, X.; Hang, C. Resveratrol Attenuates Early Brain Injury after Experimental Subarachnoid Hemorrhage via Inhibition of NLRP3 Inflammasome Activation. Front. Neurosci. 2017, 11, 611. [Google Scholar] [CrossRef]
- Zou, P.; Liu, X.; Li, G.; Wang, Y. Resveratrol pretreatment attenuates traumatic brain injury in rats by suppressing NLRP3 inflammasome activation via SIRT1. Mol. Med. Rep. 2018, 17, 3212–3217. [Google Scholar] [CrossRef]
- He, Q.; Li, Z.; Wang, Y.; Hou, Y.; Li, L.; Zhao, J. Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int. Immunopharmacol. 2017, 50, 208–215. [Google Scholar] [CrossRef]
- Sadeghi, A.; Seyyed Ebrahimi, S.S.; Golestani, A.; Meshkani, R. Resveratrol ameliorates palmitate-induced inflammation in skeletal muscle cells by attenuating oxidative stress and JNK/NF-κB pathway in a SIRT1-independent mechanism. J. Cel. Biochem. 2017, 118, 2654–2663. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, Q.; Wang, M.; Liang, M.; Yang, X.; Xu, X.; Zou, H.; Qiu, J. Activation of Sirt1 by resveratrol inhibits TNF-α induced inflammation in fibroblasts. PLoS ONE 2011, 6, e27081. [Google Scholar] [CrossRef]
- Li, D.; Liu, N.; Zhao, H.H.; Zhang, X.; Kawano, H.; Liu, L.; Zhao, L.; Li, H.P. Interactions between Sirt1 and MAPKs regulate astrocyte activation induced by brain injury in vitro and in vivo. J. Neuroinflamm. 2017, 14, 67. [Google Scholar] [CrossRef]
- Yang, H.; Gu, Z.T.; Li, L.; Maegele, M.; Zhou, B.Y.; Li, F.; Zhao, M.; Zhao, K.S. SIRT1 plays a neuroprotective role in traumatic brain injury in rats via inhibiting the p38 MAPK pathway. Acta Pharmacol. Sin. 2017, 38, 168–181. [Google Scholar] [CrossRef]
- Hung, C.H.; Chan, S.H.; Chu, P.M.; Tsai, K.L. Quercetin is a potent anti-atherosclerotic compound by activation of SIRT1 signaling under oxLDL stimulation. Mol. Nutr. Food Res. 2015, 59, 1905–1917. [Google Scholar] [CrossRef]
- Hu, T.; Shi, J.J.; Fang, J.; Wang, Q.; Chen, Y.B.; Zhang, S.J. Quercetin ameliorates diabetic encephalopathy through SIRT1/ER stress pathway in db/db mice. Aging 2020, 12, 7015–7029. [Google Scholar] [CrossRef]
- Chen, D.L.; Yang, K.Y. Berberine Alleviates Oxidative Stress in Islets of Diabetic Mice by Inhibiting miR-106b Expression and Up-Regulating SIRT1. J. Cell Biochem. 2017, 118, 4349–4357. [Google Scholar] [CrossRef]
- Wu, Y.Z.; Zhang, L.; Wu, Z.X.; Shan, T.T.; Xiong, C. Berberine Ameliorates Doxorubicin-Induced Cardiotoxicity via a SIRT1/p66Shc-Mediated Pathway. Oxid. Med. Cell Longev. 2019, 2019, 2150394. [Google Scholar] [CrossRef]
- Yang, Y.; Duan, W.; Lin, Y.; Yi, W.; Liang, Z.; Yan, J.; Wang, N.; Deng, C.; Zhang, S.; Li, Y.; et al. SIRT1 activation by curcumin pretreatment attenuates mitochondrial oxidative damage induced by myocardial ischemia reperfusion injury. Free Radic. Biol. Med. 2013, 65, 667–679. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, J.; Ye, B.; Wang, Q.; Xie, X.; Shen, H. Protective effect of curcumin on TNBS-induced intestinal inflammation is mediated through the JAK/STAT pathway. BMC Complement. Altern. 2016, 16, 299. [Google Scholar] [CrossRef]
- Habtemariam, S. The Molecular Pharmacology of Phloretin: Anti-Inflammatory Mechanisms of Action. Biomedicines 2023, 11, 143. [Google Scholar] [CrossRef]
- Habtemariam, S. The Nrf2/HO-1 Axis as Targets for Flavanones: Neuroprotection by Pinocembrin, Naringenin, and Eriodictyol. Oxid. Med. Cell Longev. 2019, 2019, 4724920. [Google Scholar] [CrossRef]
- Chen, J.; Huang, Z.; Cao, X.; Chen, X.; Zou, T.; You, J. Plant-Derived Polyphenols as Nrf2 Activators to Counteract Oxidative Stress and Intestinal Toxicity Induced by Deoxynivalenol in Swine: An Emerging Research Direction. Antioxidants 2022, 11, 2379. [Google Scholar] [CrossRef] [PubMed]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Nuñez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 2011, 187, 613–617. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Kong, R.; Sun, L.; Li, H.; Wang, D. The role of NLRP3 inflammasome in the pathogenesis of rheumatic disease. Autoimmunity 2022, 55, 1–7. [Google Scholar] [CrossRef]
- Lu, X.; Tan, Q.; Ma, J.; Zhang, J.; Yu, P. Emerging Role of LncRNA Regulation for NLRP3 Inflammasome in Diabetes Complications. Front. Cell. Dev. Biol. 2022, 9, 792401. [Google Scholar] [CrossRef]
- Vafaei, S.; Taheri, H.; Hajimomeni, Y.; Fakhre Yaseri, A.; Abolhasani Zadeh, F. The role of NLRP3 inflammasome in colorectal cancer: Potential therapeutic target. Clin. Transl. Oncol. 2022, 24, 1881–1889. [Google Scholar] [CrossRef]
- Liang, T.; Zhang, Y.; Wu, S.; Chen, Q.; Wang, L. The Role of NLRP3 Inflammasome in Alzheimer’s Disease and Potential Therapeutic Targets. Front. Pharmacol. 2022, 13, 845185. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, G.; Gurley, E.C.; Zhou, H. Flavonoid apigenin inhibits lipopolysaccharide-induced inflammatory response through multiple mechanisms in macrophages. PLoS ONE 2014, 9, e107072. [Google Scholar]
- Lee, H.E.; Yang, G.; Kim, N.D.; Jeong, S.; Jung, Y.; Choi, J.Y.; Park, H.H.; Lee, J.Y. Targeting ASC in NLRP3 inflammasome by caffeic acid phenethyl ester: A novel strategy to treat acute gout. Sci. Rep. 2016, 6, 38622. [Google Scholar] [CrossRef]
- Kong, F.; Ye, B.; Cao, J.; Cai, X.; Lin, L.; Huang, S.; Huang, W.; Huang, Z. Curcumin represses NLRP3 inflammasome activation via TLR4/MyD88/NF-κB and P2X7R signaling in PMA-induced macrophages. Front. Pharmacol. 2016, 7, 369. [Google Scholar] [CrossRef]
- Chang, Y.P.; Ka, S.M.; Hsu, W.H.; Chen, A.; Chao, L.K.; Lin, C.C.; Hsieh, C.C.; Chen, M.C.; Chiu, H.W.; Ho, C.L.; et al. Resveratrol inhibits NLRP3 inflammasome activation by preserving mitochondrial integrity and augmenting autophagy. J. Cell Physiol. 2015, 230, 1567–1579. [Google Scholar] [CrossRef]
- Tufekci, K.U.; Eltutan, B.I.; Isci, K.B.; Genc, S. Resveratrol Inhibits NLRP3 Inflammasome-Induced Pyroptosis and miR-155 Expression in Microglia Through Sirt1/AMPK Pathway. Neurotox. Res. 2021, 39, 1812–1829. [Google Scholar] [CrossRef]
- Xue, Y.; Du, M.; Zhu, M.J. Quercetin suppresses NLRP3 inflammasome activation in epithelial cells triggered by Escherichia coli O157:H7. Free Radic. Biol. Med. 2017, 108, 760–769. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).