

BZD9L1 Differentially Regulates Sirtuins in Liver-Derived Cells by Inducing Reactive Oxygen Species


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Treatment
2.3. Cell Viability Assay
2.4. ROS Assay
2.5. Mito Stress Test Assay
2.6. Western Blot
2.7. Statistical Analyses
3. Results
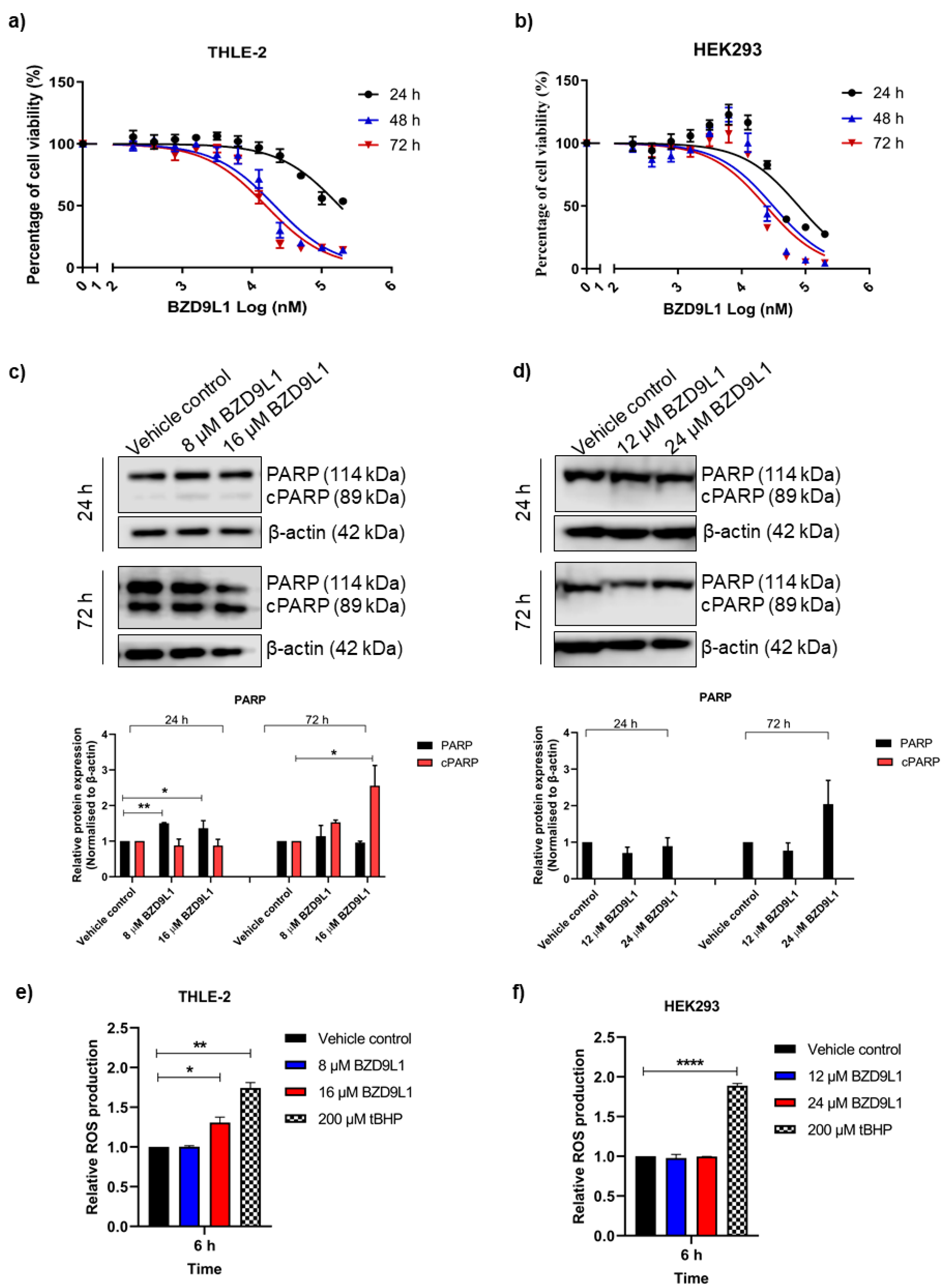
3.1. BZD9L1 Reduced THLE-2 and HEK293 Cell Viability but Induced ROS Only in THLE-2 Cells
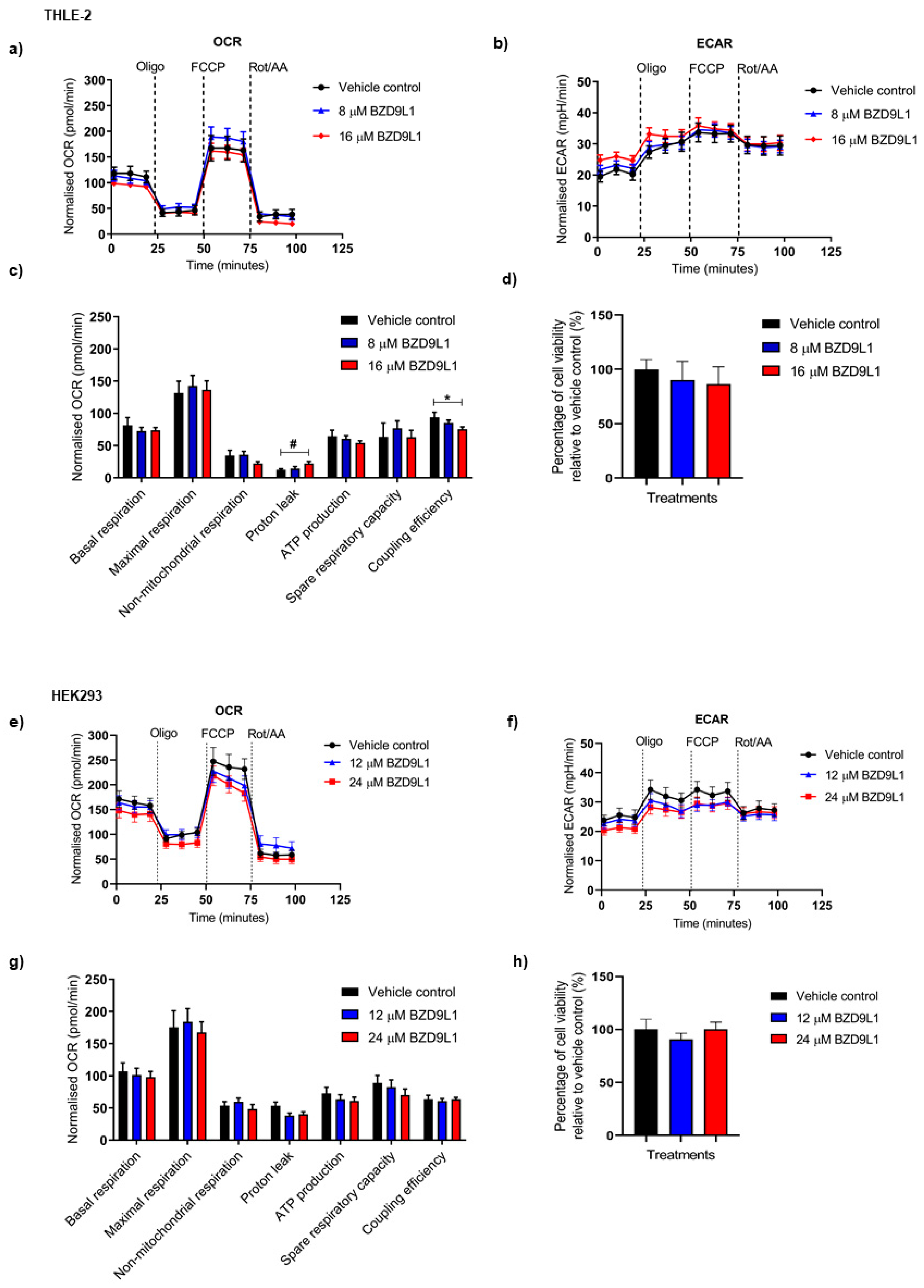
3.2. Effects of BZD9L1 on the Bioenergetics Profile of THLE-2 and HEK293 Cells
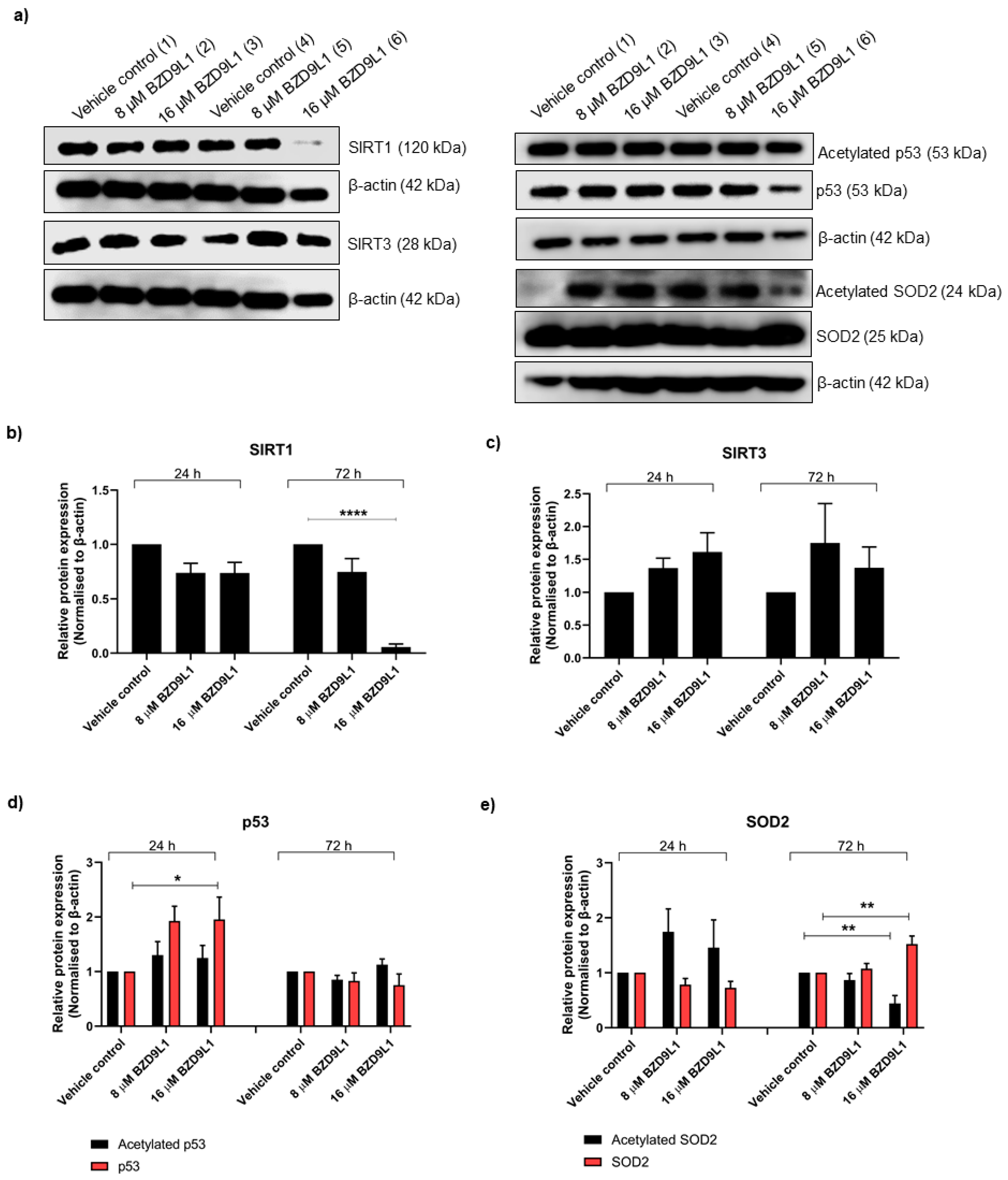
3.3. Effects of BZD9L1 on the Regulation of Sirtuins in THLE-2 and HEK293 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mayr, J.A. Lipid metabolism in mitochondrial membranes. J. Inherit. Metab. Dis. 2015, 38, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Piel, R.B., 3rd; Dailey, H.A., Jr.; Medlock, A.E. The mitochondrial heme metabolon: Insights into the complex(ity) of heme synthesis and distribution. Mol. Genet. Metab. 2019, 128, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Roca-Agujetas, V.; de Dios, C.; Lestón, L.; Marí, M.; Morales, A.; Colell, A. Recent Insights into the Mitochondrial Role in Autophagy and Its Regulation by Oxidative Stress. Oxid. Med. Cell. Longev. 2019, 2019, 3809308. [Google Scholar] [CrossRef] [PubMed]
- Will, Y.; Shields, J.E.; Wallace, K.B. Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions. Biology 2019, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, C.M.; Duarte, F.V.; Teodoro, J.S.; Varela, A.T.; Rolo, A.P. Chapter 51—Biomarkers of mitochondrial dysfunction and toxicity. In Biomarkers in Toxicology; Gupta, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 847–861. [Google Scholar]
- Carafa, V.; Russo, R.; Della Torre, L.; Cuomo, F.; Dell’Aversana, C.; Sarno, F.; Sgueglia, G.; Di Donato, M.; Rotili, D.; Mai, A.; et al. The Pan-Sirtuin Inhibitor MC2494 Regulates Mitochondrial Function in a Leukemia Cell Line. Front. Oncol. 2020, 10, 820. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Lin, K.H.; Huang, C.J.; Wei, A.C. MitoTox: A comprehensive mitochondrial toxicity database. BMC Bioinform. 2021, 22, 369. [Google Scholar] [CrossRef]
- Dykens, J.A.; Will, Y. Mitochondrial Toxicity. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 349–353. [Google Scholar]
- Wang, R.; Novick, S.J.; Mangum, J.B.; Queen, K.; Ferrick, D.A.; Rogers, G.W.; Stimmel, J.B. The acute extracellular flux (XF) assay to assess compound effects on mitochondrial function. J. Biomol. Screen. 2015, 20, 422–429. [Google Scholar] [CrossRef]
- Yépez, V.A.; Kremer, L.S.; Iuso, A.; Gusic, M.; Kopajtich, R.; Koňaříková, E.; Nadel, A.; Wachutka, L.; Prokisch, H.; Gagneur, J. OCR-Stats: Robust estimation and statistical testing of mitochondrial respiration activities using Seahorse XF Analyzer. PLoS ONE 2018, 13, e0199938. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Jaber, S.M.; Yadava, N.; Polster, B.M. Mapping mitochondrial respiratory chain deficiencies by respirometry: Beyond the Mito Stress Test. Exp. Neurol. 2020, 328, 113282. [Google Scholar] [CrossRef]
- Alhazzazi, T.Y.; Kamarajan, P.; Joo, N.; Huang, J.-Y.; Verdin, E.; D’Silva, N.J.; Kapila, Y.L. Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer 2011, 117, 1670–1678. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Mok, P.Y.; Subramaniam, A.V.; Oon, C.E. Chapter 9—The bifunctional roles of sirtuins and their therapeutic potential in cancer. In Sirtuin Biology in Cancer and Metabolic Disease; Maiese, K., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 153–177. [Google Scholar]
- Huang, J.Y.; Hirschey, M.D.; Shimazu, T.; Ho, L.; Verdin, E. Mitochondrial sirtuins. Biochim. Biophys. Acta 2010, 1804, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.J.; Lee, Y.T.; Yeong, K.Y.; Petersen, S.H.; Kono, K.; Tan, S.C.; Oon, C.E. Anticancer activities of a benzimidazole compound through sirtuin inhibition in colorectal cancer. Future Med. Chem. 2018, 10, 2039–2057. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.J.; Lee, Y.T.; Petersen, S.H.; Kaur, G.; Kono, K.; Tan, S.C.; Majid, A.M.S.A.; Oon, C.E. BZD9L1 sirtuin inhibitor as a potential adjuvant for sensitization of colorectal cancer cells to 5-fluorouracil. Ther. Adv. Med. Oncol. 2019, 11, 1758835919878977. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.J.; Lee, Y.T.; Mancera, R.L.; Oon, C.E. BZD9L1 sirtuin inhibitor: Identification of key molecular targets and their biological functions in HCT116 colorectal cancer cells. Life Sci. 2021, 284, 119747. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Mok, P.Y.; Kaur, G.; Sreenivasan, S.; Falasca, M.; Oon, C.E. Sex-divergent expression of cytochrome P450 and SIRTUIN 1-7 proteins in toxicity evaluation of a benzimidazole-derived epigenetic modulator in mice. Toxicol. Appl. Pharmacol. 2022, 445, 116039. [Google Scholar] [CrossRef]
- Zhong, L.; Mostoslavsky, R. Fine tuning our cellular factories: Sirtuins in mitochondrial biology. Cell Metab. 2011, 13, 621–626. [Google Scholar] [CrossRef]
- Fanibunda, S.E.; Deb, S.; Maniyadath, B.; Tiwari, P.; Ghai, U.; Gupta, S.; Figueiredo, D.; Weisstaub, N.; Gingrich, J.A.; Vaidya, A.D.B.; et al. Serotonin regulates mitochondrial biogenesis and function in rodent cortical neurons via the 5-HT2A receptor and SIRT1—PGC-1α axis. Proc. Natl. Acad. Sci. USA 2019, 116, 11028–11037. [Google Scholar] [CrossRef]
- Kwon, S.J.; Lee, D.W.; Shah, D.A.; Ku, B.; Jeon, S.Y.; Solanki, K.; Ryan, J.D.; Clark, D.S.; Dordick, J.S.; Lee, M.-Y. High-throughput and combinatorial gene expression on a chip for metabolism-induced toxicology screening. Nat. Commun. 2014, 5, 3739. [Google Scholar] [CrossRef]
- Capes-Davis, A.; Bairoch, A.; Barrett, T.; Burnett, E.C.; Dirks, W.G.; Hall, E.M.; Healy, L.; Kniss, D.A.; Korch, C.; Liu, Y.; et al. Cell Lines as Biological Models: Practical Steps for More Reliable Research. Chem. Res. Toxicol. 2019, 32, 1733–1736. [Google Scholar] [CrossRef]
- Guo, L.; Dial, S.; Shi, L.; Branham, W.; Liu, J.; Fang, J.L.; Green, B.; Deng, H.; Kaput, J.; Ning, B. Similarities and differences in the expression of drug-metabolizing enzymes between human hepatic cell lines and primary human hepatocytes. Drug Metab. Dispos. 2011, 39, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Little, A.C.; Kovalenko, I.; Goo, L.E.; Hong, H.S.; Kerk, S.A.; Yates, J.A.; Purohit, V.; Lombard, D.B.; Merajver, S.D.; Lyssiotis, C.A. High-content fluorescence imaging with the metabolic flux assay reveals insights into mitochondrial properties and functions. Commun. Biol. 2020, 3, 271. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Gu, W. SIRT1: Regulator of p53 Deacetylation. Genes Cancer 2013, 4, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Eakins, J.; Bauch, C.; Woodhouse, H.; Park, B.; Bevan, S.; Dilworth, C.; Walker, P. A combined in vitro approach to improve the prediction of mitochondrial toxicants. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2016, 34, 161–170. [Google Scholar] [CrossRef]
- Rana, P.; Aleo, M.D.; Gosink, M.; Will, Y. Evaluation of in Vitro Mitochondrial Toxicity Assays and Physicochemical Properties for Prediction of Organ Toxicity Using 228 Pharmaceutical Drugs. Chem. Res. Toxicol. 2019, 32, 156–167. [Google Scholar] [CrossRef]
- Cortassa, S.; O’Rourke, B.; Aon, M.A. Redox-Optimized ROS Balance and the relationship between mitochondrial respiration and ROS. Biochim. Et Biophys. Acta (BBA)—Bioenerg. 2014, 1837, 287–295. [Google Scholar] [CrossRef]
- Nanayakkara, G.K.; Wang, H.; Yang, X. Proton leak regulates mitochondrial reactive oxygen species generation in endothelial cell activation and inflammation—A novel concept. Arch. Biochem. Biophys. 2019, 662, 68–74. [Google Scholar] [CrossRef]
- Dranka, B.P.; Benavides, G.A.; Diers, A.R.; Giordano, S.; Zelickson, B.R.; Reily, C.; Zou, L.; Chatham, J.C.; Hill, B.G.; Zhang, J.; et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free. Radic. Biol. Med. 2011, 51, 1621–1635. [Google Scholar] [CrossRef]
- Gnaiger, E.; Boushel, R.; Søndergaard, H.; Munch-Andersen, T.; Damsgaard, R.; Hagen, C.; Díez-Sánchez, C.; Ara, I.; Wright-Paradis, C.; Schrauwen, P.; et al. Mitochondrial coupling and capacity of oxidative phosphorylation in skeletal muscle of Inuit and Caucasians in the arctic winter. Scand. J. Med. Sci. Sports 2015, 25 (Suppl. 4), 126–134. [Google Scholar] [CrossRef]
- Cheng, J.; Nanayakkara, G.; Shao, Y.; Cueto, R.; Wang, L.; Yang, W.Y.; Tian, Y.; Wang, H.; Yang, X. Mitochondrial Proton Leak Plays a Critical Role in Pathogenesis of Cardiovascular Diseases. Adv. Exp. Med. Biol. 2017, 982, 359–370. [Google Scholar] [CrossRef]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.R., Jr.; Ballinger, S.; Dell’Italia, L.; Jianhua, Z.; Darley-Usmar, V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef]
- Marchetti, D.; Reiland, J.; Erwin, B.; Roy, M. Inhibition of heparanase activity and heparanase-induced angiogenesis by suramin analogues. Int. J. Cancer 2003, 104, 167–174. [Google Scholar] [CrossRef]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. 2020, 34, 13106–13124. [Google Scholar] [CrossRef] [PubMed]
- Pour, P.A.; Kenney, M.C.; Kheradvar, A. Bioenergetics Consequences of Mitochondrial Transplantation in Cardiomyocytes. J. Am. Heart Assoc. 2020, 9, e014501. [Google Scholar] [CrossRef] [PubMed]
- Will, Y.; Dykens, J. Mitochondrial toxicity assessment in industry--a decade of technology development and insight. Expert Opin Drug Metab. Toxicol. 2014, 10, 1061–1067. [Google Scholar] [CrossRef]
- van der Stel, W.; Carta, G.; Eakins, J.; Darici, S.; Delp, J.; Forsby, A.; Bennekou, S.H.; Gardner, I.; Leist, M.; Danen, E.H.J.; et al. Multiparametric assessment of mitochondrial respiratory inhibition in HepG2 and RPTEC/TERT1 cells using a panel of mitochondrial targeting agrochemicals. Arch. Toxicol. 2020, 94, 2707–2729. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Chapter Sixteen—Analysis and Interpretation of Microplate-Based Oxygen Consumption and pH Data. In Methods in Enzymology; Murphy, A.N., Chan, D.C., Eds.; Academic Press: Oxford, UK, 2014; Volume 547, pp. 309–354. [Google Scholar]
- Chan, G.K.; Kleinheinz, T.L.; Peterson, D.; Moffat, J.G. A simple high-content cell cycle assay reveals frequent discrepancies between cell number and ATP and MTS proliferation assays. PLoS ONE 2013, 8, e63583. [Google Scholar] [CrossRef]
- Kam, Y.; Rogers, G.W.; Jastromb, N.; Dranka, B.P. Methods and Strategies for Normalizing XF Metabolic Data to Cellular Parameters. Available online: https://www.agilent.com/cs/library/technicaloverviews/public/Methods_and_Strategies_for_Normalizing_Tech_Overview_022118.pdf (accessed on 5 June 2021).
- de Moura, M.B.; Uppala, R.; Zhang, Y.; Van Houten, B.; Goetzman, E.S. Overexpression of mitochondrial sirtuins alters glycolysis and mitochondrial function in HEK293 cells. PLoS ONE 2014, 9, e106028. [Google Scholar] [CrossRef]
- Lombard, D.B.; Alt, F.W.; Cheng, H.-L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef]
- Pfeifer, A.M.; Cole, K.E.; Smoot, D.T.; Weston, A.; Groopman, J.D.; Shields, P.G.; Vignaud, J.M.; Juillerat, M.; Lipsky, M.M.; Trump, B.F. Simian virus 40 large tumor antigen-immortalized normal human liver epithelial cells express hepatocyte characteristics and metabolize chemical carcinogens. Proc. Natl. Acad. Sci. USA 1993, 90, 5123–5127. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cai, Z. SIRT3 regulates mitochondrial biogenesis in aging-related diseases. J. Biomed. Res. 2022, 37, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Buler, M.; Aatsinki, S.-M.; Izzi, V.; Hakkola, J. Metformin Reduces Hepatic Expression of SIRT3, the Mitochondrial Deacetylase Controlling Energy Metabolism. PLoS ONE 2012, 7, e49863. [Google Scholar] [CrossRef] [PubMed]
- Kokoszka, J.E.; Coskun, P.; Esposito, L.A.; Wallace, D.C. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. USA 2001, 98, 2278–2283. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef]
- Candas, D.; Li, J.J. MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxid. Redox Signal. 2014, 20, 1599–1617. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Shen, Y.; Wu, Q.; Shi, J.; Zhou, S. Regulation of SIRT3 on mitochondrial functions and oxidative stress in Parkinson’s disease. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 132, 110928. [Google Scholar] [CrossRef]
- Hjelmeland, A.B.; Patel, R.P. SOD2 acetylation and deacetylation: Another tale of Jekyll and Hyde in cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 23376–23378. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Garg, N.J. Sirtuin Control of Mitochondrial Dysfunction, Oxidative Stress, and Inflammation in Chagas Disease Models. Front. Cell. Infect. Microbiol. 2021, 11, 110928. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; He, H.; Zuo, Z.; Xu, Z.; Wei, Z.; Deng, J. The role of different SIRT1-mediated signaling pathways in toxic injury. Cell. Mol. Biol. Lett. 2019, 24, 36. [Google Scholar] [CrossRef] [PubMed]
- Alam, F.; Syed, H.; Amjad, S.; Baig, M.; Khan, T.A.; Rehman, R. Interplay between oxidative stress, SIRT1, reproductive and metabolic functions. Curr. Res. Physiol. 2021, 4, 119–124. [Google Scholar] [CrossRef]
- Buler, M.; Andersson, U.; Hakkola, J. Who watches the watchmen? Regulation of the expression and activity of sirtuins. FASEB J. 2016, 30, 3942–3960. [Google Scholar] [CrossRef]
- Takahashi, E.; Yamaoka, Y. Simple and inexpensive technique for measuring oxygen consumption rate in adherent cultured cells. J. Physiol. Sci. 2017, 67, 731–737. [Google Scholar] [CrossRef]
- Cirit, M.; Stokes, C.L. Maximizing the impact of microphysiological systems with in vitro-in vivo translation. Lab Chip 2018, 18, 1831–1837. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Duration | Cell Viability (IC50), µM | |
|---|---|---|
| THLE-2 | HEK293 | |
| 24 h | 178.92 ± 0.049 | 77.72 ± 0.083 |
| 48 h | 21.11 ± 0.052 | 31.00 ± 0.089 |
| 72 h | 15.75 ± 0.051 | 23.74 ± 0.072 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.T.; Tan, Y.J.; Oon, C.E. BZD9L1 Differentially Regulates Sirtuins in Liver-Derived Cells by Inducing Reactive Oxygen Species. Biomedicines 2023, 11, 3059. https://doi.org/10.3390/biomedicines11113059
Lee YT, Tan YJ, Oon CE. BZD9L1 Differentially Regulates Sirtuins in Liver-Derived Cells by Inducing Reactive Oxygen Species. Biomedicines. 2023; 11(11):3059. https://doi.org/10.3390/biomedicines11113059
Chicago/Turabian StyleLee, Yeuan Ting, Yi Jer Tan, and Chern Ein Oon. 2023. "BZD9L1 Differentially Regulates Sirtuins in Liver-Derived Cells by Inducing Reactive Oxygen Species" Biomedicines 11, no. 11: 3059. https://doi.org/10.3390/biomedicines11113059
APA StyleLee, Y. T., Tan, Y. J., & Oon, C. E. (2023). BZD9L1 Differentially Regulates Sirtuins in Liver-Derived Cells by Inducing Reactive Oxygen Species. Biomedicines, 11(11), 3059. https://doi.org/10.3390/biomedicines11113059