Fibronectin Type III Domain Containing 3B as a Potential Prognostic and Therapeutic Biomarker for Glioblastoma

 , , and
, , and
Abstract
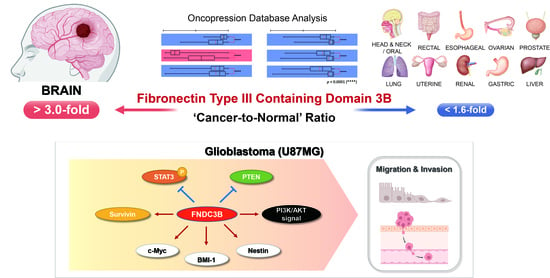
1. Introduction
2. Materials and Methods
2.1. Gene Expression and Prognosis Analysis
2.2. Cell Cultures
2.3. Small Interfering RNA Transfection
2.4. Cell Viability Assay
2.5. Quantitative Reverse Transcription Polymerase Chain Reaction
2.6. Cell Migration Assays
2.7. Cell Adhesion Assay
2.8. Three-Dimensional Spheroid Invasion Assays
2.9. Proteome Profiler Phospho-Kinase Array
2.10. Western Blot Analysis
2.11. Network Analysis Using the MetaCore Database
2.12. Statistical Analysis
3. Results
3.1. FNDC3B Expression and Related Prognosis Analysis Using the Oncopression Database
3.1.1. FNDC3B Expression in Various Organ Cancers
3.1.2. FNDC3B Expression in Human Brain Pathologies and Brain Tumors
3.1.3. FNDC3B as a Prognostic Factor for Glioblastoma
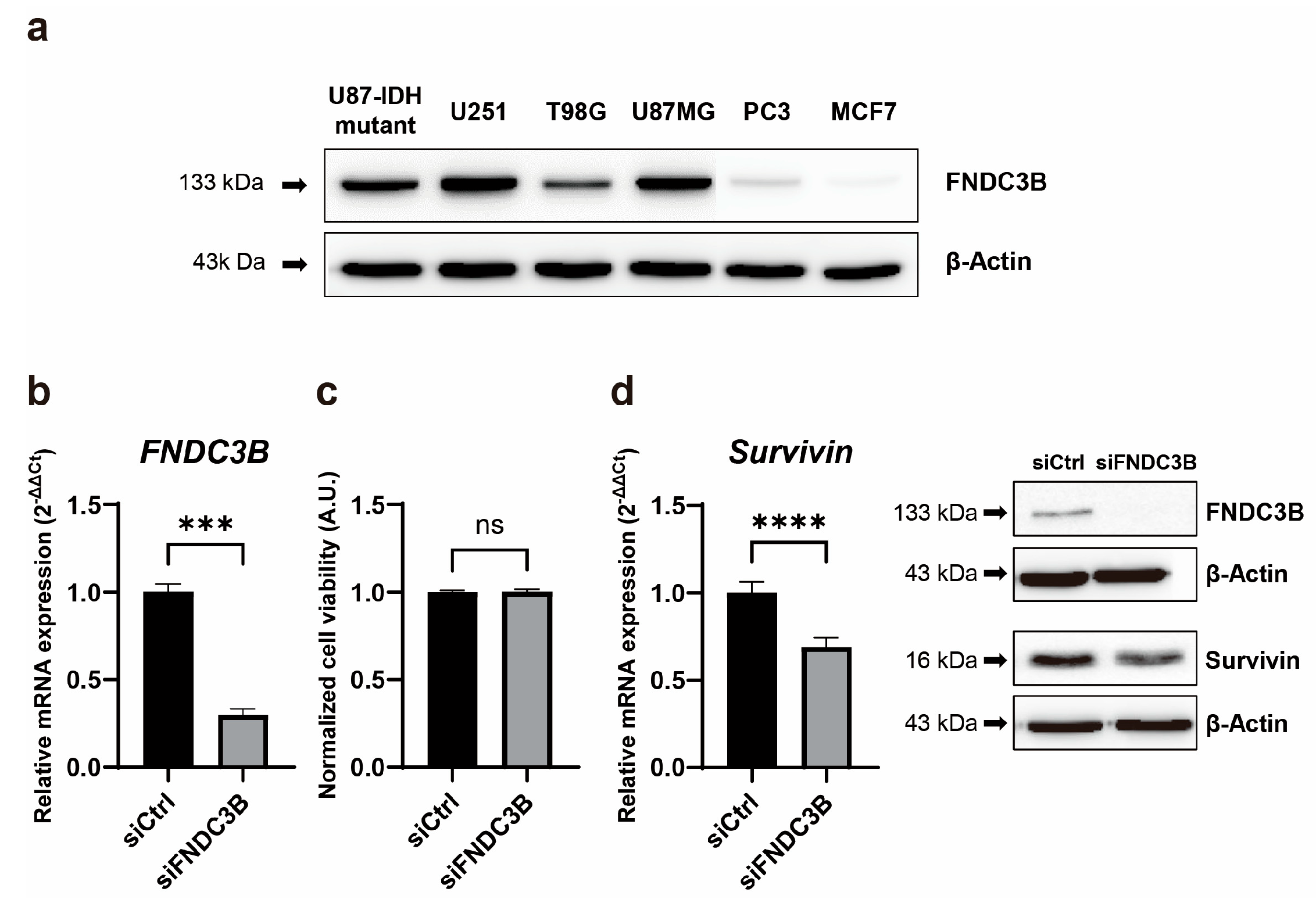
3.2. FNDC3B Expression and Relations with Survivin Expression in GBM Cells
3.3. Inhibition of Cell Migration through FNDC3B Silencing in GBM Cells
3.4. Inhibition of Cell Invasion and Stemness through FNDC3B Silencing in GBM Cells
3.5. Investigation of FNDC3B-Related Signaling Pathways in GBM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef]
- Babi, A.; Menlibayeva, K.; Bex, T.; Kuandykova, S.; Akshulakov, S. The Current State of Adult Glial Tumor Patients’ Care in Kazakhstan: Challenges in Diagnosis and Patterns in Survival Outcomes. Biomedicines 2023, 11, 886. [Google Scholar] [CrossRef]
- Newlands, E.S.; Stevens, M.F.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Karachi, A.; Dastmalchi, F.; Mitchell, D.A.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro Oncol. 2018, 20, 1566–1572. [Google Scholar] [CrossRef]
- Silva, J.A.; Colquhoun, A. Effect of Polyunsaturated Fatty Acids on Temozolomide Drug-Sensitive and Drug-Resistant Glioblastoma Cells. Biomedicines 2023, 11, 779. [Google Scholar] [CrossRef]
- Feldheim, J.; Kessler, A.F.; Monoranu, C.M.; Ernestus, R.I.; Lohr, M.; Hagemann, C. Changes of O(6)-Methylguanine DNA Methyltransferase (MGMT) Promoter Methylation in Glioblastoma Relapse-A Meta-Analysis Type Literature Review. Cancers 2019, 11, 1837. [Google Scholar] [CrossRef]
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef]
- Liu, Y.; Lang, F.; Chou, F.J.; Zaghloul, K.A.; Yang, C. Isocitrate Dehydrogenase Mutations in Glioma: Genetics, Biochemistry, and Clinical Indications. Biomedicines 2020, 8, 294. [Google Scholar] [CrossRef]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells: Significance in Origin, Pathogenesis and Treatment of Glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef]
- Vargas-Toscano, A.; Nickel, A.C.; Li, G.; Kamp, M.A.; Muhammad, S.; Leprivier, G.; Fritsche, E.; Barker, R.A.; Sabel, M.; Steiger, H.J.; et al. Rapalink-1 Targets Glioblastoma Stem Cells and Acts Synergistically with Tumor Treating Fields to Reduce Resistance against Temozolomide. Cancers 2020, 12, 3859. [Google Scholar] [CrossRef]
- Cai, Y.; Gu, W.T.; Cheng, K.; Jia, P.F.; Li, F.; Wang, M.; Zhang, W.F.; Qiu, J.T.; Wu, Z.B.; Zhao, W.G. Knockdown of TRIM32 inhibits tumor growth and increases the therapeutic sensitivity to temozolomide in glioma in a p53-dependent and -independent manner. Biochem. Biophys. Res. Commun. 2021, 550, 134–141. [Google Scholar] [CrossRef]
- Castro, B.A.; Aghi, M.K. Bevacizumab for glioblastoma: Current indications, surgical implications, and future directions. Neurosurg. Focus. 2014, 37, E9. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Bevacizumab for the treatment of recurrent glioblastoma. Clin. Med. Insights Oncol. 2011, 5, 117–129. [Google Scholar] [CrossRef]
- Siegelin, M.D.; Raskett, C.M.; Gilbert, C.A.; Ross, A.H.; Altieri, D.C. Sorafenib exerts anti-glioma activity in vitro and in vivo. Neurosci. Lett. 2010, 478, 165–170. [Google Scholar] [CrossRef]
- Lee, A.; Arasaratnam, M.; Chan, D.L.H.; Khasraw, M.; Howell, V.M.; Wheeler, H. Anti-epidermal growth factor receptor therapy for glioblastoma in adults. Cochrane Database Syst. Rev. 2020, 5, CD013238. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef]
- Nafe, R.; Hattingen, E. The Spectrum of Molecular Pathways in Gliomas-An Up-to-Date Review. Biomedicines 2023, 11, 2281. [Google Scholar] [CrossRef]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neurooncol 2021, 151, 41–53. [Google Scholar] [CrossRef]
- Yu, M.W.; Quail, D.F. Immunotherapy for Glioblastoma: Current Progress and Challenges. Front. Immunol. 2021, 12, 676301. [Google Scholar] [CrossRef]
- El Atat, O.; Naser, R.; Abdelkhalek, M.; Habib, R.A.; El Sibai, M. Molecular targeted therapy: A new avenue in glioblastoma treatment. Oncol. Lett. 2023, 25, 46. [Google Scholar] [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Burster, T.; Traut, R.; Yermekkyzy, Z.; Mayer, K.; Westhoff, M.A.; Bischof, J.; Knippschild, U. Critical View of Novel Treatment Strategies for Glioblastoma: Failure and Success of Resistance Mechanisms by Glioblastoma Cells. Front. Cell Dev. Biol. 2021, 9, 695325. [Google Scholar] [CrossRef]
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma targeted therapy: Insight into future of molecular approaches. Mol. Cancer 2022, 21, 39. [Google Scholar] [CrossRef]
- Kishimoto, K.; Kato, A.; Osada, S.; Nishizuka, M.; Imagawa, M. Fad104, a positive regulator of adipogenesis, negatively regulates osteoblast differentiation. Biochem. Biophys. Res. Commun. 2010, 397, 187–191. [Google Scholar] [CrossRef]
- Lin, C.H.; Lin, Y.W.; Chen, Y.C.; Liao, C.C.; Jou, Y.S.; Hsu, M.T.; Chen, C.F. FNDC3B promotes cell migration and tumor metastasis in hepatocellular carcinoma. Oncotarget 2016, 7, 49498–49508. [Google Scholar] [CrossRef][Green Version]
- Han, B.; Wang, H.; Zhang, J.; Tian, J. FNDC3B is associated with ER stress and poor prognosis in cervical cancer. Oncol. Lett. 2020, 19, 406–414. [Google Scholar] [CrossRef]
- Cheng, C.K.; Wang, A.Z.; Wong, T.H.Y.; Wan, T.S.K.; Cheung, J.S.; Raghupathy, R.; Chan, N.P.H.; Ng, M.H.L. FNDC3B is another novel partner fused to RARA in the t(3;17)(q26;q21) variant of acute promyelocytic leukemia. Blood 2017, 129, 2705–2709. [Google Scholar] [CrossRef]
- Li, Y.; Yang, J.; Wang, H.; Qiao, W.; Guo, Y.; Zhang, S.; Guo, Y. FNDC3B, Targeted by miR-125a-5p and miR-217, Promotes the Proliferation and Invasion of Colorectal Cancer Cells via PI3K/mTOR Signaling. Onco Targets Ther. 2020, 13, 3501–3510. [Google Scholar] [CrossRef]
- Fucci, C.; Resnati, M.; Riva, E.; Perini, T.; Ruggieri, E.; Orfanelli, U.; Paradiso, F.; Cremasco, F.; Raimondi, A.; Pasqualetto, E.; et al. The Interaction of the Tumor Suppressor FAM46C with p62 and FNDC3 Proteins Integrates Protein and Secretory Homeostasis. Cell Rep. 2020, 32, 108162. [Google Scholar] [CrossRef]
- Stangeland, B.; Mughal, A.A.; Grieg, Z.; Sandberg, C.J.; Joel, M.; Nygård, S.; Meling, T.; Murrell, W.; Vik Mo, E.O.; Langmoen, I.A. Combined expressional analysis, bioinformatics and targeted proteomics identify new potential therapeutic targets in glioblastoma stem cells. Oncotarget 2015, 6, 26192–26215. [Google Scholar] [CrossRef]
- Wang, G.H.; Wang, L.Y.; Zhang, C.; Zhang, P.; Wang, C.H.; Cheng, S. MiR-1225-5p acts as tumor suppressor in glioblastoma via targeting FNDC3B. Open Med. 2020, 15, 872–881. [Google Scholar] [CrossRef]
- Lee, J.; Choi, C. Oncopression: Gene expression compendium for cancer with matched normal tissues. Bioinformatics 2017, 33, 2068–2070. [Google Scholar] [CrossRef]
- Piccolo, S.R.; Withers, M.R.; Francis, O.E.; Bild, A.H.; Johnson, W.E. Multiplatform single-sample estimates of transcriptional activation. Proc. Natl. Acad. Sci. USA 2013, 110, 17778–17783. [Google Scholar] [CrossRef]
- Zaykin, D.V. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J. Evol. Biol. 2011, 24, 1836–1841. [Google Scholar] [CrossRef]
- Vanamala, J.; Radhakrishnan, S.; Reddivari, L.; Bhat, V.B.; Ptitsyn, A. Resveratrol suppresses human colon cancer cell proliferation and induces apoptosis via targeting the pentose phosphate and the talin-FAK signaling pathways-A proteomic approach. Proteome Sci. 2011, 9, 49. [Google Scholar] [CrossRef]
- Hwang, P.I.; Wu, H.B.; Wang, C.D.; Lin, B.L.; Chen, C.T.; Yuan, S.; Wu, G.; Li, K.C. Tissue-specific gene expression templates for accurate molecular characterization of the normal physiological states of multiple human tissues with implication in development and cancer studies. BMC Genom. 2011, 12, 439. [Google Scholar] [CrossRef]
- O’ Driscoll, L.; Linehan, R.; Clynes, M. Survivin: Role in normal cells and in pathological conditions. Curr. Cancer Drug Targets 2003, 3, 131–152. [Google Scholar] [CrossRef]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T., Jr.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.C.; et al. Human glioblastoma-derived cancer stem cells: Establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef]
- Molina, J.R.; Hayashi, Y.; Stephens, C.; Georgescu, M.M. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia 2010, 12, 453–463. [Google Scholar] [CrossRef]
- Kumar, S.U.; Kumar, D.T.; Siva, R.; Doss, C.G.P.; Zayed, H. Integrative Bioinformatics Approaches to Map Potential Novel Genes and Pathways Involved in Ovarian Cancer. Front. Bioeng. Biotechnol. 2019, 7, 391. [Google Scholar] [CrossRef]
- De la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; Depinho, R.A.; Bonni, A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes. Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef]
- Zhang, H.F.; Lai, R. STAT3 in Cancer-Friend or Foe? Cancers 2014, 6, 1408–1440. [Google Scholar] [CrossRef]
- Wang, J.; Lv, X.; Guo, X.; Dong, Y.; Peng, P.; Huang, F.; Wang, P.; Zhang, H.; Zhou, J.; Wang, Y.; et al. Feedback activation of STAT3 limits the response to PI3K/AKT/mTOR inhibitors in PTEN-deficient cancer cells. Oncogenesis 2021, 10, 8. [Google Scholar] [CrossRef]
- Yoshida, G.J. Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 2018, 37, 173. [Google Scholar] [CrossRef]
- Legler, J.M.; Ries, L.A.; Smith, M.A.; Warren, J.L.; Heineman, E.F.; Kaplan, R.S.; Linet, M.S. Cancer surveillance series [corrected]: Brain and other central nervous system cancers: Recent trends in incidence and mortality. J. Natl. Cancer Inst. 1999, 91, 1382–1390. [Google Scholar] [CrossRef]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and Therapeutic Biomarkers in Glioblastoma: Current Status and Future Perspectives. Biomed. Res. Int. 2017, 2017, 8013575. [Google Scholar] [CrossRef]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2020, 10, 574012. [Google Scholar] [CrossRef]
- Zhu, H.; Ren, X.; Ding, W. A pan cancerous analysis of FNDC3B in human multiple tumors. Res. Sq. 2023, preprint. [Google Scholar] [CrossRef]
- Xu, H.; Hu, Y.; Qiu, W. Potential mechanisms of microRNA-129-5p in inhibiting cell processes including viability, proliferation, migration and invasiveness of glioblastoma cells U87 through targeting FNDC3B. Biomed. Pharmacother. 2017, 87, 405–411. [Google Scholar] [CrossRef]
- Katoh, D.; Nishizuka, M.; Osada, S.; Imagawa, M. Fad104, a positive regulator of adipocyte differentiation, suppresses invasion and metastasis of melanoma cells by inhibition of STAT3 activity. PLoS ONE 2015, 10, e0117197. [Google Scholar] [CrossRef]
- Aigner, P.; Just, V.; Stoiber, D. STAT3 isoforms: Alternative fates in cancer? Cytokine 2019, 118, 27–34. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ung, T.T.; Li, S.; Sah, D.K.; Park, S.Y.; Lian, S.; Jung, Y.D. Lithocholic Acid Induces miR21, Promoting PTEN Inhibition via STAT3 and ERK-1/2 Signaling in Colorectal Cancer Cells. Int. J. Mol. Sci. 2021, 22, 10209. [Google Scholar] [CrossRef]
- Hashemi, M.; Etemad, S.; Rezaei, S.; Ziaolhagh, S.; Rajabi, R.; Rahmanian, P.; Abdi, S.; Koohpar, Z.K.; Rafiei, R.; Raei, B.; et al. Progress in targeting PTEN/PI3K/Akt axis in glioblastoma therapy: Revisiting molecular interactions. Biomed. Pharmacother. 2023, 158, 114204. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ | Number of Cancer Samples | Number of Normal Samples | ‘Cancer-to-Normal’ Ratio | p Value |
|---|---|---|---|---|
| Brain | 2517 | 723 | 3.0287 ± 1.2028 | <0.0001 |
| Head and Neck | 360 | 119 | 1.5969 ± 0.2944 | <0.0001 |
| Oral | 309 | 167 | 1.4436 ± 0.3410 | <0.0001 |
| Thyroid | 338 | 197 | 1.0261 ± 0.1225 | ns |
| Esophageal | 120 | 21 | 1.4027 ± 0.2779 | <0.0001 |
| Lung | 2502 | 650 | 1.1854 ± 0.1820 | <0.0001 |
| Breast | 5516 | 471 | 1.0113 ± 0.2339 | <0.05 |
| Gastric | 934 | 110 | 1.1142 ± 0.1079 | <0.0001 |
| Liver | 524 | 322 | 1.1138 ± 0.1453 | <0.0001 |
| Pancreatic | 240 | 98 | 1.0509 ± 0.2307 | <0.05 |
| Colon | 2449 | 500 | 1.0653 ± 0.1315 | <0.0001 |
| Rectal | 205 | 21 | 1.4152 ± 0.0964 | <0.0001 |
| Adrenal | 355 | 50 | 0.8813 ± 0.1592 | <0.0001 |
| Renal | 504 | 195 | 1.1643 ± 0.2109 | <0.0001 |
| Ureter | 58 | 45 | 1.0410 ± 1.0008 | ns |
| Bladder | 260 | 11 | 1.0001 ± 0.0995 | ns |
| Ovarian | 1146 | 92 | 1.3800 ± 0.3783 | <0.0001 |
| Uterine | 387 | 192 | 1.1760 ± 0.1723 | <0.0001 |
| Prostate | 257 | 75 | 1.3595 ± 0.5624 | <0.0001 |
| Skin | 454 | 488 | 1.0501 ± 0.3296 | <0.0001 |
| Pathology | Number of Samples | Mean | Standard Deviation | Contrast to Normal (Fold) | Contrast to Normal, p Value |
|---|---|---|---|---|---|
| Normal | 723 | 0.1948 | 0.1155 | - | - |
| AD | 227 | 0.1966 | 0.0954 | 1.0096 | ns |
| PD | 51 | 0.2177 | 0.1447 | 1.1179 | ns |
| MDD | 134 | 0.2145 | 0.0472 | 1.1011 | <0.0001 |
| Epilepsy | 43 | 0.3716 | 0.1525 | 1.9079 | <0.0001 |
| Grade 1 | 74 | 0.5973 | 0.2193 | 3.0665 | <0.0001 |
| Grade 2 | 134 | 0.4330 | 0.1606 | 2.2234 | <0.0001 |
| Grade 3 | 132 | 0.5297 | 0.1756 | 2.7197 | <0.0001 |
| Grade 4 | 865 | 0.7034 | 0.2075 | 3.6113 | <0.0001 |
| Grade 4 MGMT methylated | 44 | 0.5797 | 0.1633 | 2.9764 | <0.0001 |
| Grade 4 MGMT unmethylated | 34 | 0.6155 | 0.1447 | 3.1600 | <0.0001 |
| Grade 4 IDH- mutated | 12 | 0.7016 | 0.2143 | 3.6020 | <0.0001 |
| Grade 4 IDH- wild type | 37 | 0.8123 | 0.1725 | 4.1705 | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, H.; Yun, M.; Kwon, T.-H.; Bang, M.; Lee, J.; Lee, Y.S.; Ko, H.Y.; Chong, K. Fibronectin Type III Domain Containing 3B as a Potential Prognostic and Therapeutic Biomarker for Glioblastoma. Biomedicines 2023, 11, 3168. https://doi.org/10.3390/biomedicines11123168
Kwon H, Yun M, Kwon T-H, Bang M, Lee J, Lee YS, Ko HY, Chong K. Fibronectin Type III Domain Containing 3B as a Potential Prognostic and Therapeutic Biomarker for Glioblastoma. Biomedicines. 2023; 11(12):3168. https://doi.org/10.3390/biomedicines11123168
Chicago/Turabian StyleKwon, Hyukjun, Minji Yun, Taek-Hyun Kwon, Minji Bang, Jungsul Lee, Yeo Song Lee, Hae Young Ko, and Kyuha Chong. 2023. "Fibronectin Type III Domain Containing 3B as a Potential Prognostic and Therapeutic Biomarker for Glioblastoma" Biomedicines 11, no. 12: 3168. https://doi.org/10.3390/biomedicines11123168
APA StyleKwon, H., Yun, M., Kwon, T.-H., Bang, M., Lee, J., Lee, Y. S., Ko, H. Y., & Chong, K. (2023). Fibronectin Type III Domain Containing 3B as a Potential Prognostic and Therapeutic Biomarker for Glioblastoma. Biomedicines, 11(12), 3168. https://doi.org/10.3390/biomedicines11123168