
Dysuricemia

 ,
,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. History of Dysuricemia
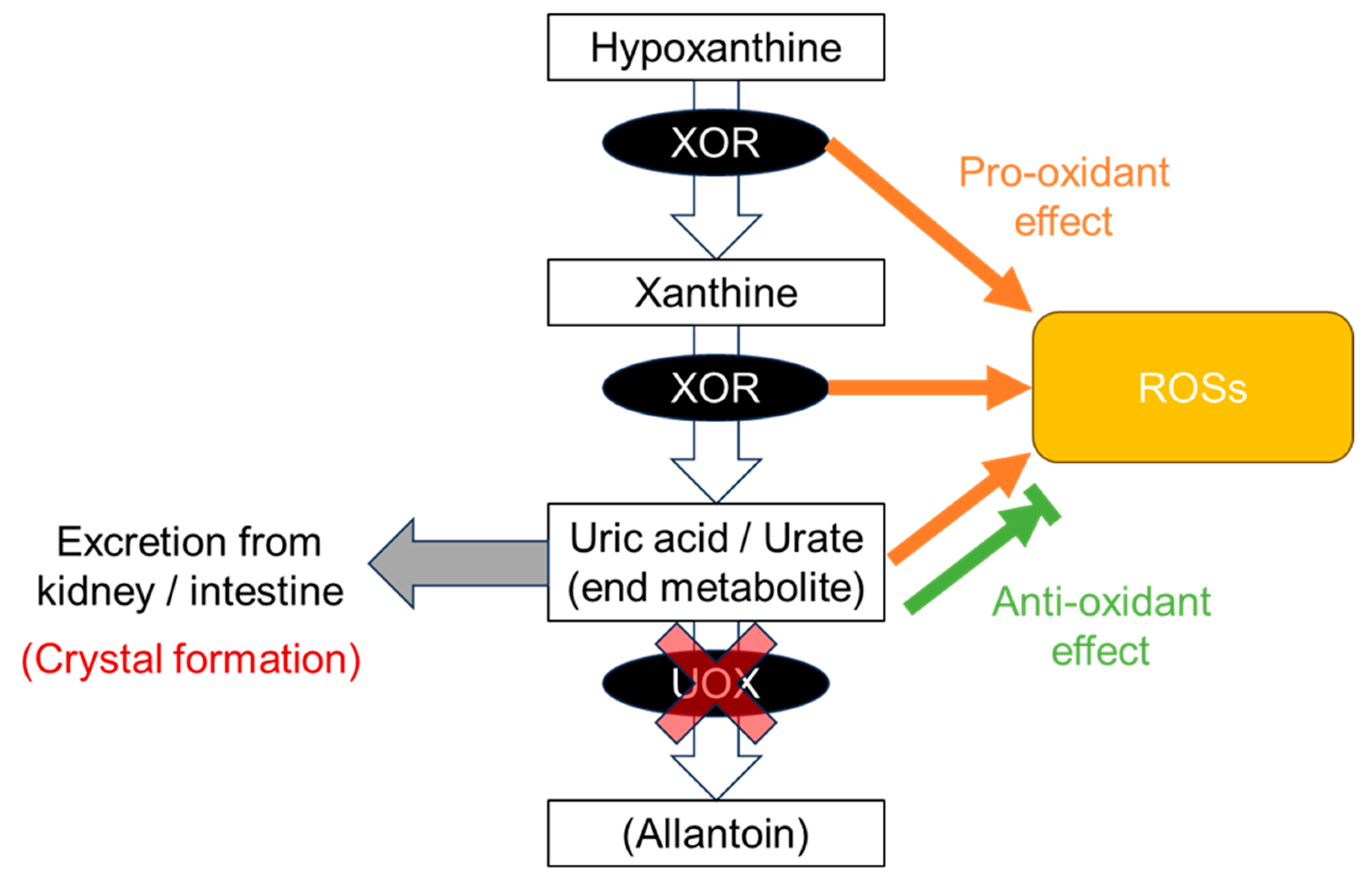
3. Production of Uric Acid and Dysuricemia
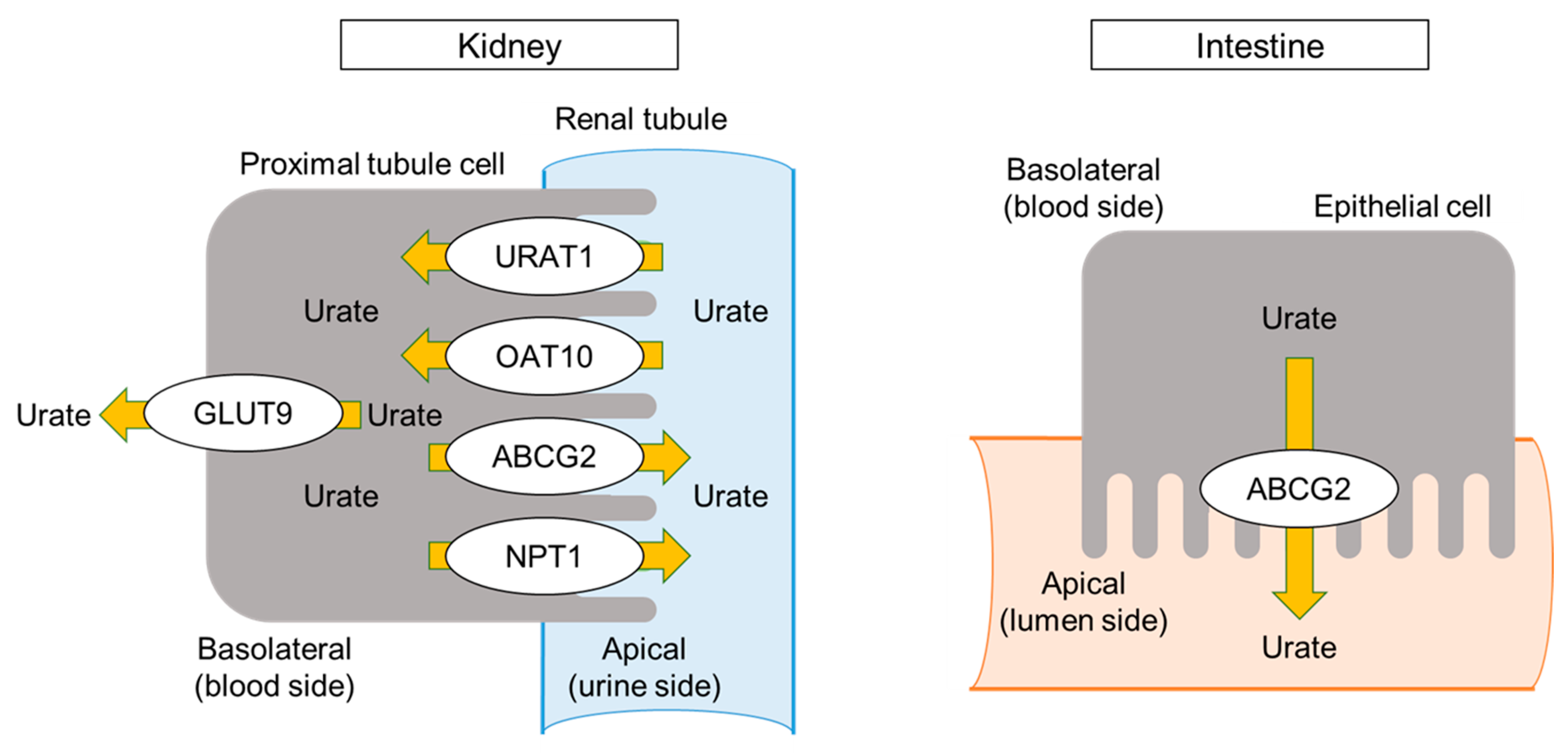
4. Urate Transporters and Dysuricemia
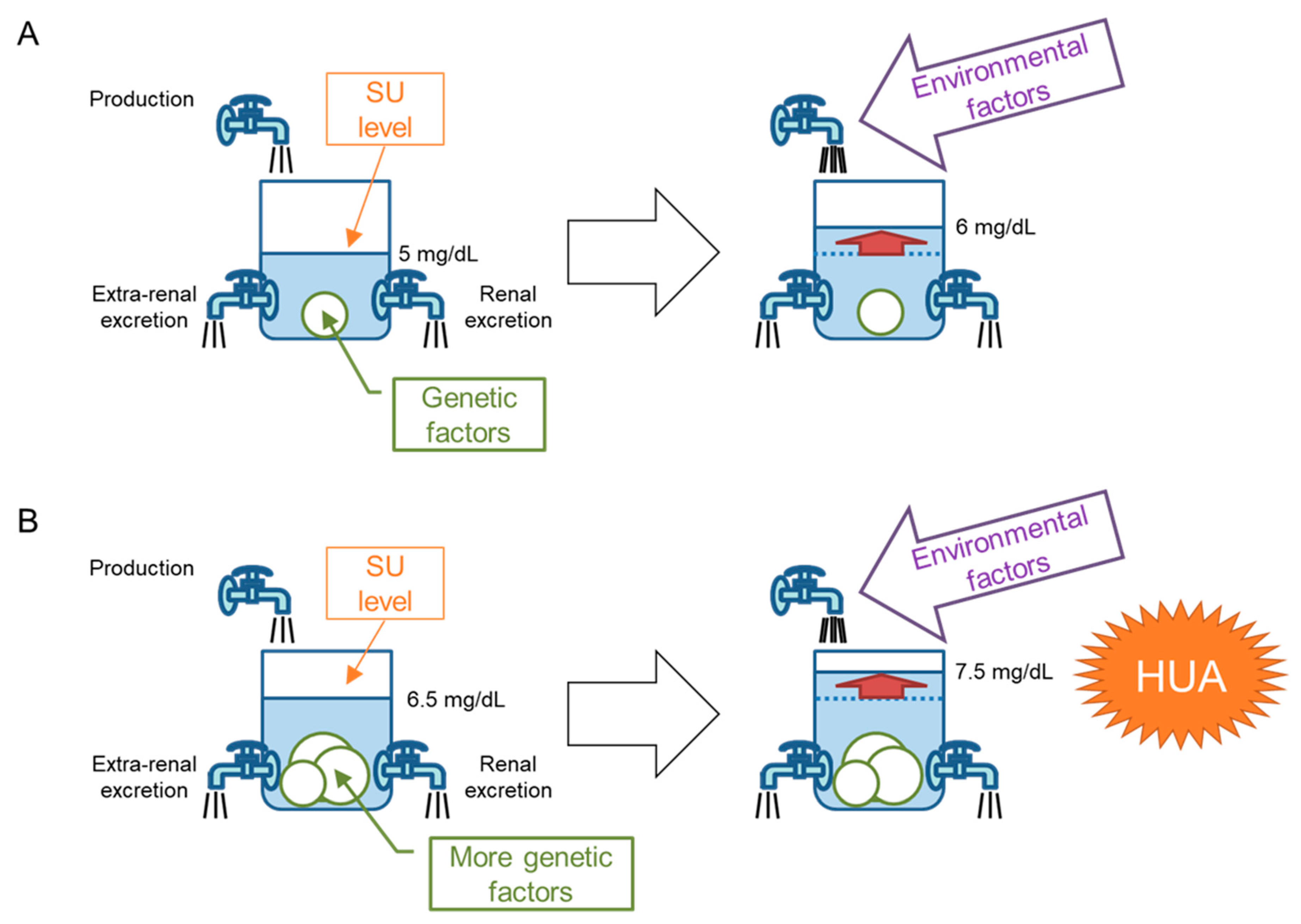
5. “Bucket-and-Balls” Theory for Hyperuricemia
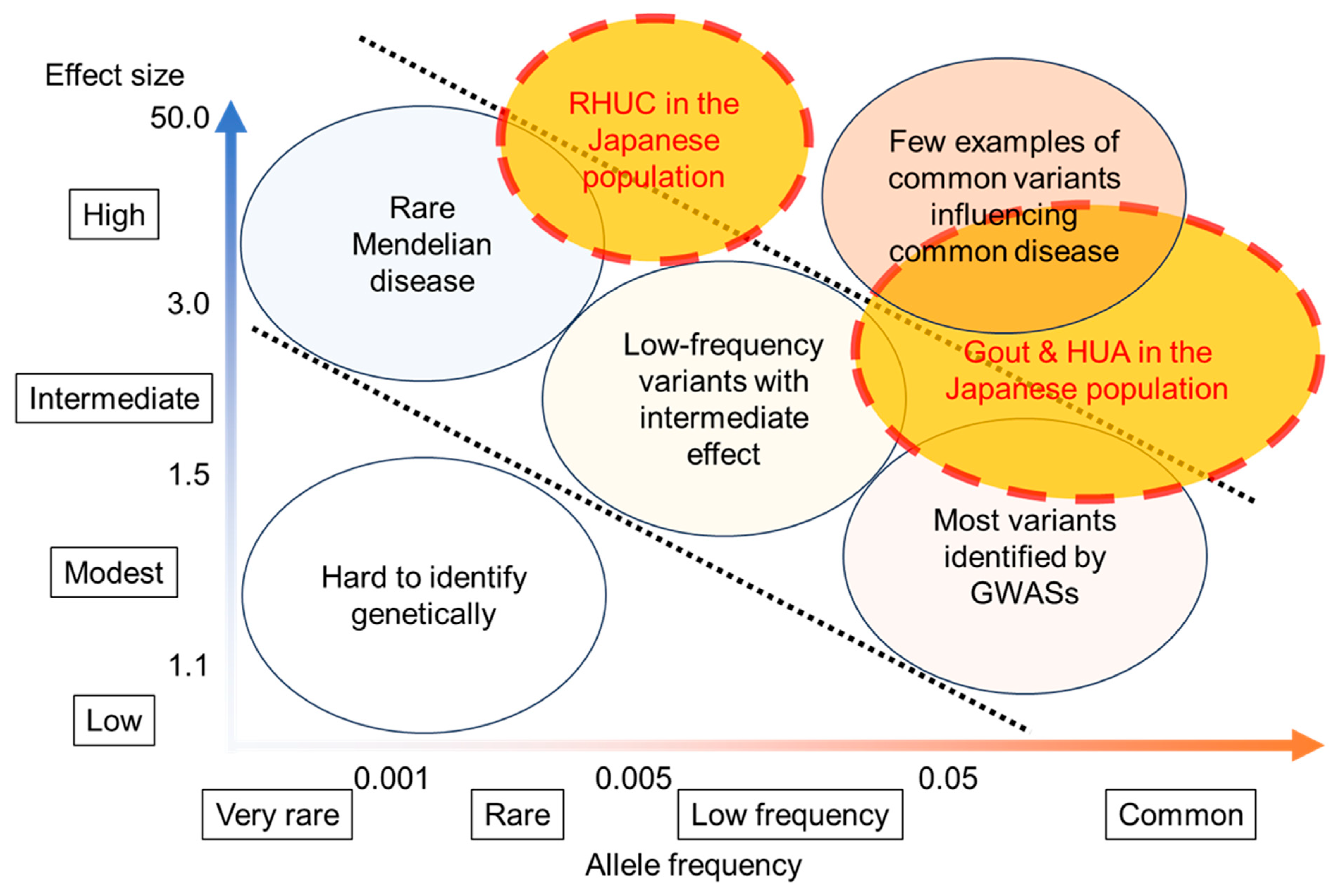
6. Genetic Factors That Favor Dysuricemia in the Japanese Population
7. Secondary Dysuricemia
8. Relationship between Diseases and Dysuricemia
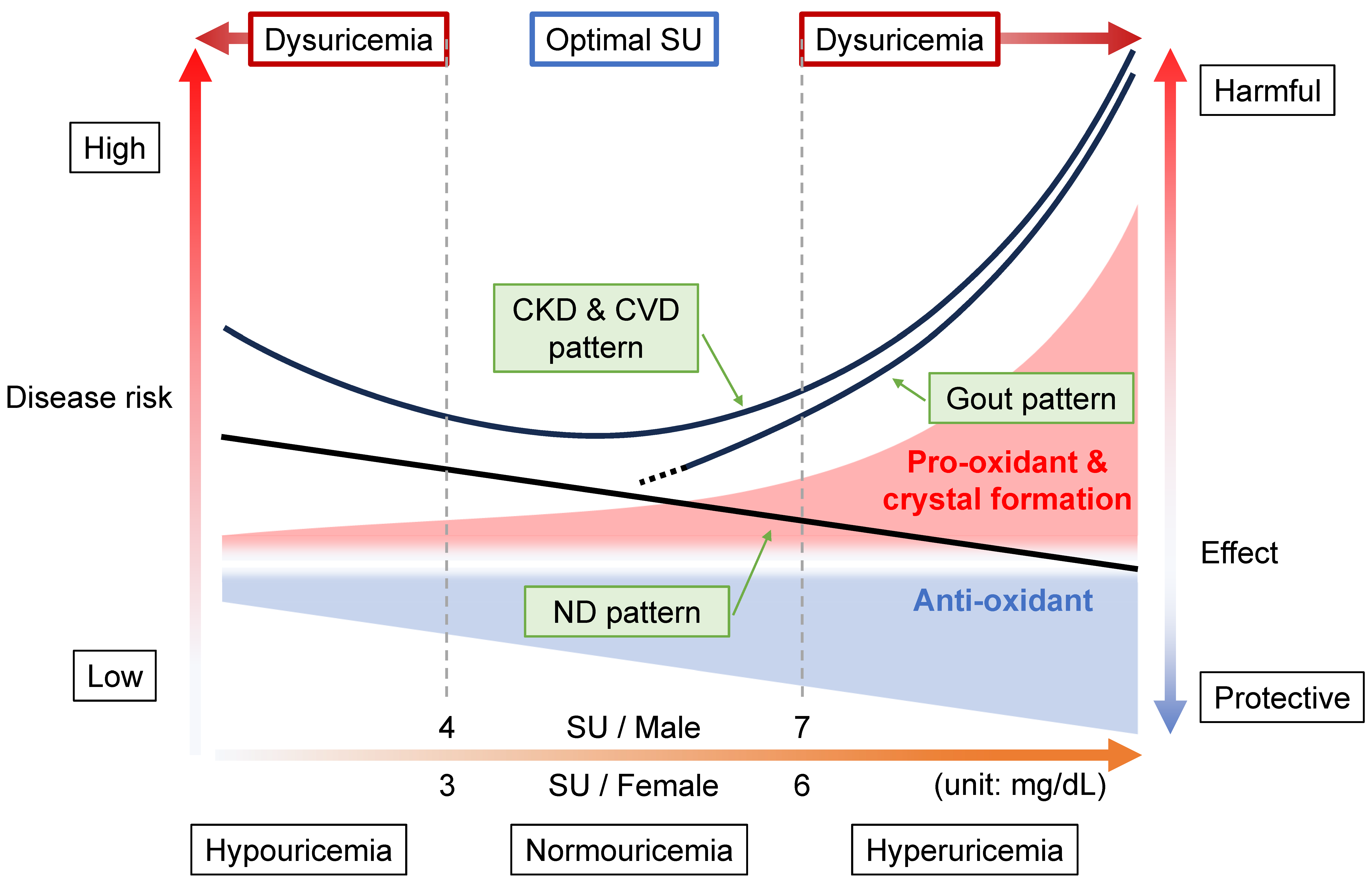
8.1. Gout Pattern: Crystal Formation and Pro-Oxidative Effects
8.2. ND Pattern: Anti-Oxidative Effects
8.3. CKD and CVD Pattern: Combination with Gout and ND Patterns
8.4. Range of Normouricemia as Optimal SU Level
9. Conclusions with Future Research/Clinical Questions on Dysuricemia
- ➢
- What causes EIAKI in RHUC patients? One hypothetical mechanism of EIAKI suggests that lowered anti-oxidative effect in RHUC patients causes renal vasopressin by exercise-induced ROSs from XOR; based on this hypothesis, some case studies report the effectiveness of allopurinol or febuxostat (XOR inhibitors) administration in preventing EIAKI [106,107,108,109]. However, convincing evidence for their efficacy is lacking [8];
- ➢
- While some epidemiological studies on NDs support the “ND pattern”, several other studies (described above) do not. Research into the effects of low SU against NDs should be conducted to elucidate the effects of urate on NDs;
- ➢
- Which comes first, dysuricemia, CKD, or CVD? It is also possible that SU is simply a marker of these diseases. Further investigation of their causality by SU should be determined from the viewpoint of the anti-oxidative, pro-oxidative, and crystal-forming effects of urate;
- ➢
- Why are females more vulnerable to SU? It is known that female hormones decrease SU levels [5]. Our previous studies do, in fact, reveal sex differences in SU in the order of 1–1.5 mg/dL (60–90 μmol/L) [26,54]. The optimal SU range differs between the sexes by 1 mg/dL (60 μmol/L), so females can be concluded to be more vulnerable to SU at the same SU level as males.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jordheim, L.P.; Peters, G.J. Recent updates on purine and pyrimidine metabolism in physiological and pathological settings. Nucleosides Nucleotides Nucleic Acids 2020, 39, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.W.; Lee, C.C.; Muzny, D.M.; Caskey, C.T. Urate oxidase: Primary structure and evolutionary implications. Proc. Natl. Acad. Sci. USA 1989, 86, 9412–9416. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.W.; Muzny, D.M.; Lee, C.C.; Caskey, C.T. Two independent mutational events in the loss of urate oxidase during hominoid evolution. J. Mol. Evol. 1992, 34, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Choi, H.K.; Joosten, L.A.B.; Khanna, P.P.; Matsuo, H.; Perez-Ruiz, F.; Stamp, L.K. Gout. Nat. Rev. Dis. Primers 2019, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Gosling, A.L.; Gaffo, A.; Abhishek, A. Gout. Lancet 2021, 397, 1843–1855. [Google Scholar] [CrossRef]
- O’Keefe, J.H., Jr.; Cordain, L.; Harris, W.H.; Moe, R.M.; Vogel, R. Optimal low-density lipoprotein is 50 to 70 mg/dl: Lower is better and physiologically normal. J. Am. Coll. Cardiol. 2004, 43, 2142–2146. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine oxidoreductase: One enzyme for multiple physiological tasks. Redox Biol. 2021, 41, 101882. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Matsuo, H.; Ohtahara, A.; Ogino, K.; Hakoda, M.; Hamada, T.; Hosoyamada, M.; Yamaguchi, S.; Hisatome, I.; Ichida, K.; et al. Clinical practice guideline for renal hypouricemia (1st edition). Hum. Cell 2019, 32, 83–87. [Google Scholar] [CrossRef]
- Loeb, J.N. The influence of temperature on the solubility of monosodium urate. Arthritis Rheum. 1972, 15, 189–192. [Google Scholar] [CrossRef]
- Nuki, G.; Simkin, P.A. A concise history of gout and hyperuricemia and their treatment. Arthritis Res. Ther. 2006, 8 (Suppl. S1), S1. [Google Scholar] [CrossRef][Green Version]
- Hippocrates. The Genuine Works of Hippocrates; Wood: New York, NY, USA, 1886; Volumes I–II. [Google Scholar]
- McCarty, D.J. A historical note: Leeuwenhoek’s description of crystals from a gouty tophus. Arthritis Rheum. 1970, 13, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Wollaston, W.H. XVII. On gouty and urinary concretions. Philos. Trans. R. Soc. Lond. 1997, 87, 386–400. [Google Scholar] [CrossRef]
- Garrod, A.B. Observations on certain pathological conditions of the blood and urine, in gout, rheumatism, and Bright’s disease. Med. Chir. Trans. 1848, 31, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Dehlin, M.; Jacobsson, L.; Roddy, E. Global epidemiology of gout: Prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 2020, 16, 380–390. [Google Scholar] [CrossRef]
- Gosling, A.L.; Matisoo-Smith, E.; Merriman, T.R. Hyperuricaemia in the Pacific: Why the elevated serum urate levels? Rheumatol. Int. 2014, 34, 743–757. [Google Scholar] [CrossRef]
- Nakayama, A.; Nakatochi, M.; Kawamura, Y.; Yamamoto, K.; Nakaoka, H.; Shimizu, S.; Higashino, T.; Koyama, T.; Hishida, A.; Kuriki, K.; et al. Subtype-specific gout susceptibility loci and enrichment of selection pressure on ABCG2 and ALDH2 identified by subtype genome-wide meta-analyses of clinically defined gout patients. Ann. Rheum. Dis. 2020, 79, 657–665. [Google Scholar] [CrossRef]
- Praetorius, E.; Kirk, J.E. Hypouricemia: With evidence for tubular elimination of uric acid. J. Lab. Clin. Med. 1950, 35, 865–868. [Google Scholar]
- Greene, M.L.; Marcus, R.; Aurbach, G.D.; Kazam, E.S.; Seegmiller, J.E. Hypouricemia due to isolated renal tubular defect. Dalmatian dog mutation in man. Am. J. Med. 1972, 53, 361–367. [Google Scholar] [CrossRef]
- Akaoka, I.; Nishizawa, T.; Yano, E.; Takeuchi, A.; Nishida, Y. Familial hypouricaemia due to renal tubular defect of urate transport. Ann. Clin. Res. 1975, 7, 318–324. [Google Scholar]
- Ishikawa, I. Acute renal failure with severe loin pain and patchy renal ischemia after anaerobic exercise in patients with or without renal hypouricemia. Nephron 2002, 91, 559–570. [Google Scholar] [CrossRef]
- Suzuki, T.; Kidoguchi, K.; Hayashi, A. Genetic heterogeneity of familial hypouricemia due to isolated renal tubular defect. Jinrui Idengaku Zasshi 1981, 26, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Cha, D.H.; Gee, H.Y.; Cachau, R.; Choi, J.M.; Park, D.; Jee, S.H.; Ryu, S.; Kim, K.K.; Won, H.H.; Limou, S.; et al. Contribution of SLC22A12 on hypouricemia and its clinical significance for screening purposes. Sci. Rep. 2019, 9, 14360. [Google Scholar] [CrossRef] [PubMed]
- Stiburkova, B.; Bohatá, J.; Pavelcová, K.; Tasic, V.; Plaseska-Karanfilska, D.; Cho, S.-K.; Potočnaková, L.; Šaligová, J. Renal hypouricemia 1: Rare disorder as common disease in Eastern Slovakia Roma population. Biomedicines 2021, 9, 1607. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ma, L.; Zhou, J.; Song, Z.; Zhang, J.; Wang, K.; Chen, B.; Pan, D.; Li, Z.; Li, C.; et al. Renal hypouricemia caused by novel compound heterozygous mutations in the SLC22A12 gene: A case report with literature review. BMC Med. Genet. 2018, 19, 142. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Kawamura, Y.; Toyoda, Y.; Shimizu, S.; Kawaguchi, M.; Aoki, Y.; Takeuchi, K.; Okada, R.; Kubo, Y.; Imakiire, T.; et al. Genetic epidemiological analysis of hypouricaemia from 4993 Japanese on non-functional variants of URAT1/SLC22A12 gene. Rheumatology 2022, 61, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Ichida, K.; Hosoyamada, M.; Kamatani, N.; Kamitsuji, S.; Hisatome, I.; Shibasaki, T.; Hosoya, T. Age and origin of the G774A mutation in SLC22A12 causing renal hypouricemia in Japanese. Clin. Genet. 2008, 74, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Matsuo, H.; Abhishek, A.; Ichida, K.; Shinomiya, N.; Members of Guideline Development Committee of Clinical Practice Guideline for Renal Hypouricaemia. First clinical practice guideline for renal hypouricaemia: A rare disorder that aided the development of urate-lowering drugs for gout. Rheumatology 2021, 60, 3961–3963. [Google Scholar] [CrossRef]
- Saksela, M.; Lapatto, R.; Raivio, K.O. Xanthine oxidoreductase gene expression and enzyme activity in developing human tissues. Biol. Neonate 1998, 74, 274–280. [Google Scholar] [CrossRef]
- Hille, R.; Nishino, T. Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. FASEB J. 1995, 9, 995–1003. [Google Scholar] [CrossRef]
- Carcassi, A.; Marcolongo, R., Jr.; Marinello, E.; Riario-Sforza, G.; Boggiano, C. Liver xanthine oxidase in gouty patients. Arthritis Rheum. 1969, 12, 17–20. [Google Scholar] [CrossRef]
- Elion, G.B. Enzymatic and metabolic studies with allopurinol. Ann. Rheum. Dis. 1966, 25, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Kurajoh, M.; Fukumoto, S.; Murase, T.; Nakamura, T.; Ishihara, T.; Go, H.; Yamamoto, K.; Nakatani, S.; Tsuda, A.; Morioka, T.; et al. Insulin Resistance Associated with Plasma Xanthine Oxidoreductase Activity Independent of Visceral Adiposity and Adiponectin Level: MedCity21 Health Examination Registry. Int. J. Endocrinol. 2019, 2019, 1762161. [Google Scholar] [CrossRef] [PubMed]
- Kurajoh, M.; Fukumoto, S.; Emoto, M.; Murase, T.; Nakamura, T.; Ishihara, T.; Go, H.; Yamamoto, K.; Nakatani, S.; Tsuda, A.; et al. Independent association of plasma xanthine oxidoreductase activity with serum uric acid level based on stable isotope-labeled xanthine and liquid chromatography/triple quadrupole mass spectrometry: MedCity21 health examination registry. Clin. Chem. Lab. Med. 2020, 58, 780–786. [Google Scholar] [CrossRef]
- Kurajoh, M.; Fukumoto, S.; Akari, S.; Murase, T.; Nakamura, T.; Takahashi, K.; Yoshida, H.; Nakatani, S.; Tsuda, A.; Morioka, T.; et al. Possible role of insulin resistance in activation of plasma xanthine oxidoreductase in health check-up examinees. Sci. Rep. 2022, 12, 10281. [Google Scholar] [CrossRef] [PubMed]
- Dent, C.E.; Philpot, G.R. Xanthinuria, an inborn error (or deviation) of metabolism. Lancet 1954, 266, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Sekine, M.; Okamoto, K.; Ichida, K. Association of Mutations Identified in Xanthinuria with the Function and Inhibition Mechanism of Xanthine Oxidoreductase. Biomedicines 2021, 9, 1723. [Google Scholar] [CrossRef] [PubMed]
- Ichida, K.; Amaya, Y.; Kamatani, N.; Nishino, T.; Hosoya, T.; Sakai, O. Identification of two mutations in human xanthine dehydrogenase gene responsible for classical type I xanthinuria. J. Clin. Investig. 1997, 99, 2391–2397. [Google Scholar] [CrossRef]
- Ichida, K.; Matsumura, T.; Sakuma, R.; Hosoya, T.; Nishino, T. Mutation of human molybdenum cofactor sulfurase gene is responsible for classical xanthinuria type II. Biochem. Biophys. Res. Commun. 2001, 282, 1194–1200. [Google Scholar] [CrossRef]
- Kusano, T.; Nishino, T.; Okamoto, K.; Hille, R.; Nishino, T. The mechanism and significance of the conversion of xanthine dehydrogenase to xanthine oxidase in mammalian secretory gland cells. Redox Biol. 2023, 59, 102573. [Google Scholar] [CrossRef]
- Yoshida, S.; Kurajoh, M.; Fukumoto, S.; Murase, T.; Nakamura, T.; Yoshida, H.; Hirata, K.; Inaba, M.; Emoto, M. Association of plasma xanthine oxidoreductase activity with blood pressure affected by oxidative stress level: MedCity21 health examination registry. Sci. Rep. 2020, 10, 4437. [Google Scholar] [CrossRef]
- Nakatani, S.; Ishimura, E.; Murase, T.; Nakamura, T.; Nakatani, A.; Toi, N.; Nishide, K.; Uedono, H.; Tsuda, A.; Kurajoh, M.; et al. Plasma Xanthine Oxidoreductase Activity Associated with Glycemic Control in Patients with Pre-Dialysis Chronic Kidney Disease. Kidney Blood Press. Res. 2021, 46, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, S.; Nasu, T.; Satoh, M.; Kotozaki, Y.; Tanno, K.; Tanaka, F.; Asahi, K.; Ohmomo, H.; Kikuchi, H.; Kobayashi, T.; et al. Association between Plasma Xanthine Oxidoreductase Activity and the Renal Function in a General Japanese Population: The Tohoku Medical Megabank Community-Based Cohort Study. Kidney Blood Press. Res. 2022, 47, 722–728. [Google Scholar] [CrossRef]
- Watanabe, K.; Watanabe, T.; Otaki, Y.; Murase, T.; Nakamura, T.; Kato, S.; Tamura, H.; Nishiyama, S.; Takahashi, H.; Arimoto, T.; et al. Gender Differences in the Impact of Plasma Xanthine Oxidoreductase Activity on Coronary Artery Spasm. J. Clin. Med. 2021, 10, 5550. [Google Scholar] [CrossRef] [PubMed]
- Kurajoh, M.; Fukumoto, S.; Akari, S.; Murase, T.; Nakamura, T.; Ihara, Y.; Imai, T.; Nagata, Y.; Morioka, T.; Mori, K.; et al. Association of plasma xanthine oxidoreductase activity with vascular endothelial function independent of serum uric acid level: MedCity21 health examination registry. Int. J. Cardiol. Heart Vasc. 2023, 48, 101264. [Google Scholar] [CrossRef] [PubMed]
- Oki, R.; Hamasaki, Y.; Komaru, Y.; Miyamoto, Y.; Matsuura, R.; Akari, S.; Nakamura, T.; Murase, T.; Doi, K.; Nangaku, M. Plasma xanthine oxidoreductase is associated with carotid atherosclerosis in stable kidney transplant recipients. Nephrology 2022, 27, 363–370. [Google Scholar] [CrossRef]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef]
- Matsuo, H.; Chiba, T.; Nagamori, S.; Nakayama, A.; Domoto, H.; Phetdee, K.; Wiriyasermkul, P.; Kikuchi, Y.; Oda, T.; Nishiyama, J.; et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am. J. Hum. Genet. 2008, 83, 744–751. [Google Scholar] [CrossRef]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S.; et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: A function-based genetic analysis in a Japanese population. Sci. Transl. Med. 2009, 1, 5ra11. [Google Scholar] [CrossRef]
- Chiba, T.; Matsuo, H.; Kawamura, Y.; Nagamori, S.; Nishiyama, T.; Wei, L.; Nakayama, A.; Nakamura, T.; Sakiyama, M.; Takada, T.; et al. NPT1/SLC17A1 is a renal urate exporter in humans and its common gain-of-function variant decreases the risk of renal underexcretion gout. Arthritis Rheumatol. 2015, 67, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Higashino, T.; Morimoto, K.; Nakaoka, H.; Toyoda, Y.; Kawamura, Y.; Shimizu, S.; Nakamura, T.; Hosomichi, K.; Nakayama, A.; Ooyama, K.; et al. Dysfunctional missense variant of OAT10/SLC22A13 decreases gout risk and serum uric acid levels. Ann. Rheum. Dis. 2020, 79, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.; Nakayama, A.; Shimizu, S.; Toyoda, Y.; Nishida, Y.; Hishida, A.; Katsuura-Kamano, S.; Shibuya, K.; Tamura, T.; Kawaguchi, M.; et al. A Proposal for Practical Diagnosis of Renal Hypouricemia: Evidenced from Genetic Studies of Nonfunctional Variants of URAT1/SLC22A12 among 30,685 Japanese Individuals. Biomedicines 2021, 9, 1012. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Kottgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Kottgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef]
- Matsuo, H.; Ichida, K.; Takada, T.; Nakayama, A.; Nakashima, H.; Nakamura, T.; Kawamura, Y.; Takada, Y.; Yamamoto, K.; Inoue, H.; et al. Common dysfunctional variants in ABCG2 are a major cause of early-onset gout. Sci. Rep. 2013, 3, 2014. [Google Scholar] [CrossRef] [PubMed]
- Maliepaard, M.; Scheffer, G.L.; Faneyte, I.F.; van Gastelen, M.A.; Pijnenborg, A.C.; Schinkel, A.H.; van De Vijver, M.J.; Scheper, R.J.; Schellens, J.H. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001, 61, 3458–3464. [Google Scholar] [PubMed]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef]
- Matsuo, H.; Tsunoda, T.; Ooyama, K.; Sakiyama, M.; Sogo, T.; Takada, T.; Nakashima, A.; Nakayama, A.; Kawaguchi, M.; Higashino, T.; et al. Hyperuricemia in acute gastroenteritis is caused by decreased urate excretion via ABCG2. Sci. Rep. 2016, 6, 31003. [Google Scholar] [CrossRef]
- Matsuo, H.; Nakayama, A.; Sakiyama, M.; Chiba, T.; Shimizu, S.; Kawamura, Y.; Nakashima, H.; Nakamura, T.; Takada, Y.; Oikawa, Y.; et al. ABCG2 dysfunction causes hyperuricemia due to both renal urate underexcretion and renal urate overload. Sci. Rep. 2014, 4, 3755. [Google Scholar] [CrossRef]
- Sakiyama, M.; Matsuo, H.; Nagamori, S.; Ling, W.; Kawamura, Y.; Nakayama, A.; Higashino, T.; Chiba, T.; Ichida, K.; Kanai, Y.; et al. Expression of a human NPT1/SLC17A1 missense variant which increases urate export. Nucleosides Nucleotides Nucleic Acids 2016, 35, 536–542. [Google Scholar] [CrossRef]
- Toyoda, Y.; Kawamura, Y.; Nakayama, A.; Morimoto, K.; Shimizu, S.; Tanahashi, Y.; Tamura, T.; Kondo, T.; Kato, Y.; Ichida, K.; et al. OAT10/SLC22A13 Acts as a Renal Urate Re-Absorber: Clinico-Genetic and Functional Analyses with Pharmacological Impacts. Front. Pharmacol. 2022, 13, 842717. [Google Scholar] [CrossRef]
- Hisatome, I.; Ichida, K.; Mineo, I.; Ohtahara, A.; Ogino, K.; Kuwabara, M.; Ishizaka, N.; Uchida, S.; Kurajoh, M.; Kohagura, K.; et al. Japanese Society of Gout and Uric & Nucleic Acids 2019 Guidelines for Management of Hyperuricemia and Gout (3rd Edition). Gout Uric Nucleic Acids 2020, 44, 1018–1029. [Google Scholar] [CrossRef]
- Nakayama, A.; Matsuo, H.; Nakaoka, H.; Nakamura, T.; Nakashima, H.; Takada, Y.; Oikawa, Y.; Takada, T.; Sakiyama, M.; Shimizu, S.; et al. Common dysfunctional variants of ABCG2 have stronger impact on hyperuricemia progression than typical environmental risk factors. Sci. Rep. 2014, 4, 5227. [Google Scholar] [CrossRef]
- Matsuo, H.; Yamamoto, K.; Nakaoka, H.; Nakayama, A.; Sakiyama, M.; Chiba, T.; Takahashi, A.; Nakamura, T.; Nakashima, H.; Takada, Y.; et al. Genome-wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Ann. Rheum. Dis. 2016, 75, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Z.; Liu, S.; Wang, C.; Han, L.; Cui, L.; Zhou, J.; Zou, H.; Liu, Z.; Chen, J.; et al. Genome-wide association analysis identifies three new risk loci for gout arthritis in Han Chinese. Nat. Commun. 2015, 6, 7041. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Nakaoka, H.; Yamamoto, K.; Sakiyama, M.; Shaukat, A.; Toyoda, Y.; Okada, Y.; Kamatani, Y.; Nakamura, T.; Takada, T.; et al. GWAS of clinically defined gout and subtypes identifies multiple susceptibility loci that include urate transporter genes. Ann. Rheum. Dis. 2017, 76, 869–877. [Google Scholar] [CrossRef]
- Kawamura, Y.; Nakaoka, H.; Nakayama, A.; Okada, Y.; Yamamoto, K.; Higashino, T.; Sakiyama, M.; Shimizu, T.; Ooyama, H.; Ooyama, K.; et al. Genome-wide association study revealed novel loci which aggravate asymptomatic hyperuricaemia into gout. Ann. Rheum. Dis. 2019, 78, 1430–1437. [Google Scholar] [CrossRef]
- Toyoda, Y.; Nakayama, A.; Nakatochi, M.; Kawamura, Y.; Nakaoka, H.; Yamamoto, K.; Shimizu, S.; Ooyama, H.; Ooyama, K.; Shimizu, T.; et al. Genome-wide meta-analysis between renal overload type and renal underexcretion type of clinically defined gout in Japanese populations. Mol. Genet. Metab. 2022, 136, 186–189. [Google Scholar] [CrossRef]
- Kottgen, A.; Albrecht, E.; Teumer, A.; Vitart, V.; Krumsiek, J.; Hundertmark, C.; Pistis, G.; Ruggiero, D.; O’Seaghdha, C.M.; Haller, T.; et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2013, 45, 145–154. [Google Scholar] [CrossRef]
- Nakatochi, M.; Kanai, M.; Nakayama, A.; Hishida, A.; Kawamura, Y.; Ichihara, S.; Akiyama, M.; Ikezaki, H.; Furusyo, N.; Shimizu, S.; et al. Genome-wide meta-analysis identifies multiple novel loci associated with serum uric acid levels in Japanese individuals. Commun. Biol. 2019, 2, 115. [Google Scholar] [CrossRef]
- Nakatochi, M.; Toyoda, Y.; Kanai, M.; Nakayama, A.; Kawamura, Y.; Hishida, A.; Mikami, H.; Matsuo, K.; Takezaki, T.; Momozawa, Y.; et al. An X chromosome-wide meta-analysis based on Japanese cohorts revealed that non-autosomal variations are associated with serum urate. Rheumatology 2021, 60, 4430–4432. [Google Scholar] [CrossRef] [PubMed]
- Tin, A.; Marten, J.; Halperin Kuhns, V.L.; Li, Y.; Wuttke, M.; Kirsten, H.; Sieber, K.B.; Qiu, C.; Gorski, M.; Yu, Z.; et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 2019, 51, 1459–1474. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Nakatochi, M.; Nakayama, A.; Kawamura, Y.; Nakaoka, H.; Wakai, K.; Matsuo, K.; Matsuo, H.; Japan Gout Genomics, C. SNP-based heritability estimates of gout and its subtypes determined by genome-wide association studies of clinically defined gout. Rheumatology 2023, 62, e144–e146. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.; Hirschhorn, J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef]
- Pineda, C.; Soto-Fajardo, C.; Mendoza, J.; Gutierrez, J.; Sandoval, H. Hypouricemia: What the practicing rheumatologist should know about this condition. Clin. Rheumatol. 2020, 39, 135–147. [Google Scholar] [CrossRef]
- Latourte, A.; Dumurgier, J.; Paquet, C.; Richette, P. Hyperuricemia, Gout, and the Brain—An Update. Curr. Rheumatol. Rep. 2021, 23, 82. [Google Scholar] [CrossRef]
- Lu, N.; Dubreuil, M.; Zhang, Y.; Neogi, T.; Rai, S.K.; Ascherio, A.; Hernan, M.A.; Choi, H.K. Gout and the risk of Alzheimer’s disease: A population-based, BMI-matched cohort study. Ann. Rheum. Dis. 2016, 75, 547–551. [Google Scholar] [CrossRef]
- Hong, J.Y.; Lan, T.Y.; Tang, G.J.; Tang, C.H.; Chen, T.J.; Lin, H.Y. Gout and the risk of dementia: A nationwide population-based cohort study. Arthritis Res. Ther. 2015, 17, 139. [Google Scholar] [CrossRef]
- Matsuo, H.; Tomiyama, H.; Satake, W.; Chiba, T.; Onoue, H.; Kawamura, Y.; Nakayama, A.; Shimizu, S.; Sakiyama, M.; Funayama, M.; et al. ABCG2 variant has opposing effects on onset ages of Parkinson’s disease and gout. Ann. Clin. Transl. Neurol. 2015, 2, 302–306. [Google Scholar] [CrossRef]
- Weisskopf, M.G.; O’Reilly, E.; Chen, H.; Schwarzschild, M.A.; Ascherio, A. Plasma urate and risk of Parkinson’s disease. Am. J. Epidemiol. 2007, 166, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhong, S.; Liang, Y.; Zhang, X.; Zhang, R.; Kang, K.; Qu, H.; Xu, Y.; Zhao, C.; Zhao, M. Serum Uric Acid and the Risk of Dementia: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2021, 13, 625690. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Drory, V.E. Influence of serum uric acid levels on prognosis and survival in amyotrophic lateral sclerosis: A meta-analysis. J. Neurol. 2014, 261, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Dehlin, M.; Sandstrom, T.Z.; Jacobsson, L.T. Incident Gout: Risk of Death and Cause-Specific Mortality in Western Sweden: A Prospective, Controlled Inception Cohort Study. Front. Med. 2022, 9, 802856. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Zhong, X.; Xu, T.; Xu, T.; Zhang, Y. Sex-Specific Relationship Between Serum Uric Acid and Risk of Stroke: A Dose-Response Meta-Analysis of Prospective Studies. J. Am. Heart Assoc. 2017, 6, e005042. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liang, Y.; Lin, J.; Zhang, X.; Qu, H.; Xu, J.; Zhao, C.; Zhao, M. Serum uric acid concentrations and risk of intracerebral hemorrhage: A systematic review and meta-analysis. Atherosclerosis 2018, 275, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Richette, P.; Doherty, M.; Pascual, E.; Barskova, V.; Becce, F.; Castaneda-Sanabria, J.; Coyfish, M.; Guillo, S.; Jansen, T.L.; Janssens, H.; et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann. Rheum. Dis. 2017, 76, 29–42. [Google Scholar] [CrossRef]
- Coneys, R.; Storm, C.S.; Kia, D.A.; Almramhi, M.; Wood, N.W. Mendelian Randomisation Finds No Causal Association between Urate and Parkinson’s Disease Progression. Mov. Disord. 2021, 36, 2182–2187. [Google Scholar] [CrossRef]
- Kobylecki, C.J.; Nordestgaard, B.G.; Afzal, S. Plasma urate and risk of Parkinson’s disease: A mendelian randomization study. Ann. Neurol. 2018, 84, 178–190. [Google Scholar] [CrossRef]
- Lee, Y.H. Gout and the risk of Alzheimer’s disease: A Mendelian randomization study. Int. J. Rheum. Dis. 2019, 22, 1046–1051. [Google Scholar] [CrossRef]
- Parkinson Study Group, S.-P.D.I.; Schwarzschild, M.A.; Ascherio, A.; Casaceli, C.; Curhan, G.C.; Fitzgerald, R.; Kamp, C.; Lungu, C.; Macklin, E.A.; Marek, K.; et al. Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 2021, 326, 926–939. [Google Scholar] [CrossRef]
- Nardi, V.; Franchi, F.; Prasad, M.; Fatica, E.M.; Alexander, M.P.; Bois, M.C.; Lam, J.; Singh, R.J.; Meyer, F.B.; Lanzino, G.; et al. Uric Acid Expression in Carotid Atherosclerotic Plaque and Serum Uric Acid Are Associated with Cerebrovascular Events. Hypertension 2022, 79, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Kusano, T.; Ehirchiou, D.; Matsumura, T.; Chobaz, V.; Nasi, S.; Castelblanco, M.; So, A.; Lavanchy, C.; Acha-Orbea, H.; Nishino, T.; et al. Targeted knock-in mice expressing the oxidase-fixed form of xanthine oxidoreductase favor tumor growth. Nat. Commun. 2019, 10, 4904. [Google Scholar] [CrossRef] [PubMed]
- Konta, T.; Ichikawa, K.; Kawasaki, R.; Fujimoto, S.; Iseki, K.; Moriyama, T.; Yamagata, K.; Tsuruya, K.; Narita, I.; Kondo, M.; et al. Association between serum uric acid levels and mortality: A nationwide community-based cohort study. Sci. Rep. 2020, 10, 6066. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Schillaci, G.; Reboldi, G.; Santeusanio, F.; Porcellati, C.; Brunetti, P. Relation between serum uric acid and risk of cardiovascular disease in essential hypertension. The PIUMA study. Hypertension 2000, 36, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Kamei, K.; Konta, T.; Hirayama, A.; Ichikawa, K.; Kubota, I.; Fujimoto, S.; Iseki, K.; Moriyama, T.; Yamagata, K.; Tsuruya, K.; et al. Associations between serum uric acid levels and the incidence of nonfatal stroke: A nationwide community-based cohort study. Clin. Exp. Nephrol. 2017, 21, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Satoh, M.; Tatsumi, Y.; Murakami, T.; Muroya, T.; Hirose, T.; Ohkubo, T.; Mori, T.; Hozawa, A.; Metoki, H. Detailed association between serum uric acid levels and the incidence of chronic kidney disease stratified by sex in middle-aged adults. Atherosclerosis 2021, 330, 107–113. [Google Scholar] [CrossRef]
- Kuwabara, M.; Niwa, K.; Ohtahara, A.; Hamada, T.; Miyazaki, S.; Mizuta, E.; Ogino, K.; Hisatome, I. Prevalence and complications of hypouricemia in a general population: A large-scale cross-sectional study in Japan. PLoS ONE 2017, 12, e0176055. [Google Scholar] [CrossRef]
- Cho, S.K.; Chang, Y.; Kim, I.; Ryu, S. U-Shaped Association Between Serum Uric Acid Level and Risk of Mortality: A Cohort Study. Arthritis Rheumatol. 2018, 70, 1122–1132. [Google Scholar] [CrossRef]
- Hu, L.; Hu, G.; Xu, B.P.; Zhu, L.; Zhou, W.; Wang, T.; Bao, H.; Cheng, X. U-Shaped Association of Serum Uric Acid with All-Cause and Cause-Specific Mortality in US Adults: A Cohort Study. J. Clin. Endocrinol. Metab. 2020, 105, e597–e609. [Google Scholar] [CrossRef]
- Russo, E.; Viazzi, F.; Pontremoli, R.; Barbagallo, C.M.; Bombelli, M.; Casiglia, E.; Cicero, A.F.G.; Cirillo, M.; Cirillo, P.; Desideri, G.; et al. Serum Uric Acid and Kidney Disease Measures Independently Predict Cardiovascular and Total Mortality: The Uric Acid Right for Heart Health (URRAH) Project. Front. Cardiovasc. Med. 2021, 8, 713652. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Chubachi, S.; Namkoong, H.; Otake, S.; Nakagawara, K.; Tanaka, H.; Lee, H.; Morita, A.; Watase, M.; Kusumoto, T.; et al. U-shaped association between abnormal serum uric acid levels and COVID-19 severity: Reports from the Japan COVID-19 Task Force. Int. J. Infect. Dis. 2022, 122, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Namkoong, H.; Edahiro, R.; Takano, T.; Nishihara, H.; Shirai, Y.; Sonehara, K.; Tanaka, H.; Azekawa, S.; Mikami, Y.; Lee, H.; et al. DOCK2 is involved in the host genetics and biology of severe COVID-19. Nature 2022, 609, 754–760. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, J.D.; Dalbeth, N.; Mikuls, T.; Brignardello-Petersen, R.; Guyatt, G.; Abeles, A.M.; Gelber, A.C.; Harrold, L.R.; Khanna, D.; King, C.; et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis Care Res. 2020, 72, 744–760. [Google Scholar] [CrossRef] [PubMed]
- Yeun, J.Y.; Hasbargen, J.A. Renal hypouricemia: Prevention of exercise-induced acute renal failure and a review of the literature. Am. J. Kidney Dis. 1995, 25, 937–946. [Google Scholar] [CrossRef]
- Bhasin, B.; Stiburkova, B.; De Castro-Pretelt, M.; Beck, N.; Bodurtha, J.N.; Atta, M.G. Hereditary renal hypouricemia: A new role for allopurinol? Am. J. Med. 2014, 127, e3–e4. [Google Scholar] [CrossRef]
- Sanchis-Gomar, F.; Pareja-Galeano, H.; Perez-Quilis, C.; Santos-Lozano, A.; Fiuza-Luces, C.; Garatachea, N.; Lippi, G.; Lucia, A. Effects of allopurinol on exercise-induced muscle damage: New therapeutic approaches? Cell Stress Chaperones 2015, 20, 3–13. [Google Scholar] [CrossRef]
- Aizawa, C.; Okabe, M.; Takahashi, D.; Sagasaki, M.; Watanabe, M.; Fujimoto, T.; Yoshioka, Y.; Katsuma, A.; Kimura, A.; Miyamoto, D.; et al. Possible Use of Non-purine Selective Xanthine Oxidoreductase Inhibitors for Prevention of Exercise-induced Acute Kidney Injury Associated with Renal Hypouricemia. Intern. Med. 2023, 62, 2725–2730. [Google Scholar] [CrossRef]
- Biomedicines. Special Issue “Dysuricemia: Recent Advances in Urate Research from Hypouricemia to Hyperuricemia/Gout”. Available online: https://www.mdpi.com/journal/biomedicines/special_issues/V3XF0F6I99 (accessed on 5 August 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uric Acid Production | Urate Excretion | Production and Excretion | |
|---|---|---|---|
| Hyperuricemia | Renal overload type (overproduction type and extra-renal underexcretion type *) | Renal underexcretion type | Combined type |
|
|
| |
| Hypouricemia | Underproduction type | Overexcretion type | Combined type |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, A.; Kurajoh, M.; Toyoda, Y.; Takada, T.; Ichida, K.; Matsuo, H. Dysuricemia. Biomedicines 2023, 11, 3169. https://doi.org/10.3390/biomedicines11123169
Nakayama A, Kurajoh M, Toyoda Y, Takada T, Ichida K, Matsuo H. Dysuricemia. Biomedicines. 2023; 11(12):3169. https://doi.org/10.3390/biomedicines11123169
Chicago/Turabian StyleNakayama, Akiyoshi, Masafumi Kurajoh, Yu Toyoda, Tappei Takada, Kimiyoshi Ichida, and Hirotaka Matsuo. 2023. "Dysuricemia" Biomedicines 11, no. 12: 3169. https://doi.org/10.3390/biomedicines11123169
APA StyleNakayama, A., Kurajoh, M., Toyoda, Y., Takada, T., Ichida, K., & Matsuo, H. (2023). Dysuricemia. Biomedicines, 11(12), 3169. https://doi.org/10.3390/biomedicines11123169