Abstract
Stem cell-based therapies (SCT) to treat neurodegenerative disorders have promise but clinical trials have only recently begun, and results are not expected for several years. While most SCTs largely lead to a symptomatic therapeutic effect by replacing lost cell types, there may also be disease-modifying therapeutic effects. In fact, SCT may complement a multi-drug, subtype-specific therapeutic approach, consistent with the idea of precision medicine, which matches molecular therapies to biological subtypes of disease. In this narrative review, we examine published and ongoing trials in SCT in Parkinson’s Disease, atypical parkinsonian disorders, Huntington’s disease, amyotrophic lateral sclerosis, and spinocerebellar ataxia in humans. We discuss the benefits and pitfalls of using this treatment approach within the spectrum of disease-modification efforts in neurodegenerative diseases. SCT may hold greater promise in the treatment of neurodegenerative disorders, but much research is required to determine the feasibility, safety, and efficacy of these complementary aims of therapeutic efforts.
1. Introduction
Neurodegenerative disorders result from a complex interplay between genes and environment. Due to their complexity and our limited understanding of the underlying pathological mechanisms, modelling these diseases in the laboratory has proven to be extremely challenging. Many cell and animal models have provided important mechanistic insight into neurodegenerative diseases but, thus far, there is a disconnect between therapeutic successes in animal models and those in clinical trials in humans.
Human stem cells (pluripotent and multipotent) are increasingly being studied as models or therapies for human disease. In most cases, stem-cell based therapies (SCTs) for neurodegenerative disorders rely on replacing lost cell types (e.g., replacing degenerated cells with new ones), thus exerting a symptomatic therapeutic effect. However, while cell replacement may provide rescue and neurorestorative effects, it is likely that disease-modification should rely on precision medicine approaches, matching molecular therapies to biological subtypes of disease. Within the precision medicine approach, which involves combination of multi-drug treatments, rather than a monotherapy, SCT may play a significant contribution in the treatment of neurodegenerative disorders [1].
Over the years, there has been significant hope that SCT would lead to curative therapies in these diseases, but unfortunately, that has not been observed thus far. We propose that, unless it addresses the inciting etiology, which is expected to vary among affected individuals, it will never be completely curative. However, with the recent development of newer and more effective cell lineages, differentiation processes, and grafting techniques, the once-imagined regenerative utopia may still be possible. Many review articles have been written on the possible utilization of stem cell therapies in various animal models and/or in patients with dementia, but very few have specifically discussed this topic in patients with movement disorders [2,3,4,5,6]. This narrative review will cover the basics of SCT in neurodegenerative movement disorders, such as Parkinson’s Disease (PD), atypical parkinsonian disorders (APD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and spinocerebellar ataxia (SCA). We will also discuss the benefits and pitfalls of using this approach.
Stem cells can be classified on their intrinsic ability to differentiate into the end organism [7]. There are five main categories of stem cells: totipotent, pluripotent, multipotent, oligopotent, and unipotent (Glossary). Stem cells can also be categorized as embryonic stem cells (ESCs)—cells derived during early development—and adult stem cells— rare, undifferentiated cells present in many adult tissues [8]. Special attention has been given to pluripotent ESCs, which can differentiate into any embryonic cell; initial trials required harvesting it at the blastocyte stage, but, in 2007, induced adult pluripotent stem cells (iPSCs) were artificially reprogrammed back from human fibroblasts or blood cells [9]. The ability to develop iPSCs, a non-embryonic source of multipurposed cells, was a breakthrough that avoided many ethical pitfalls as they can be derived from the patient’s own cells (e.g., autologous stem cells), and they avoid the risk of immunological rejections that are associated with non-autologous or heterologous stem cells [10]. Mesenchymal cells derived from the mesoderm and neuroectoderm were initially obtained from bone marrow; common sources now include adipose tissue, placenta, and umbilical cord, and they have the ability to differentiate into cell types from all three embryonic layers [11]. Interestingly, they can grow towards inflammation through the expression of chemokine receptors, making it an attractive candidate for cell loss, secondary to inflammatory conditions [12]. Totipotent cells are infrequently used in research as they are difficult to isolate and, once again, ethical questions arise. The above provides a simplified description of these categories to understand the following concepts.
2. Methods
In the present narrative review, we searched PubMed until 10 November 2022, to identify key articles to screen for main results and for relevant bibliography. We mainly focused on the more informative studies in humans, with valid and clear methodology, and with the more recent dates of publication, if multiple similar studies were available. Single case reports were not considered, unless otherwise specified. Particular attention was given to multiple publications from the same trial with different follow-up periods.
In Parkinson’s disease (PD), we used the terms “Parkinsonism” OR “Parkinson’s Disease”, as well as “Stem Cell” OR “Allogenic” OR “autologous” OR “Transplantation”. We also applied the same search terms to search in ‘www.clincaltrial.gov’, and we selected “ongoing trials”. The search terms were purposely non-specific to allow for a greater number of results so that they could be filtered individually. This resulted in 60 trials. A large number were removed for non-stem cell related mechanisms, fecal transplants, other neurodegenerative conditions, and incomplete and self-terminated studies. This resulted in 15 studies (Table 1). In multiple system atrophy (MSA), progressive supranuclear palsy (PSP), cortico-basal syndrome (CBS), and dementia with Lewy bodies (DLB) we used the following search terms: “Multiple Systems Atrophy”, “MSA”, “Progressive Supranuclear Palsy”, “PSP”, “Corticobasal Degeneration”, “Corticobasal syndrome”, “CBD”, “CBS”, and “stem cell OR autologous OR transplant”. In HD we used the following search terms: “Huntington’s Disease” OR “Huntington’s Chorea” and “stem cell OR autologous OR transplant”. In ALS we used the following search terms: “ALS”, “Amyotrophic Lateral Sclerosis”, “motor neuron disease”, and “stem cell OR autologous OR transplant”. In SCA we used the following search terms: “spinocerebellar ataxia”, “SCA”, and “stem cell OR autologous OR transplant”.

Table 1.
Main published and current studies exploring stem cell treatments in patients with idiopathic Parkinson’s disease.
3. Parkinson’s Disease
PD is a neurodegenerative syndrome which results in a loss of dopaminergic neurons, leading to nigrostriatal degeneration [17]. The pathogenesis of neuronal degeneration in PD likely involves the polymerization of alpha-synuclein, with a subsequent loss of normal, soluble synuclein and degeneration. As such, PD belongs to the category of “synucleinopathies” [18]. While serotonin and acetylcholine are involved to some extent, the mainstay of therapy has always been and continues to be dopamine replacement [19]. The loss of dopaminergic neurons is mainly located in the substantia nigra (SN)-pars compacta and its projections to the striatum. SCT cannot address the disease-causative mechanisms but can replace dopaminergic-producing cells.
Stem cell transplants in PD started in the mid-1990s, with variable results. Olanow et al. showed that fetal stem cell transplants could improve motor symptoms (as measured by the Unified Parkinson’s Disease Rating Scale (UPDRS)-part III) up to 9 months after the transplant, and that this effect was not maintained at 12 or 24 months (e.g., primary endpoint not met) [14]. This transient improvement in UPDRS scores coincided with the duration of immune-suppressant post-transplant, suggesting a loss of the transplanted tissue. This was more evident in patients with somewhat less severe PD (UPDRS-III score < 49) when compared to those with more advanced PD (UPDRS-III score > 49). Overall, PD patients in early SCT trials had a range of responses; from no response, disabling dyskinesias (mainly due to heterogeneous fetal grafts containing both dopaminergic and serotonergic cells) to discontinuation of oral levodopa medication, it was hard to predict how patients would respond [20]. Failure of these trials was attributed to factors like including non-motor predominant patients, insufficient amount of transplanted tissue, older age, and more diffuse loss of dopaminergic neurons. Those who had dopamine neuronal loss, restricted to putamen, benefited more from this treatment [21]. A few encouraging single case reports have suggested that if the graft survives after immune-suppression discontinuation, and patients are properly selected, the effects of transplanted dopaminergic-producing cells could be tangible up to 20 years after the transplant [22,23]. However, these are observations based on single cases and it is hard to generalize their effect. Additionally, after decades of failures of SCT in PD [13,15,16,21] (Table 1), experts tried to review and devise new strategies for clinical trials. TRANSNEURO is a current, ongoing multicenter trial that involves implantation of allogenic human iPSCs-derived into the putamen and addresses some of these past limitations [16]. Preliminary data at 36 months on 11 subjects suggests the absence of disabling dyskinesias, continued deterioration of motor signs per Movement Disorders Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) part III, lack of evidence for association between disease duration and clinical outcomes, and no major cognitive problems [16]. Separately, Aspen Neuroscience (ANPD001) has started recruitment for their autologous, iPSCs-derived SCT for idiopathic PD. It will avoid the need for immunosuppressants but does come at the cost of developing personalized individual cell lines for each individual. The feasibility of this approach has already been demonstrated in a single patient in whom PET imaging showed graft survival and a 6% reduction of levodopa requirement at 24 months [24].
Currently, there are fifteen ongoing clinical trials on parkinsonism and SCT (Table 1). Out of these, thirteen deal specifically with PD, suggesting that momentum continues within this field. Limitations have included choosing the right stem cell source, creating a cost-effective process to derive cell lineage in sufficient quantities, proper patient selection (currently restricted to motor-predominant and levodopa-responsive individuals in “earlier” stages), appropriate placement of the graft with verification of synapse connection to host networks, and finally, ensuring the longevity of grafts. Of interest, ongoing pre-clinical and phase I trials are mainly using iPSCs or ESC, whereas phase II trials also use mesenchymal stem cells (MSC).
4. Atypical Parkinsonian Disorders
APD are a broad number of conditions with PD-like phenotypes and include MSA, PSP, CBS, and DLB [25]. APD are primarily characterized by the combination of parkinsonism with additional motor and non-motor features that are beyond the “classical” spectrum of idiopathic PD, with a more aggressive disease progression. From a pathophysiological standpoint, APD consist of the “synucleinopaties”, such as MSA and DLB (with the common hallmark of soluble α-synuclein loss with corresponding insoluble α-synuclein accumulation), and “tauopathies” (characterized by soluble tau loss with corresponding insoluble tau accumulation), which include PSP and CBS [18,25]. The main studies exploring the effects of stem cells in APD are presented in Table 2.
Table 2.
Main studies exploring stem cell treatments in patients with atypical parkinsonian syndromes.
MSA. Studies on stem cells in MSA patients have suggested a putative transient disease-modifying role of stem cells [30]. However, these studies have been conducted in a small number of patients (mainly MSA-cerebellar subtype and not on MSA-parkinsonian subtype) and in single centers, and without a double-blinded approach, implanting intravenous or intraarterial MSC [26,30]. These studies have not been followed by others on wider number of patients and other MSA subtypes; additionally, some of these studies have been published more than a decade ago without further confirmation studies [26], thus suggesting limited applications of stem cells for MSA patients. Furthermore, as multiple systems and cell types are affected in MSA, different cell types may be needed for a stem-cell based therapeutic approach in this condition [31].
PSP. It has been documented in PSP that bone marrow MSC can be safely used with a possible beneficial effect (or, at least, with stabilization of disease progression), after having excluded the placebo effect [27]. The rationale of MSC in PSP is not to replace diseased neurons, but rather to minimize the consequences of neural cell deterioration by using stem cells as treatment [28]. Single-case reports have documented encouraging results with intraarterial autologous adipose tissue-derived MSC [32] and umbilical cord blood stem cell transplantation [33] in patients with PSP.
CBS and DLB. Studies specifically conducted in patients with CBS and DLB are lacking. We only found a case series describing the outcomes of the intravenous administration of granulocyte colony stimulating factor (GCSF) (which stimulates the differentiation of hematopoietic stem cells) in patients with MSA, PSP, and CBS (n = 2). Patients with CBS showed improvement (n = 1) or stability (n = 1) in motor scales over the study period (3 months), but no follow-up was available [29].
5. Huntington’s Disease
HD is a neurodegenerative autosomal dominant condition associated with a CAG repeat expansion in the HTT gene, with the number of repeats shown to be inversely correlated with the disease severity and age at onset [34]. However, the recent failure of trials of the antisense oligonucleotide tominersen, aimed at reducing the “toxic” level of huntingtin (HTT) protein, possibly suggests that the “causal” role of HTT in this monogenic disease is hypothetical, and thus, that further studies are required to determine the neurodegenerative mechanisms associated with HD [35].
The clinical spectrum of HD includes motor (chorea, dystonia, ataxia) and cognitive-behavioral symptoms, due to a dysfunction in the striatum [36]. Only symptomatic treatments are available for HD. Some studies have tested the role of intrastriatal transplantation of fetal striatal neuroblast cells as a possible symptomatic, and in some cases, even putative rescue/neurorestorative disease-modifying treatment, showing some promising results at different follow-up periods [37,38,39,40,41,42,43] (Table 3). Nevertheless, as in APD, these open-label single-center studies were not followed by other trials on a broader number of participants, thus bringing into question the real effectiveness of their therapeutic approach. However, most studies have documented the overall safety of the transplant procedures in HD patients.
Table 3.
Main studies exploring stem cell treatments in patients with Huntington’s disease.
6. Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder characterized by the dysfunction of both upper and lower motor neurons, leading to muscle weakness and paralysis, and eventual death [44]. Studies in ALS models have suggested that the primary pathophysiological target should be the environment of the motor neuron rather than the motor neuron itself [45]. Studies with different stem cell types in ALS patients have documented variable results over years, but overall, stem cell transplantation may delay ALS progression, improving quality of life (Table 4) [46,47,48,49,50,51,52]. Although multiple molecules (e.g., Vascular endothelial growth factors—VEGF, angiopoietin-related growth factor—ANG, and transforming growth factor beta—TGF-β) have been investigated, we are still missing effective biological markers to predict the efficacy of MSC transplants in patients with ALS [53]. Importantly, female sex and a positive therapeutic response to the first stem cell infusion are predictors of the efficacy of treatment with MSC in ALS patients [54]. Interestingly, the rate of disease progression seems to be an important predictor of therapeutic response to MSC in ALS patients, meaning rapid progressors have a better therapeutic response [55]. A recent study has published preliminary results on 18 ALS patients implanted, in the spinal cord, with human neural progenitor cells transduced with GDNF, because animal studies showed their ability to differentiate to astrocytes, thus protecting motor neurons [56]. These preliminary data showed encouraging results in terms of safety and tolerability at 12 months in ALS patients; however, and more interestingly, data from autoptic studies in a subgroup of patients suggested graft survival and satisfactory GDNF production. In sum, the future applications of gene therapy, combined with stem cell infusion, will consist of bilateral cortical and spinal cord infusions of STC, transduced with GDNF, to have a trophic effect for first and second order motor neurons [57].
Table 4.
Main studies exploring stem cell treatments in patients with amyotrophic lateral sclerosis.
7. Spinocerebellar Ataxia
SCA consists of a heterogeneous group of autosomal-dominant neurodegenerative conditions associated with ataxia, often caused by exonic CAG trinucleotide repeat expansions, which cause long polyglutamine chains [34,58]. More than forty types of SCA have been described up to date, many of them with associated movement disorders [58]. Among possible disease-modifying therapies, treatments with MSC have been proposed for SCA with the idea of stimulating plasticity and cell differentiation in the cerebellum [59]. However, only a few studies have explicitly tested MSC in patients with SCA, with limited evidence [60,61,62] (Table 5). Based on the results of a recent systematic review and meta-analysis of stem cell therapy in SCA, we can say that there is low evidence for recommending stem cells for these heterogeneous conditions, and that trials with large sample sizes and a lower risk of bias are still missing [63].
Table 5.
Main studies exploring stem cell treatments in patients with spinocerebellar ataxia.
8. Current Challenges and Future Directions
In the present review, we have performed a narrative review of the known studies in human stem cell transplantation for patients with neurodegenerative movement disorders. Many of the examined trials have investigated the role of SCT for symptomatic treatment, while other studies have tried to investigate the possible disease-modifying role of SCT through the production of growing/trophic factors.
The limited results of these trials can be attributed to multiple issues. First, the complexity of the various diseases makes it difficult to generate animal models that accurately reproduce the full diversity of disease features. Studies on animal models using iPSC have been useful to address early-onset neurodevelopmental diseases (e.g., Rett or Dravet syndromes, spinal muscular atrophy, or Friedreich’s ataxia), but not late-onset neurodegenerative disorders, which are the focus of this review. This is due to intrinsic problems of iPSC, namely the lack of maturation of the iPSC-derived cell lines [64]. Additionally, the existing iPSC models are only able to reproduce cells that show the behavior and features of the fetal cells. Aging should be considered as an independent factor to allow the in vitro modeling of the late-onset neurodegenerative diseases [65]. An accurate reproduction of late-onset neurodegeneration is as difficult as it is challenging to physiologically induce ‘aging’ into these animal models. [65]. Lastly, animal models are models of a disease construct, as we have defined it, not of people affected (e.g., PD patients, HD patients, etc.). In the last decades, the scientific field has significantly progressed, thus offering a great knowledge about disease mechanisms, but very little is known about the molecular biology of people affected by these diseases. Hence, we should be aware that animal models may not always be suitable to keep testing our hypotheses.
Another challenge in SCT is delivering the cells to their target sites within the central nervous system (CNS) in a safe and consistent way. Peripheral delivery of SC through intravenous infusions can lead to sequestration in tissues and organs, like the spleen, liver, and lung, thus preventing their passage through the blood brain barrier (BBB) [66,67]. Intraarterial injections are more effective for neurological conditions [68], but this increased efficacy comes at the cost of potential safety issues, such as microvascular complications and higher mortality [69]. Other trials are currently proposing CT/MRI-guided neurosurgical injections of STC that have the advantage of reaching the target structures precisely, and less adverse events (mainly hemorrhages). On this basis, a n = 1 patient-funded, compassionate use personalized trial with autologous iPSCs has been recently conceived, and preliminary clinical results at 24 months are highly encouraging. As they require substantial resources, SCT trials have commonly occurred in single centers where expertise and resources can be concentrated. Recent ongoing trials, such as the TRANSNEURO, have attempted to account for this and they have illuminated the challenges faced by multiple trials, primarily with recruitment and timing stem cell obtainment and implantation [16].
To partially overcome these limitations, an innovative phase I/II clinical trial, using human ESC-derived midbrain dopamine neurons (MSK-DA01) for patients with advanced PD and older age (>74) [70], has recently been approved by the FDA (Dec 2020; NCT04802733). The results are not yet available. As with many trials in neurodegeneration, patient selection and the measurement of disease modification is of utmost importance. Many studies have only examined SCT in earlier stages of PD, in a certain phenotypic subtype of the disease in MSA (e.g., MSA-C versus MSA-P), or in APD, in the very advanced stages, as a “rescue” strategy in patients who are already in poor health. We believe that the correct selection of patients should not be based on clinical/phenotypical criteria, but rather on biological and molecular ones. The highly encouraging results of clinical trials in ALS have proven that female sex, rapid disease progression, and positive response to the first SCT infusion are good predictors of effectiveness [54,58]. Additionally, patients with motor-predominant PD and dopaminergic loss, confined to the putamen, tend to respond much better to SCT than those without these features [20]. True disease-modification effect is hard to measure in neurodegenerative disease due to the lack of precise biomarkers [71]. Moreover, it is difficult to distinguish between an early symptomatic therapeutic effect and a true disease-modifying effect, at least in early (untreated) disease stages [72]. The tested endpoints may be highly variable and include the time-to-milestones of disease progression, the change in the MDS-UPDRS (or other clinical scores), and modification in a given imaging biomarker of metabolic function (e.g., dopaminergic in parkinsonian syndromes) [72,73]. Future disease-modifying trials may abandon the subjective clinical scales and questionnaires in favor of the objective evaluations offered by technology-based objective measures, imaging, and/or new molecular markers [71,73,74,75].
A final consideration for SCT is related to their ethical and social aspects. In 2016, the International Society for Stem Cell Research (ISSCR) created a guideline to regulate the rigorous scientific inquiry and careful ethical deliberations regarding SCT and regenerative medicine [76]. These guidelines contain the ethical principles for guiding both basic and clinical SCT research by regulating and codifying the integrity of research, patients’ welfare, respect for research participants, transparency, and finally, social justice. The discovery of iPSCs helped avoid some of the previously raised issues, but these guidelines will continue to play a large role as induced totipotent cells became a reality [77]. Lastly, researchers are increasingly debating the ethical ramifications of performing sham procedures. This includes procedures, such as sham neurosurgical injections, in double blinded trials. There is a possibility that researchers may abandon this process, opting for open label, single arm trials.
9. Final Remarks
Overall, interest in SCT has waxed and waned over more than thirty years, but there is a recent, significant renewed interest. We have since realized that the etiopathogenesis of neurodegenerative diseases is far more complex than previously thought and that each entity might not represent one unifying diagnosis, but rather, the gross pathology represents the end-product of various, unique etiologies [78]. Nevertheless, gold-standard diagnosis continues to rely on pathology [79,80]. Clinical and prognostic heterogeneity is the rule rather than the exception and affects patient selection and the ability of each body to accept the grafts. There is significant work underway in hopes of uncovering biomarkers that may better help identifying these various etiologies, perhaps leading to curative treatments in very select individuals. As displayed in the Figure 1, success in SCT will involve choosing the appropriate stem cell line from which to derive iPSCs, creating a streamlined, cost-effective, and scalable method in which to produce stem cells, ensuring safe delivery of stem cells past the BBB, in sufficient quantities, to the ideal location with synapses connection and growth, clinically relevant measurement of graft success, and lastly, appropriate patient selection and representation in trials. Despite the still limited knowledge on the mechanisms of neurodegeneration, we believe that future SCT may help in tackling some specific subtypes of neurodegenerative diseases. To this aim, we should move away from the old clinicopathology-based nosology of neurodegenerative disorders and focus on their underlining biological mechanisms [80,81]. It remains to be seen what future studies will uncover; if, indeed, we are able to accomplish the above, it may prove to be an ideal symptomatic therapy for certain individuals. As an example, around 10% of pathology-proven PD cases are unresponsive to dopaminergic treatment, and an additional 12% have a modest response [82]; these patients, to our knowledge, could be suitable candidates to symptomatic SCT based on dopaminergic neuronal transplantation. Other than that, SCT may also be useful for disease-modifying treatment options, which rely on precision medicine approaches. In fact, SCT may hold greater promise in the treatment of neurodegenerative disorders as part of a multi-drug, subtype-specific therapeutic approach, but much research is required to determine the feasibility, safety, and efficacy of these complementary aims of therapeutic efforts (Figure 1).
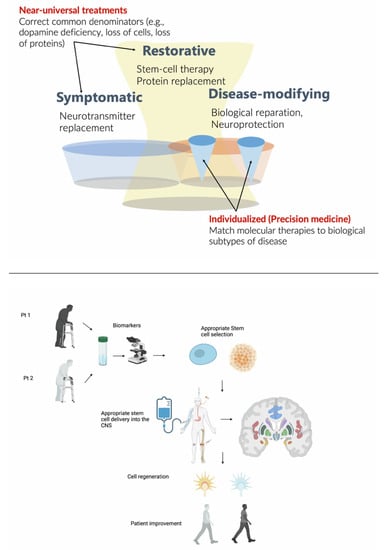
Figure 1.
Schematic representation of the main theme of the review.
Upper panel: Stem cell therapies (SCT) may have both symptomatic and disease-modifying effects. The diagram also illustrates a major difference between symptomatic treatments, which are largely universal, versus disease-modifying treatments, which encompass both rescue and restorative treatments and precision medicine, which must be individualized, requiring biological subtyping. Lower panel: Stem cell therapies in the future should be based on biological markers of disease progression and administered through autologous transplantation, possibly directly injected into the brain target sites. This approach will hopefully allow cell regeneration and ultimately, a possible restorative disease-modifying effect improving patients’ symptoms. Created with BioRender.com.
10. Glossary
- Totipotent cells can differentiate into any cell in the body—both embryonic and extra-embryonic tissue, such as the placenta. In mammals, they consist of the zygote (fertilized egg) and the first blastomeres cells (up to stages 4–8 cells).
- Pluripotent cells include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). Pluripotent cells can differentiate in any embryonic cell but cannot differentiate any extra-embryonic tissue. It can form all three germ layers and is formed from the inner cell mass of a blastocyst; these are specifically named embryonic stem cells (ESC).
- Multipotent cells can differentiate into a family of cells that, in many cases, belong to the same tissue (e.g., these cells can develop only into specialized cell types). Examples include Neural Stem Cells (NSCs), which can become any cell part of the central nervous system.
- Oligopotent cells can differentiate into a limited number of different cells.
- Unipotent cells can generate a single type of cell.
Author Contributions
Conceptualization, L.M., J.S. and C.C.; methodology, L.M. and J.S.; investigation, L.M. and J.S.; data curation, L.M. and J.S.; writing—original draft preparation, L.M and J.S.; writing—review and editing, T.F.O. and C.C.; supervision, C.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank the valuable feedback from Jeanne Loring and Alberto Espay, which contributed to improving this manuscript. Alberto Espay also contributed to the figure creation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Espay, A.J. Your After-Visit Summary-May 29, 2042. Lancet Neurol. 2022, 21, 412–413. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Herrero, M.; Soto-Rojas, L.O.; Reyes-Sabater, H.; Garcés-Ramirez, L.; de la Cruz López, F.; Villanueva-Fierro, I.; Luna-Muñoz, J. Current Status and Challenges of Stem Cell Treatment for Alzheimer’s Disease. J. Alzheimers Dis. 2021, 84, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Guttikonda, S.R.; Sikkema, L.; Tchieu, J.; Saurat, N.; Walsh, R.M.; Harschnitz, O.; Ciceri, G.; Sneeboer, M.; Mazutis, L.; Setty, M.; et al. Fully defined human pluripotent stem cell-derived microglia and tri-culture system model C3 production in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 343–354. [Google Scholar] [CrossRef]
- García-León, J.A.; Cabrera-Socorro, A.; Eggermont, K.; Swijsen, A.; Terryn, J.; Fazal, R.; Nami, F.; Ordovás, L.; Quiles, A.; Lluis, F.; et al. Generation of a human induced pluripotent stem cell-based model for tauopathies combining three microtubule-associated protein TAU mutations which displays several phenotypes linked to neurodegeneration. Alzheimers Dement. 2018, 14, 1261–1280. [Google Scholar] [CrossRef]
- Lines, G.; Casey, J.M.; Preza, E.; Wray, S. Modelling frontotemporal dementia using patient-derived induced pluripotent stem cells. Mol. Cell Neurosci. 2020, 109, 103553. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Kao, A.W.; Karydas, A.; Onanuga, K.; Martinez, R.; Argouarch, A.; Wang, C.; Huang, C.; Sohn, P.D.; Bowles, K.R.; et al. A Comprehensive Resource for Induced Pluripotent Stem Cells from Patients with Primary Tauopathies. Stem Cell Rep. 2019, 13, 939–955. [Google Scholar] [CrossRef]
- Adami, R.; Bottai, D. Spinal Muscular Atrophy Modeling and Treatment Advances by Induced Pluripotent Stem Cells Studies. Stem Cell Rev. Rep. 2019, 15, 795–813. [Google Scholar] [CrossRef]
- Prochazkova, M.; Chavez, M.G.; Prochazka, J.; Felfy, H.; Mushegyan, V.; Klein, O.D. Chapter 18—Embryonic Versus Adult Stem Cells. In Stem Cell Biology and Tissue Engineering in Dental Sciences; Ajaykumar Vishwakarma, P.S., Shi, S., Ramalingam, M., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 249–262. [Google Scholar]
- Zhang, X.; Hu, D.; Shang, Y.; Qi, X. Using induced pluripotent stem cell neuronal models to study neurodegenerative diseases. Biochim Biophys Acta Mol. Basis Dis. 2020, 1866, 165431. [Google Scholar] [CrossRef]
- Omole, A.E.; Fakoya, A.O.J. Ten years of progress and promise of induced pluripotent stem cells: Historical origins, characteristics, mechanisms, limitations, and potential applications. PeerJ 2018, 6, e4370. [Google Scholar] [CrossRef]
- Ferroni, L.; Gardin, C.; Tocco, I.; Epis, R.; Casadei, A.; Vindigni, V.; Mucci, G.; Zavan, B. Potential for neural differentiation of mesenchymal stem cells. Adv. Biochem Eng. Biotechnol 2013, 129, 89–115. [Google Scholar] [CrossRef]
- Hernández, R.; Jiménez-Luna, C.; Perales-Adán, J.; Perazzoli, G.; Melguizo, C.; Prados, J. Differentiation of Human Mesenchymal Stem Cells towards Neuronal Lineage: Clinical Trials in Nervous System Disorders. Biomol. Ther. (Seoul) 2020, 28, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Freed, C.R.; Greene, P.E.; Breeze, R.E.; Tsai, W.Y.; DuMouchel, W.; Kao, R.; Dillon, S.; Winfield, H.; Culver, S.; Trojanowski, J.Q.; et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N. Engl. J. Med. 2001, 344, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Goetz, C.G.; Kordower, J.H.; Stoessl, A.J.; Sossi, V.; Brin, M.F.; Shannon, K.M.; Nauert, G.M.; Perl, D.P.; Godbold, J.; et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann. Neurol. 2003, 54, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Tang, C.; Chaly, T.; Greene, P.; Breeze, R.; Fahn, S.; Freed, C.; Dhawan, V.; Eidelberg, D. Dopamine cell implantation in Parkinson’s disease: Long-term clinical and (18)F-FDOPA PET outcomes. J. Nucl. Med. 2010, 51, 7–15. [Google Scholar] [CrossRef]
- Barker, R.A. Designing stem-cell-based dopamine cell replacement trials for Parkinson’s disease. Nat. Med. 2019, 25, 1045–1053. [Google Scholar] [CrossRef]
- Uwishema, O.; Onyeaka, H.; Badri, R.; Yücel, A.N.; Korkusuz, A.K.; Ajagbe, A.O.; Abuleil, A.; Chaaya, C.; Alhendawi, B.H.M.; Chalhoub, E. The understanding of Parkinson’s disease through genetics and new therapies. Brain Behav. 2022, 12, e2577. [Google Scholar] [CrossRef]
- Marsili, L.; Rizzo, G.; Colosimo, C. Diagnostic Criteria for Parkinson’s Disease: From James Parkinson to the Concept of Prodromal Disease. Front. Neurol. 2018, 9, 156. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. Jama 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Gravitz, L. The promise and potential of stem cells in Parkinson’s disease. Nature 2021, 597, 8–10. [Google Scholar] [CrossRef]
- Lindvall, O.; Rehncrona, S.; Brundin, P.; Gustavii, B.; Astedt, B.; Widner, H.; Lindholm, T.; Björklund, A.; Leenders, K.L.; Rothwell, J.C.; et al. Human fetal dopamine neurons grafted into the striatum in two patients with severe Parkinson’s disease. A detailed account of methodology and a 6-month follow-up. Arch. Neurol. 1989, 46, 615–631. [Google Scholar] [CrossRef]
- Piccini, P.; Brooks, D.J.; Björklund, A.; Gunn, R.N.; Grasby, P.M.; Rimoldi, O.; Brundin, P.; Hagell, P.; Rehncrona, S.; Widner, H.; et al. Dopamine release from nigral transplants visualized in vivo in a Parkinson’s patient. Nat. Neurosci. 1999, 2, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Kefalopoulou, Z.; Politis, M.; Piccini, P.; Mencacci, N.; Bhatia, K.; Jahanshahi, M.; Widner, H.; Rehncrona, S.; Brundin, P.; Björklund, A.; et al. Long-term clinical outcome of fetal cell transplantation for Parkinson disease: Two case reports. JAMA Neurol. 2014, 71, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Marsili, L.; Bologna, M.; Kojovic, M.; Berardelli, A.; Espay, A.J.; Colosimo, C. Dystonia in atypical parkinsonian disorders. Parkinsonism Relat. Disord. 2019, 66, 25–33. [Google Scholar] [CrossRef]
- Lee, P.H.; Lee, J.E.; Kim, H.S.; Song, S.K.; Lee, H.S.; Nam, H.S.; Cheong, J.W.; Jeong, Y.; Park, H.J.; Kim, D.J.; et al. A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann. Neurol. 2012, 72, 32–40. [Google Scholar] [CrossRef]
- Giordano, R.; Canesi, M.; Isalberti, M.; Isaias, I.U.; Montemurro, T.; Viganò, M.; Montelatici, E.; Boldrin, V.; Benti, R.; Cortelezzi, A.; et al. Autologous mesenchymal stem cell therapy for progressive supranuclear palsy: Translation into a phase I controlled, randomized clinical study. J. Transl. Med. 2014, 12, 14. [Google Scholar] [CrossRef]
- Canesi, M.; Giordano, R.; Lazzari, L.; Isalberti, M.; Isaias, I.U.; Benti, R.; Rampini, P.; Marotta, G.; Colombo, A.; Cereda, E.; et al. Finding a new therapeutic approach for no-option Parkinsonisms: Mesenchymal stromal cells for progressive supranuclear palsy. J. Transl. Med. 2016, 14, 127. [Google Scholar] [CrossRef]
- Pezzoli, G.; Tesei, S.; Canesi, M.; Sacilotto, G.; Vittorio, M.; Mizuno, Y.; Mochizuki, H.; Antonini, A. The effect of repeated administrations of granulocyte colony stimulating factor for blood stem cells mobilization in patients with progressive supranuclear palsy, corticobasal degeneration and multiple system atrophy. Clin. Neurol. Neurosurg. 2010, 112, 65–67. [Google Scholar] [CrossRef]
- Chung, S.J.; Lee, T.Y.; Lee, Y.H.; Baik, K.; Jung, J.H.; Yoo, H.S.; Shim, C.J.; Eom, H.; Hong, J.Y.; Kim, D.J.; et al. Phase I Trial of Intra-arterial Administration of Autologous Bone Marrow-Derived Mesenchymal Stem Cells in Patients with Multiple System Atrophy. Stem Cells Int. 2021, 2021, 9886877. [Google Scholar] [CrossRef]
- Ndayisaba, A.; Herrera-Vaquero, M.; Wenning, G.K.; Stefanova, N. Induced pluripotent stem cells in multiple system atrophy: Recent developments and scientific challenges. Clin. Auton Res. 2019, 29, 385–395. [Google Scholar] [CrossRef]
- Choi, S.W.; Park, K.B.; Woo, S.K.; Kang, S.K.; Ra, J.C. Treatment of progressive supranuclear palsy with autologous adipose tissue-derived mesenchymal stem cells: A case report. J. Med. Case Rep. 2014, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yuan, F.; Du, Y.; Pan, T.; Wen, W.; Li, S.; Wang, L.; Lu, A. Umbilical cord blood stem cells transplantation in a patient with severe progressive supranuclear palsy: A case report. J. Med. Case Rep. 2021, 15, 574. [Google Scholar] [CrossRef] [PubMed]
- Marsili, L.; Duque, K.R.; Bode, R.L.; Kauffman, M.A.; Espay, A.J. Uncovering essential tremor genetics: The promise of long-read sequencing. Front. Neurol. 2022, 13, 821189. [Google Scholar] [PubMed]
- Tabrizi, S.J.; Estevez-Fraga, C.; van Roon-Mom, W.M.C.; Flower, M.D.; Scahill, R.I.; Wild, E.J.; Muñoz-Sanjuan, I.; Sampaio, C.; Rosser, A.E.; Leavitt, B.R. Potential disease-modifying therapies for Huntington’s disease: Lessons learned and future opportunities. Lancet Neurol. 2022, 21, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.J.; Dunnett, S.B.; Isacson, O.; Sirinathsinghji, D.J.; Björklund, A. Striatal grafts in rats with unilateral neostriatal lesions--I. Ultrastructural evidence of afferent synaptic inputs from the host nigrostriatal pathway. Neuroscience 1988, 24, 791–801. [Google Scholar] [CrossRef]
- Bachoud-Lévi, A.C.; Rémy, P.; Nguyen, J.P.; Brugières, P.; Lefaucheur, J.P.; Bourdet, C.; Baudic, S.; Gaura, V.; Maison, P.; Haddad, B.; et al. Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet 2000, 356, 1975–1979. [Google Scholar] [CrossRef]
- Bachoud-Lévi, A.C.; Gaura, V.; Brugières, P.; Lefaucheur, J.P.; Boissé, M.F.; Maison, P.; Baudic, S.; Ribeiro, M.J.; Bourdet, C.; Remy, P.; et al. Effect of fetal neural transplants in patients with Huntington’s disease 6 years after surgery: A long-term follow-up study. Lancet Neurol. 2006, 5, 303–309. [Google Scholar] [CrossRef]
- Hauser, R.A.; Furtado, S.; Cimino, C.R.; Delgado, H.; Eichler, S.; Schwartz, S.; Scott, D.; Nauert, G.M.; Soety, E.; Sossi, V.; et al. Bilateral human fetal striatal transplantation in Huntington’s disease. Neurology 2002, 58, 687–695. [Google Scholar] [CrossRef]
- Reuter, I.; Tai, Y.F.; Pavese, N.; Chaudhuri, K.R.; Mason, S.; Polkey, C.E.; Clough, C.; Brooks, D.J.; Barker, R.A.; Piccini, P. Long-term clinical and positron emission tomography outcome of fetal striatal transplantation in Huntington’s disease. J. Neurol Neurosurg Psychiatry 2008, 79, 948–951. [Google Scholar] [CrossRef]
- Paganini, M.; Biggeri, A.; Romoli, A.M.; Mechi, C.; Ghelli, E.; Berti, V.; Pradella, S.; Bucciantini, S.; Catelan, D.; Saccardi, R.; et al. Fetal striatal grafting slows motor and cognitive decline of Huntington’s disease. J. Neurol. Neurosurg Psychiatry 2014, 85, 974–981. [Google Scholar] [CrossRef]
- Barker, R.A.; Mason, S.L.; Harrower, T.P.; Swain, R.A.; Ho, A.K.; Sahakian, B.J.; Mathur, R.; Elneil, S.; Thornton, S.; Hurrelbrink, C.; et al. The long-term safety and efficacy of bilateral transplantation of human fetal striatal tissue in patients with mild to moderate Huntington’s disease. J. Neurol. Neurosurg Psychiatry 2013, 84, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Bachoud-Lévi, A.; Bourdet, C.; Brugières, P.; Nguyen, J.P.; Grandmougin, T.; Haddad, B.; Jény, R.; Bartolomeo, P.; Boissé, M.F.; Barba, G.D.; et al. Safety and tolerability assessment of intrastriatal neural allografts in five patients with Huntington’s disease. Exp. Neurol. 2000, 161, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R. Amyotrophic lateral sclerosis--are microglia killing motor neurons? N Engl. J. Med. 2006, 355, 1611–1613. [Google Scholar] [CrossRef] [PubMed]
- Martinez, H.R.; Gonzalez-Garza, M.T.; Moreno-Cuevas, J.E.; Caro, E.; Gutierrez-Jimenez, E.; Segura, J.J. Stem-cell transplantation into the frontal motor cortex in amyotrophic lateral sclerosis patients. Cytotherapy 2009, 11, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Deda, H.; Inci, M.C.; Kürekçi, A.E.; Sav, A.; Kayihan, K.; Ozgün, E.; Ustünsoy, G.E.; Kocabay, S. Treatment of amyotrophic lateral sclerosis patients by autologous bone marrow-derived hematopoietic stem cell transplantation: A 1-year follow-up. Cytotherapy 2009, 11, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.D.; Boulis, N.M.; Johe, K.; Rutkove, S.B.; Federici, T.; Polak, M.; Kelly, C.; Feldman, E.L. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: Results of a phase I trial in 12 patients. Stem Cells 2012, 30, 1144–1151. [Google Scholar] [CrossRef]
- Oh, K.W.; Moon, C.; Kim, H.Y.; Oh, S.I.; Park, J.; Lee, J.H.; Chang, I.Y.; Kim, K.S.; Kim, S.H. Phase I trial of repeated intrathecal autologous bone marrow-derived mesenchymal stromal cells in amyotrophic lateral sclerosis. Stem Cells Transl. Med. 2015, 4, 590–597. [Google Scholar] [CrossRef]
- Riley, J.; Federici, T.; Polak, M.; Kelly, C.; Glass, J.; Raore, B.; Taub, J.; Kesner, V.; Feldman, E.L.; Boulis, N.M. Intraspinal stem cell transplantation in amyotrophic lateral sclerosis: A phase I safety trial, technical note, and lumbar safety outcomes. Neurosurgery 2012, 71, 405–416, discussion 416. [Google Scholar] [CrossRef]
- Mazzini, L.; Gelati, M.; Profico, D.C.; Sorarù, G.; Ferrari, D.; Copetti, M.; Muzi, G.; Ricciolini, C.; Carletti, S.; Giorgi, C.; et al. Results from Phase I Clinical Trial with Intraspinal Injection of Neural Stem Cells in Amyotrophic Lateral Sclerosis: A Long-Term Outcome. Stem Cells Transl. Med. 2019, 8, 887–897. [Google Scholar] [CrossRef]
- Petrou, P.; Kassis, I.; Yaghmour, N.E.; Ginzberg, A.; Karussis, D. A phase II clinical trial with repeated intrathecal injections of autologous mesenchymal stem cells in patients with amyotrophic lateral sclerosis. Front. Biosci. (Landmark Ed.) 2021, 26, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, H.; Oh, K.W.; Oh, S.I.; Koh, S.H.; Baik, W.; Noh, M.Y.; Kim, K.S.; Kim, S.H. Biological markers of mesenchymal stromal cells as predictors of response to autologous stem cell transplantation in patients with amyotrophic lateral sclerosis: An investigator-initiated trial and in vivo study. Stem Cells 2014, 32, 2724–2731. [Google Scholar] [CrossRef] [PubMed]
- Barczewska, M.; Maksymowicz, S.; Zdolińska-Malinowska, I.; Siwek, T.; Grudniak, M. Umbilical Cord Mesenchymal Stem Cells in Amyotrophic Lateral Sclerosis: An Original Study. Stem Cell Rev. Rep. 2020, 16, 922–932. [Google Scholar] [CrossRef]
- Siwek, T.; Jezierska-Woźniak, K.; Maksymowicz, S.; Barczewska, M.; Sowa, M.; Badowska, W.; Maksymowicz, W. Repeat Administration of Bone Marrow-Derived Mesenchymal Stem Cells for Treatment of Amyotrophic Lateral Sclerosis. Med. Sci. Monit. 2020, 26, e927484. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H.; Johnson, J.P.; Avalos, P.; Allred, P.; Svendsen, S.; Gowing, G.; Roxas, K.; Wu, A.; Donahue, B.; Osborne, S.; et al. Transplantation of human neural progenitor cells secreting GDNF into the spinal cord of patients with ALS: A phase 1/2a trial. Nat. Med. 2022, 28, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Okano, H. A combined stem-cell-gene therapy strategy for ALS. Nat. Med. 2022, 28, 1751–1752. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Yau, W.Y.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef]
- Bang, O.Y.; Lee, J.S.; Lee, P.H.; Lee, G. Autologous mesenchymal stem cell transplantation in stroke patients. Ann. Neurol. 2005, 57, 874–882. [Google Scholar] [CrossRef]
- Dongmei, H.; Jing, L.; Mei, X.; Ling, Z.; Hongmin, Y.; Zhidong, W.; Li, D.; Zikuan, G.; Hengxiang, W. Clinical analysis of the treatment of spinocerebellar ataxia and multiple system atrophy-cerebellar type with umbilical cord mesenchymal stromal cells. Cytotherapy 2011, 13, 913–917. [Google Scholar] [CrossRef]
- Jin, J.L.; Liu, Z.; Lu, Z.J.; Guan, D.N.; Wang, C.; Chen, Z.B.; Zhang, J.; Zhang, W.Y.; Wu, J.Y.; Xu, Y. Safety and efficacy of umbilical cord mesenchymal stem cell therapy in hereditary spinocerebellar ataxia. Curr. Neurovasc. Res. 2013, 10, 11–20. [Google Scholar] [CrossRef]
- Tsai, Y.A.; Liu, R.S.; Lirng, J.F.; Yang, B.H.; Chang, C.H.; Wang, Y.C.; Wu, Y.S.; Ho, J.H.; Lee, O.K.; Soong, B.W. Treatment of Spinocerebellar Ataxia With Mesenchymal Stem Cells: A Phase I/IIa Clinical Study. Cell Transplant. 2017, 26, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Appelt, P.A.; Comella, K.; de Souza, L.; Luvizutto, G.J. Effect of stem cell treatment on functional recovery of spinocerebellar ataxia: Systematic review and meta-analysis. Cerebellum Ataxias 2021, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Sabitha, K.R.; Shetty, A.K.; Upadhya, D. Patient-derived iPSC modeling of rare neurodevelopmental disorders: Molecular pathophysiology and prospective therapies. Neurosci. Biobehav. Rev. 2021, 121, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Vera, E.; Studer, L. When rejuvenation is a problem: Challenges of modeling late-onset neurodegenerative disease. Development 2015, 142, 3085–3089. [Google Scholar] [CrossRef] [PubMed]
- Low, P.A.; Gilman, S. Are trials of intravascular infusions of autologous mesenchymal stem cells in patients with multiple system atrophy currently justified, and are they effective? Ann. Neurol. 2012, 72, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S., Jr. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first-pass effect. Stem Cells Dev. 2009, 18, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Guzman, R.; De Los Angeles, A.; Cheshier, S.; Choi, R.; Hoang, S.; Liauw, J.; Schaar, B.; Steinberg, G. Intracarotid injection of fluorescence activated cell-sorted CD49d-positive neural stem cells improves targeted cell delivery and behavior after stroke in a mouse stroke model. Stroke 2008, 39, 1300–1306. [Google Scholar] [CrossRef]
- Walczak, P.; Zhang, J.; Gilad, A.A.; Kedziorek, D.A.; Ruiz-Cabello, J.; Young, R.G.; Pittenger, M.F.; van Zijl, P.C.; Huang, J.; Bulte, J.W. Dual-modality monitoring of targeted intraarterial delivery of mesenchymal stem cells after transient ischemia. Stroke 2008, 39, 1569–1574. [Google Scholar] [CrossRef]
- Van Esch, B.; van der Zaag-Loonen, H.; Bruintjes, T.; van Benthem, P.P. Betahistine in Ménière’s Disease or Syndrome: A Systematic Review. Audiol. Neurootol. 2022, 27, 1–33. [Google Scholar] [CrossRef]
- Marsili, L.; Giannini, G.; Cortelli, P.; Colosimo, C. Early recognition and diagnosis of multiple system atrophy: Best practice and emerging concepts. Expert Rev. Neurother 2021, 21, 993–1004. [Google Scholar] [CrossRef]
- Stocchi, F. Therapy for Parkinson’s disease: What is in the pipeline? Neurotherapeutics 2014, 11, 24–33. [Google Scholar] [CrossRef]
- Marsili, L.; Mahajan, A. Clinical milestones in Parkinson’s disease: Past, present, and future. J. Neurol. Sci. 2022, 432, 120082. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Candelise, N.; Baiardi, S.; Capellari, S.; Giannini, G.; Orrù, C.D.; Antelmi, E.; Mammana, A.; Hughson, A.G.; Calandra-Buonaura, G.; et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol. 2020, 140, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Marsili, L.; Espay, A.J.; Merola, A. Future of Neurologic Examination in Clinical Practice. JAMA Neurol. 2018, 75, 383. [Google Scholar] [CrossRef] [PubMed]
- Daley, G.Q.; Hyun, I.; Apperley, J.F.; Barker, R.A.; Benvenisty, N.; Bredenoord, A.L.; Breuer, C.K.; Caulfield, T.; Cedars, M.I.; Frey-Vasconcells, J.; et al. Setting Global Standards for Stem Cell Research and Clinical Translation: The 2016 ISSCR Guidelines. Stem Cell Rep. 2016, 6, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yang, Y.; Tan, P.; Zhang, Y.; Han, M.; Yu, J.; Zhang, X.; Jia, Z.; Wang, D.; Li, Y.; et al. Induction of mouse totipotent stem cells by a defined chemical cocktail. Nature 2022. [Google Scholar] [CrossRef]
- Espay, A.J.; Kalia, L.V.; Gan-Or, Z.; Williams-Gray, C.H.; Bedard, P.L.; Rowe, S.M.; Morgante, F.; Fasano, A.; Stecher, B.; Kauffman, M.A.; et al. Disease modification and biomarker development in Parkinson disease: Revision or reconstruction? Neurology 2020, 94, 481–494. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Espay, A.J. Is Pathology Always the Diagnostic Gold Standard in Neurodegeneration? Mov. Disord. Clin. Pract. 2022, 9, 1152–1153. [Google Scholar] [CrossRef]
- Marsili, L.; Sharma, J.; Espay, A.J.; Migazzi, A.; Abdelghany, E.; Hill, E.J.; Duque, K.R.; Hagen, M.C.; Stephen, C.D.; Kovacs, G.G.; et al. Neither a Novel Tau Proteinopathy nor an Expansion of a Phenotype: Reappraising Clinicopathology-Based Nosology. Int. J. Mol. Sci. 2021, 22, 7292. [Google Scholar] [CrossRef]
- Pitz, V.; Malek, N.; Tobias, E.S.; Grosset, K.A.; Gentleman, S.; Grosset, D.G. The Levodopa Response Varies in Pathologically Confirmed Parkinson’s Disease: A Systematic Review. Mov. Disord. Clin. Pract. 2020, 7, 218–222. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).