
Metabolic Modulators in Cardiovascular Complications of Systemic Lupus Erythematosus



Abstract
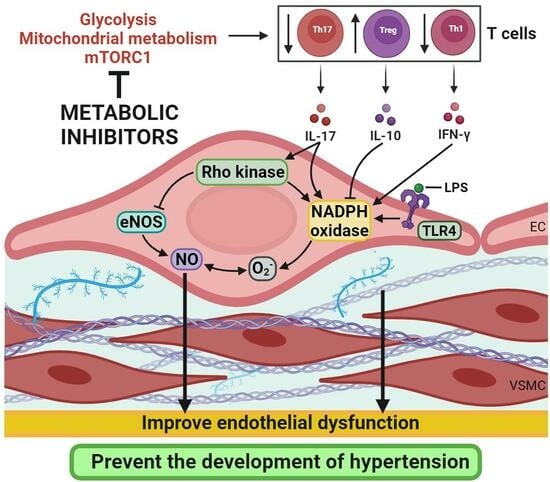
1. Introduction
2. Systemic Lupus Erythematosus and Arterial Hypertension
2.1. Role of Endothelial Function in SLE Hypertension
2.2. Immune System and Hypertension in SLE
3. The Role of Cellular Metabolism in the Pathogenesis of SLE
4. Metabolic Modulators and SLE
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsokos, G.C. Systemic Lupus Erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Gershwin, M.E.; Chang, C. Diagnostic criteria for systemic lupus erythematosus: A critical review. J. Autoimmun. 2014, 48–49, 10–13. [Google Scholar] [CrossRef]
- Akhil, A.; Bansal, R.; Anupam, K.; Tandon, A.; Bhatnagar, A. Systemic lupus erythematosus: Latest insight into etiopathogenesis. Rheumatol. Int. 2023, 43, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef]
- Blachut, D.; Przywara-Chowaniec, B.; Tomasik, A.; Kukulski, T.; Morawiec, B. Update of Potential Biomarkers in Risk Prediction and Monitoring of Atherosclerosis in Systemic Lupus Erythematosus to Prevent Cardiovascular Disease. Biomedicines 2023, 11, 2814. [Google Scholar] [CrossRef]
- Tselios, K.; Koumaras, C.; Gladman, D.D.; Urowitz, M.B. Dyslipidemia in systemic lupus erythematosus: Just another comorbidity? Semin. Arthritis Rheum. 2016, 45, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Urowitz, M.B.; Gladman, D.; Ibañez, D.; Fortin, P.; Sanchez-Guerrero, J.; Bae, S.; Clarke, A.; Bernatsky, S.; Gordon, C.; Hanly, J.; et al. Accumulation of coronary artery disease risk factors over three years: Data from an international inception cohort. Arthritis Care Res. 2008, 59, 176–180. [Google Scholar] [CrossRef]
- Urowitz, M.B.; Gladman, D.; Ibañez, D.; Fortin, P.; Sanchez-Guerrero, J.; Bae, S.; Clarke, A.; Bernatsky, S.; Gordon, C.; Hanly, J.; et al. Clinical manifestations and coronary artery disease risk factors at diagnosis of systemic lupus erythematosus: Data from an international inception cohort. Lupus 2007, 16, 731–735. [Google Scholar] [CrossRef]
- Mok, C.C. Metabolic syndrome and systemic lupus erythematosus: The connection. Expert. Rev. Clin. Immunol. 2019, 15, 765–775. [Google Scholar] [CrossRef]
- Bello, N.; Meyers, K.J.; Workman, J.; Hartley, L.; McMahon, M. Cardiovascular events and risk in patients with systemic lupus erythematosus: Systematic literature review and meta-analysis. Lupus 2023, 32, 325–341. [Google Scholar] [CrossRef]
- Esdaile, J.M.; Abrahamowicz, M.; Grodzicky, T.; Li, Y.; Panaritis, C.; Berger, R.D.; Côte, R.; Grover, S.A.; Fortin, P.R.; Clarke, A.E.; et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001, 44, 2331–2337. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J. Systemic lupus erythematosus and cardiovascular disease. J. Intern. Med. 2023, 293, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Feng, R.; Werth, V.P.; Williams, K.J. State of current management of the heightened risk for atherosclerotic cardiovascular events in an established cohort of patients with lupus erythematosus. Lupus Sci. Med. 2023, 10, e000908. [Google Scholar] [CrossRef] [PubMed]
- Tselios, K.; Koumaras, C.; Urowitz, M.B.; Gladman, D.D. Do current arterial hypertension treatment guidelines apply to systemic lupus erythematosus patients? A critical appraisal. Semin. Arthritis Rheum. 2014, 43, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Ikdahl, E.; Wibetoe, G.; Rollefstad, S.; Salberg, A.; Bergsmark, K.; Kvien, T.K.; Olsen, I.C.; Soldal, D.M.; Bakland, G.; Lexberg, Å.; et al. Guideline recommended treatment to targets of cardiovascular risk is inadequate in patients with inflammatory joint diseases. Int. J. Cardiol. 2019, 274, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J. Young Investigator Award Lecture of the APS Water and Electrolyte Homeostasis Section, 2008: The pathophysiology of hypertension in systemic lupus erythematosus, American Journal of Physiology-Regulatory. Integr. Comp. Physiol. 2009, 296, R1258–R1267. [Google Scholar] [CrossRef]
- Gómez-Guzmán, M.; Jiménez, R.; Romero, M.; Sánchez, M.; Zarzuelo, M.J.; Gómez-Morales, M.; O’Valle, F.; López-Farré, A.J.; Algieri, F.; Gálvez, J.; et al. Chronic hydroxychloroquine improves endothelial dysfunction and protects kidney in a mouse model of systemic lupus erythematosus. Hypertension 2014, 64, 330–337. [Google Scholar] [CrossRef]
- Robles-Vera, I.; La Visitación, N.D.; Toral, M.; Sánchez, M.; Gómez-Guzmán, M.; O’valle, F.; Jiménez, R.; Duarte, J.; Romero, M. Toll-like receptor 7-driven lupus autoimmunity induces hypertension and vascular alterations in mice. J. Hypertens. 2020, 38, 1322–1335. [Google Scholar] [CrossRef]
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Manzi, S.; Meilahn, E.N.; Rairie, J.E.; Conte, C.G.; Medsger, T.A.; Jansen-McWilliams, L.; D’Agostino, R.B.; Kuller, L.H. Age-specific Incidence Rates of Myocardial Infarction and Angina in Women with Systemic Lupus Erythematosus: Comparison with the Framingham Study. Am. J. Epidemiol. 1997, 145, 408–415. [Google Scholar] [CrossRef]
- Taylor, E.B.; Ryan, M.J. Understanding mechanisms of hypertension in systemic lupus erythematosus. Ther. Adv. Cardiovasc. Dis. 2017, 11, 20–32. [Google Scholar] [CrossRef]
- Small, H.Y.; Migliarino, S.; Czesnikiewicz-Guzik, M.; Guzik, T.J. Hypertension: Focus on autoimmunity and oxidative stress. Free Radic. Biol. Med. 2018, 125, 104–115. [Google Scholar] [CrossRef]
- Gonzalez-Vicente, A.; Hong, N.; Garvin, J.L. Effects of reactive oxygen species on renal tubular transport. Am. J. Physiol. Ren. Physiol. 2019, 317, F444–F455. [Google Scholar] [CrossRef]
- Burnett, R.; Ravel, G.; Descotes, J. Clinical and histopathological progression of lesions in lupus-prone (NZB×NZW) F1 mice. Exp. Toxicol. Pathol. 2004, 56, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.; Pham, G.S.; Brooks, C.D.; Dinh, V.Q.; Young-Stubbs, C.M.; Shimoura, C.G.; Mathis, K.W. Should Renal Inflammation Be Targeted While Treating Hypertension? Front. Physiol. 2022, 13, 886779. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.L.; Gilkeson, G. Mouse models of lupus: What they tell us and what they don’t. Lupus Sci. Med. 2018, 5, e000199. [Google Scholar] [CrossRef] [PubMed]
- Mathis, K.W.; Venegas-Pont, M.; Masterson, C.W.; Wasson, K.L.; Ryan, M.J. Blood pressure in a hypertensive mouse model of SLE is not salt-sensitive. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 301, R1281–R1285. [Google Scholar] [CrossRef] [PubMed]
- Herlitz, H.; Svalander, C.; Tarkowski, A.; Westberg, G. Effect of captopril on murine systemic lupus erythematosus disease. J. Hypertens. 1988, 6, S684–S686. [Google Scholar] [CrossRef]
- Herlitz, H.; Tarkowski, A.; Svalander, C.; Volkmann, R.; Westberg, G. Beneficial Effect of Captopril on Systemic Lupus erythematosus-Like Disease in MRL 1pr/1pr Mice. Int. Arch. Allergy Immunol. 1988, 85, 272–277. [Google Scholar] [CrossRef]
- De Albuquerque, D.A.; Saxena, V.; Adams, D.E.; Boivin, G.P.; Brunner, H.I.; Witte, D.P.; Singh, R.R. An ACE inhibitor reduces Th2 cytokines and TGF-β1 and TGF-β2 isoforms in murine lupus nephritis. Kidney Int. 2004, 65, 846–859. [Google Scholar] [CrossRef]
- Yokogawa, M.; Takaishi, M.; Nakajima, K.; Kamijima, R.; Fujimoto, C.; Kataoka, S.; Terada, Y.; Sano, S. Epicutaneous Application of Toll-like Receptor 7 Agonists Leads to Systemic Autoimmunity in Wild-Type Mice: A New Model of Systemic Lupus Erythematosus. Arthritis Rheumatol. 2014, 66, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Villalvazo, P.; Carriazo, S.; Rojas-Rivera, J.; Ramos, A.M.; Ortiz, A.; Perez-Gomez, M.V. Gain-of-function TLR7 and loss-of-function A20 gene variants identify a novel pathway for Mendelian lupus and lupus nephritis. Clin. Kidney J. 2022, 15, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Seto, N.L.; Carmona-Rivera, C.; Kaplan, M.J. Accelerated model of lupus autoimmunity and vasculopathy driven by toll-like receptor 7/9 imbalance. Lupus Sci. Med. 2018, 5, e000259. [Google Scholar] [CrossRef] [PubMed]
- Sandling, J.K.; Pucholt, P.; Rosenberg, L.H.; Farias, F.H.G.; Kozyrev, S.V.; Eloranta, M.-L.; Alexsson, A.; Bianchi, M.; Padyukov, L.; Bengtsson, C.; et al. Molecular pathways in patients with systemic lupus erythematosus revealed by gene-centred DNA sequencing. Ann. Rheum. Dis. 2021, 80, 109–117. [Google Scholar] [CrossRef] [PubMed]
- McClung, D.M.; Kalusche, W.J.; Jones, K.E.; Ryan, M.J.; Taylor, E.B. Hypertension and endothelial dysfunction in the pristane model of systemic lupus erythematosus. Physiol. Rep. 2021, 9, e14734. [Google Scholar] [CrossRef] [PubMed]
- Manzi, S.; Selzer, F.; Sutton-Tyrrell, K.; Fitzgerald, S.G.; Rairie, J.E.; Tracy, R.P.; Kuller, L.H. Prevalence and risk factors of carotid plaque in women with systemic lupus erythematosus. Arthritis Rheum. 1999, 42, 51–60. [Google Scholar] [CrossRef]
- Clemmer, J.S.; Hillegass, W.B.; Taylor, E.B. Antihypertensive effects of immunosuppressive therapy in autoimmune disease. J. Hum. Hypertens. 2022, 37, 300–306. [Google Scholar] [CrossRef]
- Virdis, A.; Tani, C.; Duranti, E.; Vagnani, S.; Carli, L.; Kühl, A.A.; Solini, A.; Baldini, C.; Talarico, R.; Bombardieri, S.; et al. Early treatment with hydroxychloroquine prevents the development of endothelial dysfunction in a murine model of systemic lupus erythematosus. Arthritis Res. Ther. 2015, 17, 277. [Google Scholar] [CrossRef]
- Miranda, S.; Billoir, P.; Damian, L.; Thiebaut, P.A.; Schapman, D.; Le Besnerais, M.; Jouen, F.; Galas, L.; Levesque, H.; Le Cam-Duchez, V.; et al. Hydroxychloroquine reverses the prothrombotic state in a mouse model of antiphospholipid syndrome: Role of reduced inflammation and endothelial dysfunction. PLoS ONE 2019, 14, e0212614. [Google Scholar] [CrossRef]
- Furumoto, Y.; Smith, C.K.; Blanco, L.; Zhao, W.; Brooks, S.R.; Thacker, S.G.; Zarzour, A.; Sciumè, G.; Tsai, W.L.; Trier, A.M.; et al. Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol. 2017, 69, 148–160. [Google Scholar] [CrossRef]
- Hasni, S.A.; Gupta, S.; Davis, M.; Poncio, E.; Temesgen-Oyelakin, Y.; Carlucci, P.M.; Wang, X.; Naqi, M.; Playford, M.P.; Goel, R.R.; et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat. Commun. 2021, 12, 3391. [Google Scholar] [CrossRef] [PubMed]
- Verden, A.; Dimbil, M.; Kyle, R.; Overstreet, B.; Hoffman, K.B. Analysis of Spontaneous Postmarket Case Reports Submitted to the FDA Regarding Thromboembolic Adverse Events and JAK Inhibitors. Drug Saf. 2018, 41, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Somers, E.C.; Brook, R.D.; Kehrer, C.; Pfenninger, D.; Lewis, E.; Chakrabarti, A.; Richardson, B.C.; Shelden, E.; McCune, W.J.; et al. Endothelial cell apoptosis in systemic lupus erythematosus: A common pathway for abnormal vascular function and thrombosis propensity. Blood 2004, 103, 3677–3683. [Google Scholar] [CrossRef] [PubMed]
- Nikpour, M.; Gladman, D.D.; Ibañez, D.; Bruce, I.N.; Burns, R.J.; Urowitz, M.B. Myocardial Perfusion Imaging in Assessing Risk of Coronary Events in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2009, 36, 288–294. [Google Scholar] [CrossRef][Green Version]
- Mak, A.; Kow, N.Y.; Schwarz, H.; Gong, L.; Tay, S.H.; Ling, L.H. Endothelial dysfunction in systemic lupus erythematosus—A case-control study and an updated meta-analysis and meta-regression. Sci. Rep. 2017, 7, 7320. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J.; McLemore, G.R. Hypertension and impaired vascular function in a female mouse model of systemic lupus erythematosus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R736–R742. [Google Scholar] [CrossRef]
- Romero, M.; Toral, M.; Robles-Vera, I.; Sánchez, M.; Jiménez, R.; O’Valle, F.; Rodriguez-Nogales, A.; Pérez-Vizcaino, F.; Gálvez, J.; Duarte, J. Activation of Peroxisome Proliferator Activator Receptor β/δ Improves Endothelial Dysfunction and Protects Kidney in Murine Lupus. Hypertension 2017, 69, 641–650. [Google Scholar] [CrossRef]
- Montoya, T.; Sánchez-Hidalgo, M.; Castejón, M.L.; Vazquéz-Román, M.V.; de Sotomayor, M.A.; Ortega-Vidal, J.; González, M.L.; Alarcón-de-la-Lastra, C. Oleocanthal supplemented diet improves renal damage and endothelial dysfunction in pristane-induced systemic lupus erythematosus in mice. Food Res. Int. 2023, 163, 112140. [Google Scholar] [CrossRef]
- Moleón, J.; González-Correa, C.; Robles-Vera, I.; Miñano, S.; de la Visitación, N.; Barranco, A.M.; Martín-Morales, N.; O’Valle, F.; Mayo-Martínez, L.; García, A.; et al. Targeting the gut microbiota with dietary fibers: A novel approach to prevent the development cardiovascular complications linked to systemic lupus erythematosus in a preclinical study. Gut Microbes 2023, 15, 2247053. [Google Scholar] [CrossRef]
- Nguyen, H.; Chiasson, V.L.; Chatterjee, P.; Kopriva, S.E.; Young, K.J.; Mitchell, B.M. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc. Res. 2013, 97, 696–704. [Google Scholar] [CrossRef]
- Pietrowski, E.; Bender, B.; Huppert, J.; White, R.; Luhmann, H.J.; Kuhlmann, C.R.W. Pro-Inflammatory Effects of Interleukin-17A on Vascular Smooth Muscle Cells Involve NAD(P)H- Oxidase Derived Reactive Oxygen Species. J. Vasc. Res. 2011, 48, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Kassan, M.; Galan, M.; Partyka, M.; Trebak, M.; Matrougui, K. Interleukin-10 Released by CD4 + CD25 + Natural Regulatory T Cells Improves Microvascular Endothelial Function Through Inhibition of NADPH Oxidase Activity in Hypertensive Mice. Arter. Thromb. Vasc. Biol. 2011, 31, 2534–2542. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-F.; Liu, J.T.; Wang, Y.; Xu, A.; Vanhoutte, P.M. Toll-Like Receptor 4 Mutation Protects Obese Mice Against Endothelial Dysfunction by Decreasing NADPH Oxidase Isoforms 1 and 4. Arter. Thromb. Vasc. Biol. 2013, 33, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Toral, M.; Jiménez, R.; Romero, M.; Robles-Vera, I.; Sánchez, M.; Salaices, M.; Sabio, J.M.; Duarte, J. Role of endoplasmic reticulum stress in the protective effects of PPARβ/δ activation on endothelial dysfunction induced by plasma from patients with lupus. Arthritis Res. Ther. 2017, 19, 268. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arter. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef]
- Lee, P.Y.; Li, Y.; Richards, H.B.; Chan, F.S.; Zhuang, H.; Narain, S.; Butfiloski, E.J.; Sobel, E.S.; Reeves, W.H.; Segal, M.S. Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 3759–3769. [Google Scholar] [CrossRef]
- Thacker, S.; Duquaine, D.; Park, J.; Kaplan, M. Lupus-prone New Zealand Black/New Zealand White F1 mice display endothelial dysfunction and abnormal phenotype and function of endothelial progenitor cells. Lupus 2010, 19, 288–299. [Google Scholar] [CrossRef]
- Geng, L.; Wang, S.; Li, X.; Wang, D.; Chen, H.; Chen, J.; Sun, Y.; Chen, W.; Yao, G.; Gao, X.; et al. Association between Type I interferon and depletion and dysfunction of endothelial progenitor cells in C57BL/6 mice deficient in both apolipoprotein E and Fas ligand. Curr. Res. Transl. Med. 2018, 66, 71–82. [Google Scholar] [CrossRef]
- Buie, J.J.; Renaud, L.L.; Muise-Helmericks, R.; Oates, J.C. IFN-α Negatively Regulates the Expression of Endothelial Nitric Oxide Synthase and Nitric Oxide Production: Implications for Systemic Lupus Erythematosus. J. Immunol. 2017, 199, 1979–1988. [Google Scholar] [CrossRef]
- Sacre, K.; Escoubet, B.; Pasquet, B.; Chauveheid, M.-P.; Zennaro, M.-C.; Tubach, F.; Papo, T. Increased Arterial Stiffness in Systemic Lupus Erythematosus (SLE) Patients at Low Risk for Cardiovascular Disease: A Cross-Sectional Controlled Study. PLoS ONE 2014, 9, e94511. [Google Scholar] [CrossRef]
- Shang, Q.; Tam, L.; Li, E.; Yip, G.; Yu, C. Increased arterial stiffness correlated with disease activity in systemic lupus erythematosus. Lupus 2008, 17, 1096–1102. [Google Scholar] [CrossRef]
- Diószegi, Á.; Lőrincz, H.; Kaáli, E.; Soltész, P.; Perge, B.; Varga, É.; Harangi, M.; Tarr, T. Role of Altered Metabolism of Triglyceride-Rich Lipoprotein Particles in the Development of Vascular Dysfunction in Systemic Lupus Erythematosus. Biomolecules 2023, 13, 401. [Google Scholar] [CrossRef]
- Rodriguez-Iturbe, B.; Pons, H.; Johnson, R.J. Role of the Immune System in Hypertension. Physiol. Rev. 2017, 97, 1127–1164. [Google Scholar] [CrossRef]
- Khraibi, A.A.; Norman, R.A.; Dzielak, D.J. Chronic immunosuppression attenuates hypertension in Okamoto spontaneously hypertensive rats. Am. J. Physiol.-Heart Circ. Physiol. 1984, 247, H722–H726. [Google Scholar] [CrossRef] [PubMed]
- Robles-Vera, I.; de la Visitación, N.; Sánchez, M.; Gómez-Guzmán, M.; Jiménez, R.; Moleón, J.; González-Correa, C.; Romero, M.; Yang, T.; Raizada, M.K.; et al. Mycophenolate Improves Brain–Gut Axis Inducing Remodeling of Gut Microbiota in DOCA-Salt Hypertensive Rats. Antioxidants 2020, 9, 1199. [Google Scholar] [CrossRef] [PubMed]
- Robles-Vera, I.; de la Visitación, N.; Toral, M.; Sánchez, M.; Gómez-Guzmán, M.; Jiménez, R.; Romero, M.; Duarte, J. Mycophenolate mediated remodeling of gut microbiota and improvement of gut-brain axis in spontaneously hypertensive rats. Biomed. Pharmacother. 2021, 135, 111189. [Google Scholar] [CrossRef] [PubMed]
- Tipton, A.J.; Baban, B.; Sullivan, J.C. Female spontaneously hypertensive rats have greater renal anti-inflammatory T lymphocyte infiltration than males, American Journal of Physiology-Regulatory. Integr. Comp. Physiol. 2012, 303, R359–R367. [Google Scholar] [CrossRef] [PubMed]
- Mathis, K.W.; Wallace, K.; Flynn, E.R.; Maric-Bilkan, C.; LaMarca, B.; Ryan, M.J. Preventing Autoimmunity Protects Against the Development of Hypertension and Renal Injury. Hypertension 2014, 64, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.B.; Ryan, M.J. Immunosuppression With Mycophenolate Mofetil Attenuates Hypertension in an Experimental Model of Autoimmune Disease. J. Am. Heart Assoc. 2017, 6, e005394. [Google Scholar] [CrossRef]
- Kamal, A.; Khamashta, M. The efficacy of novel B cell biologics as the future of SLE treatment: A review. Autoimmun. Rev. 2014, 13, 1094–1101. [Google Scholar] [CrossRef]
- Taylor, E.B.; Barati, M.T.; Powell, D.W.; Turbeville, H.R.; Ryan, M.J. Plasma Cell Depletion Attenuates Hypertension in an Experimental Model of Autoimmune Disease. Hypertension 2018, 71, 719–728. [Google Scholar] [CrossRef]
- Liu, Z.; Bethunaickan, R.; Huang, W.; Lodhi, U.; Solano, I.; Madaio, M.P.; Davidson, A. Interferon-α accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum. 2011, 63, 219–229. [Google Scholar] [CrossRef]
- Jacob, J.; Kelsoe, G.; Rajewsky, K.; Weiss, U. Intraclonal generation of antibody mutants in germinal centres. Nature 1991, 354, 389–392. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, I.C.M.; Toellner, K.-M.; Cunningham, A.F.; Serre, K.; Sze, D.M.-Y.; Zúñiga, E.; Cook, M.C.; Vinuesa, C.G. Extrafollicular antibody responses. Immunol. Rev. 2003, 194, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, C.G.; Cook, M.C.; Angelucci, C.; Athanasopoulos, V.; Rui, L.; Hill, K.M.; Yu, D.; Domaschenz, H.; Whittle, B.; Lambe, T.; et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 2005, 435, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Woolford, C.A.; Drill, E.; Lu, M.; Jones, E.W. Pbn1p: An essential endoplasmic reticulum membrane protein required for protein processing in the endoplasmic reticulum of budding yeast. Proc. Natl. Acad. Sci. USA 2006, 103, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Fields, M.L.; Buckler, J.L.; Reed, A.J.; Mandik-Nayak, L.; Nish, S.A.; Noelle, R.J.; Turka, L.A.; Finkelman, F.D.; Caton, A.J.; et al. The Impact of T Helper and T Regulatory Cells on the Regulation of Anti-Double-Stranded DNA B Cells. Immunity 2002, 16, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Hong, Y.-K.; Yun, H.-J.; Kim, Y.-M.; Kim, J.-R.; Yoo, W.-H. Altered frequency and migration capacity of CD4+CD25+ regulatory T cells in systemic lupus erythematosus. Rheumatology 2008, 47, 789–794. [Google Scholar] [CrossRef]
- Hsu, H.-C.; Yang, P.; Wang, J.; Wu, Q.; Myers, R.; Chen, J.; Yi, J.; Guentert, T.; Tousson, A.; Stanus, A.L.; et al. Interleukin 17–producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat. Immunol. 2008, 9, 166–175. [Google Scholar] [CrossRef]
- Taylor, E.B.; Sasser, J.M.; Maeda, K.J.; Ryan, M.J. Expansion of regulatory T cells using low-dose interleukin-2 attenuates hypertension in an experimental model of systemic lupus erythematosus. Am. J. Physiol.-Ren. Physiol. 2019, 317, F1274–F1284. [Google Scholar] [CrossRef]
- Mathis, K.W.; Taylor, E.B.; Ryan, M.J. Anti-CD3 antibody therapy attenuates the progression of hypertension in female mice with systemic lupus erythematosus. Pharmacol. Res. 2017, 120, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-C.; Xu, Z.; Li, W.; Yang, H.; Roopenian, D.C.; Morse, H.C.; Morel, L. Relative Contributions of B Cells and Dendritic Cells from Lupus-Prone Mice to CD4+ T Cell Polarization. J. Immunol. 2018, 200, 3087–3099. [Google Scholar] [CrossRef] [PubMed]
- Patrick, D.M.; de la Visitación, N.; Krishnan, J.; Chen, W.; Ormseth, M.J.; Stein, C.M.; Davies, S.S.; Amarnath, V.; Crofford, L.J.; Williams, J.M.; et al. Isolevuglandins disrupt PU.1-mediated C1q expression and promote autoimmunity and hypertension in systemic lupus erythematosus. JCI Insight 2022, 7, e136678. [Google Scholar] [CrossRef]
- Knight, J.S.; Zhao, W.; Luo, W.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Hodgin, J.B.; Eitzman, D.T.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Investig. 2013, 123, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef]
- Zegarra-Ruiz, D.F.; El Beidaq, A.; Iñiguez, A.J.; Di Ricco, M.L.; Vieira, S.M.; Ruff, W.E.; Mubiru, D.; Fine, R.L.; Sterpka, J.; Greiling, T.M.; et al. A Diet-Sensitive Commensal Lactobacillus Strain Mediates TLR7-Dependent Systemic Autoimmunity. Cell Host Microbe 2019, 25, 113–127.e6. [Google Scholar] [CrossRef]
- Mu, Q.; Zhang, H.; Liao, X.; Lin, K.; Liu, H.; Edwards, M.R.; Ahmed, S.A.; Yuan, R.; Li, L.; Cecere, T.E.; et al. Control of lupus nephritis by changes of gut microbiota. Microbiome 2017, 5, 73. [Google Scholar] [CrossRef]
- Vieira, S.M.; Hiltensperger, M.; Kumar, V.; Zegarra-Ruiz, D.; Dehner, C.; Khan, N.; Costa, F.R.C.; Tiniakou, E.; Greiling, T.; Ruff, W.; et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science (1979) 2018, 359, 1156–1161. [Google Scholar] [CrossRef]
- Ma, Y.; Xu, X.; Li, M.; Cai, J.; Wei, Q.; Niu, H. Gut microbiota promote the inflammatory response in the pathogenesis of systemic lupus erythematosus. Mol. Med. 2019, 25, 35. [Google Scholar] [CrossRef]
- Luo, X.M.; Edwards, M.R.; Mu, Q.; Yu, Y.; Vieson, M.D.; Reilly, C.M.; Ahmed, S.A.; Bankole, A.A. Gut Microbiota in Human Systemic Lupus Erythematosus and a Mouse Model of Lupus. Appl. Environ. Microbiol. 2018, 84, e02288-17. [Google Scholar] [CrossRef]
- Hevia, A.; Milani, C.; López, P.; Cuervo, A.; Arboleya, S.; Duranti, S.; Turroni, F.; González, S.; Suárez, A.; Gueimonde, M.; et al. Intestinal Dysbiosis Associated with Systemic Lupus Erythematosus. MBio 2014, 5, e01548-14. [Google Scholar] [CrossRef]
- de la Visitación, N.; Robles-Vera, I.; Toral, M.; Gómez-Guzmán, M.; Sánchez, M.; Moleón, J.; González-Correa, C.; Martín-Morales, N.; O’Valle, F.; Jiménez, R.; et al. Gut microbiota contributes to the development of hypertension in a genetic mouse model of systemic lupus erythematosus. Br. J. Pharmacol. 2021, 178, 3708–3729. [Google Scholar] [CrossRef]
- de la Visitación, N.; Robles-Vera, I.; Moleón, J.; González-Correa, C.; Aguilera-Sánchez, N.; Toral, M.; Gómez-Guzmán, M.; Sánchez, M.; Jiménez, R.; Martin-Morales, N.; et al. Gut Microbiota Has a Crucial Role in the Development of Hypertension and Vascular Dysfunction in Toll-like Receptor 7-Driven Lupus Autoimmunity. Antioxidants 2021, 10, 1426. [Google Scholar] [CrossRef]
- de la Visitación, N.; Robles-Vera, I.; Moleón-Moya, J.; Sánchez, M.; Jiménez, R.; Gómez-Guzmán, M.; González-Correa, C.; Olivares, M.; Toral, M.; Romero, M.; et al. Probiotics Prevent Hypertension in a Murine Model of Systemic Lupus Erythematosus Induced by Toll-Like Receptor 7 Activation. Nutrients 2021, 13, 2669. [Google Scholar] [CrossRef]
- de la Visitación, N.; Robles-Vera, I.; Toral, M.; Duarte, J. Protective Effects of Probiotic Consumption in Cardiovascular Disease in Systemic Lupus Erythematosus. Nutrients 2019, 11, 2676. [Google Scholar] [CrossRef]
- Toral, M.; Robles-Vera, I.; Romero, M.; Visitación, N.; Sánchez, M.; O’Valle, F.; Rodriguez-Nogales, A.; Gálvez, J.; Duarte, J.; Jiménez, R. Lactobacillus fermentum CECT5716: A novel alternative for the prevention of vascular disorders in a mouse model of systemic lupus erythematosus. FASEB J. 2019, 33, 10005–10018. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.L.; Odegard, J.M.; Bouzahzah, F.; Choi, J.-Y.; Eardley, L.D.; Zielinski, C.E.; Craft, J.E. Intrinsic T Cell Defects in Systemic Autoimmunity. Ann. N. Y. Acad. Sci. 2003, 987, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Apostolidis, S.A.; Lieberman, L.A.; Kis-Toth, K.; Crispín, J.C.; Tsokos, G.C. The Dysregulation of Cytokine Networks in Systemic Lupus Erythematosus. J. Interferon Cytokine Res. 2011, 31, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Crispín, J.C.; Kyttaris, V.C.; Juang, Y.-T.; Tsokos, G.C. How signaling and gene transcription aberrations dictate the systemic lupus erythematosus T cell phenotype. Trends Immunol. 2008, 29, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Herman, C.E.; MacIver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Alaynick, W.A.; Kondo, R.P.; Xie, W.; He, W.; Dufour, C.R.; Downes, M.; Jonker, J.W.; Giles, W.; Naviaux, R.K.; Giguère, V.; et al. ERRγ Directs and Maintains the Transition to Oxidative Metabolism in the Postnatal Heart. Cell Metab. 2007, 6, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and mTORC1 Kinase Activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef]
- Kono, M.; Yoshida, N.; Maeda, K.; Suárez-Fueyo, A.; Kyttaris, V.C.; Tsokos, G.C. Glutaminase 1 Inhibition Reduces Glycolysis and Ameliorates Lupus-like Disease in MRL/lpr Mice and Experimental Autoimmune Encephalomyelitis. Arthritis Rheumatol. 2019, 71, 1869–1878. [Google Scholar] [CrossRef] [PubMed]
- Sumikawa, M.H.; Iwata, S.; Zhang, M.; Miyata, H.; Ueno, M.; Todoroki, Y.; Nagayasu, A.; Kanda, R.; Sonomoto, K.; Torimoto, K.; et al. An enhanced mitochondrial function through glutamine metabolism in plasmablast differentiation in systemic lupus erythematosus. Rheumatology 2022, 61, 3049–3059. [Google Scholar] [CrossRef] [PubMed]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef]
- Bettencourt, I.A.; Powell, J.D. Targeting Metabolism as a Novel Therapeutic Approach to Autoimmunity, Inflammation, and Transplantation. J. Immunol. 2017, 198, 999–1005. [Google Scholar] [CrossRef]
- Morel, L. Immunometabolism in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2017, 13, 280–290. [Google Scholar] [CrossRef]
- Dufort, F.J.; Bleiman, B.F.; Gumina, M.R.; Blair, D.; Wagner, D.J.; Roberts, M.F.; Abu-Amer, Y.; Chiles, T.C. Cutting Edge: IL-4-Mediated Protection of Primary B Lymphocytes from Apoptosis via Stat6-Dependent Regulation of Glycolytic Metabolism. J. Immunol. 2007, 179, 4953–4957. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Huang, S.C.-C.; Smith, A.M.; Chang, C.-H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.W.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Del Prete, A.; Zaccagnino, P.; Di Paola, M.; Saltarella, M.; Celis, C.O.; Nico, B.; Santoro, G.; Lorusso, M. Role of mitochondria and reactive oxygen species in dendritic cell differentiation and functions. Free Radic. Biol. Med. 2008, 44, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.A.; Baker, H.V.; Brusko, T.M.; Sobel, E.S.; Morel, L. Metformin Inhibits the Type 1 IFN Response in Human CD4+ T Cells. J. Immunol. 2019, 203, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Choi, S.-C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of CD4+ T cell metabolism reverses lupus. Sci. Transl. Med. 2015, 7, 274ra18. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Choi, S.-C.; Xu, Z.; Zeumer, L.; Kanda, N.; Croker, B.P.; Morel, L. Glucose Oxidation Is Critical for CD4+ T Cell Activation in a Mouse Model of Systemic Lupus Erythematosus. J. Immunol. 2016, 196, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Moon, S.-J.; Kim, E.-K.; Seo, H.-B.; Yang, E.-J.; Son, H.-J.; Kim, J.-K.; Min, J.-K.; Park, S.-H.; Cho, M.-L. Metformin Suppresses Systemic Autoimmunity in Roquinsan/san Mice through Inhibiting B Cell Differentiation into Plasma Cells via Regulation of AMPK/mTOR/STAT3. J. Immunol. 2017, 198, 2661–2670. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.G.; Lee, J.; Hong, S.-M.; Kwok, S.-K.; Cho, M.-L.; Park, S.-H. Metformin enhances the immunomodulatory potential of adipose-derived mesenchymal stem cells through STAT1 in an animal model of lupus. Rheumatology 2020, 59, 1426–1438. [Google Scholar] [CrossRef]
- Chen, X.; Wu, D.; Wu, H.; Li, H.; Yang, C.; Su, H.; Liu, Z.; Huang, X.; Lu, X.; Huang, L.; et al. Metformin improves renal injury of MRL/lpr lupus-prone mice via the AMPK/STAT3 pathway. Lupus Sci. Med. 2022, 9, e000611. [Google Scholar] [CrossRef]
- Elshikha, A.S.; Ge, Y.; Brown, J.; Kanda, N.; Zadeh, M.; Abboud, G.; Choi, S.-C.; Silverman, G.; Garrett, T.J.; Clapp, W.L.; et al. Pharmacologic inhibition of glycolysis prevents the development of lupus by altering the gut microbiome in mice. IScience 2023, 26, 107122. [Google Scholar] [CrossRef]
- Sun, F.; Geng, S.; Wang, H.; Wang, H.; Liu, Z.; Wang, X.; Li, T.; Wan, W.; Lu, L.; Teng, X.; et al. Effects of metformin on disease flares in patients with systemic lupus erythematosus: Post hoc analyses from two randomised trials. Lupus Sci. Med. 2020, 7, e000429. [Google Scholar] [CrossRef]
- Bona, N.; Pezzarini, E.; Balbi, B.; Daniele, S.M.; Rossi, M.F.; Monje, A.L.; Basiglio, C.; Pelusa, H.F.; Arriaga, S.M.M. Oxidative stress, inflammation and disease activity biomarkers in lupus nephropathy. Lupus 2020, 29, 311–323. [Google Scholar] [CrossRef]
- Perl, A. mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann. N. Y. Acad. Sci. 2015, 1346, 33–44. [Google Scholar] [CrossRef] [PubMed]
- García-González, M.; Gómez-Bernal, F.; Quevedo-Abeledo, J.C.; Fernández-Cladera, Y.; González-Rivero, A.F.; López-Mejías, R.; Díaz-González, F.; González-Gay, M.Á.; Ferraz-Amaro, I. The complement system is linked to insulin resistance in patients with systemic lupus erythematosus. Clin Exp Rheumatol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Hallajzadeh, J.; Khoramdad, M.; Izadi, N.; Karamzad, N.; Almasi-Hashiani, A.; Ayubi, E.; Qorbani, M.; Pakzad, R.; Sullman, M.J.M.; Safiri, S. The association between metabolic syndrome and its components with systemic lupus erythematosus: A comprehensive systematic review and meta-analysis of observational studies. Lupus 2018, 27, 899–912. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, D.; Vincent, F.; Hoi, A.; Morand, E. Associations of metabolic syndrome in SLE. Lupus Sci. Med. 2020, 7, e000436. [Google Scholar] [CrossRef] [PubMed]
- Urbain, F.; Ponnaiah, M.; Ichou, F.; Lhomme, M.; Materne, C.; Galier, S.; Haroche, J.; Frisdal, E.; Mathian, A.; Durand, H.; et al. Impaired metabolism predicts coronary artery calcification in women with systemic lupus erythematosus. EBioMedicine 2023, 96, 104802. [Google Scholar] [CrossRef]
- Warner, L.M.; Adams, L.M.; Sehgal, S.N. Rapamycin Prolongs Survival and Arrests Pathophysiologic Changes in Murine Systemic Lupus Erythematosus. Arthritis Rheum. 1994, 37, 289–297. [Google Scholar] [CrossRef]
- Fernandez, D.; Bonilla, E.; Mirza, N.; Niland, B.; Perl, A. Rapamycin reduces disease activity and normalizes T cell activation–induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2983–2988. [Google Scholar] [CrossRef]
- Suwannaroj, S.; Lagoo, A.; Keisler, D.; McMurray, R.W. Antioxidants suppress mortality in the female NZB × NZW F1 mouse model of systemic lupus erythematosus (SLE). Lupus 2001, 10, 258–265. [Google Scholar] [CrossRef]
- Lai, Z.-W.; Hanczko, R.; Bonilla, E.; Caza, T.N.; Clair, B.; Bartos, A.; Miklossy, G.; Jimah, J.; Doherty, E.; Tily, H.; et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012, 64, 2937–2946. [Google Scholar] [CrossRef]
- McDonald, G.; Deepak, S.; Miguel, L.; Hall, C.J.; Isenberg, D.A.; Magee, A.I.; Butters, T.; Jury, E.C. Normalizing glycosphingolipids restores function in CD4+ T cells from lupus patients. J. Clin. Investig. 2014, 124, 712–724. [Google Scholar] [CrossRef]
- Telang, S.; Clem, B.F.; Klarer, A.C.; Clem, A.L.; Trent, J.O.; Bucala, R.; Chesney, J. Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. J. Transl. Med. 2012, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, P.; de Vries, M.R.; Peeters, B.; Guns, P.-J.; De Meyer, G.R.Y.; Quax, P.H.A.; Martinet, W. PFKFB3 gene deletion in endothelial cells inhibits intraplaque angiogenesis and lesion formation in a murine model of venous bypass grafting. Angiogenesis 2022, 25, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Z.-S.; Tang, S.-Q.; Xing, T.; Zhou, Y.; Lv, M.; Fu, H.-X.; Wang, Y.; Xu, L.-P.; Zhang, X.-H.; Lee, H.-Y.; et al. The glycolytic enzyme PFKFB3 determines bone marrow endothelial progenitor cell damage after chemotherapy and irradiation. Haematologica 2022, 107, 2365–2380. [Google Scholar] [CrossRef] [PubMed]
- Abbasifard, M.; Khorramdelazad, H.; Rostamian, A.; Rezaian, M.; Askari, P.S.; Sharifi, G.T.K.; Parizi, M.K.; Sharifi, M.T.K.; Najafizadeh, S.R. Effects of N-acetylcysteine on systemic lupus erythematosus disease activity and its associated complications: A randomized double-blind clinical trial study. Trials 2023, 24, 129. [Google Scholar] [CrossRef]
- Blanco, L.P.; Pedersen, H.L.; Wang, X.; Lightfoot, Y.L.; Seto, N.; Carmona-Rivera, C.; Yu, Z.; Hoffmann, V.; Yuen, P.S.T.; Kaplan, M.J. Improved Mitochondrial Metabolism and Reduced Inflammation Following Attenuation of Murine Lupus with Coenzyme Q10 Analog Idebenone. Arthritis Rheumatol. 2020, 72, 454–464. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Key Points |
|---|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miñano, S.; González-Correa, C.; Moleón, J.; Duarte, J. Metabolic Modulators in Cardiovascular Complications of Systemic Lupus Erythematosus. Biomedicines 2023, 11, 3142. https://doi.org/10.3390/biomedicines11123142
Miñano S, González-Correa C, Moleón J, Duarte J. Metabolic Modulators in Cardiovascular Complications of Systemic Lupus Erythematosus. Biomedicines. 2023; 11(12):3142. https://doi.org/10.3390/biomedicines11123142
Chicago/Turabian StyleMiñano, Sofía, Cristina González-Correa, Javier Moleón, and Juan Duarte. 2023. "Metabolic Modulators in Cardiovascular Complications of Systemic Lupus Erythematosus" Biomedicines 11, no. 12: 3142. https://doi.org/10.3390/biomedicines11123142
APA StyleMiñano, S., González-Correa, C., Moleón, J., & Duarte, J. (2023). Metabolic Modulators in Cardiovascular Complications of Systemic Lupus Erythematosus. Biomedicines, 11(12), 3142. https://doi.org/10.3390/biomedicines11123142