

Intercellular Molecular Crosstalk Networks within Invasive and Immunosuppressive Tumor Microenvironment Subtypes Associated with Clinical Outcomes in Four Cancer Types
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Single-Cell RNA-Seq Data Processing and Cell Type Annotation
2.3. Identification of subTMEs
2.4. Calculation of the Score of subTMEs
2.5. Classification of subTMEs for Bulk RNA-Seq Data
2.6. Construction of the Cox Proportional Risk Regression Model and Prognostic Analysis
2.7. Cell-Cell Interaction Analysis
2.8. Gene Signature Score
2.9. Spatial Transcriptome Data Analysis
2.10. Statistical Analysis
3. Results
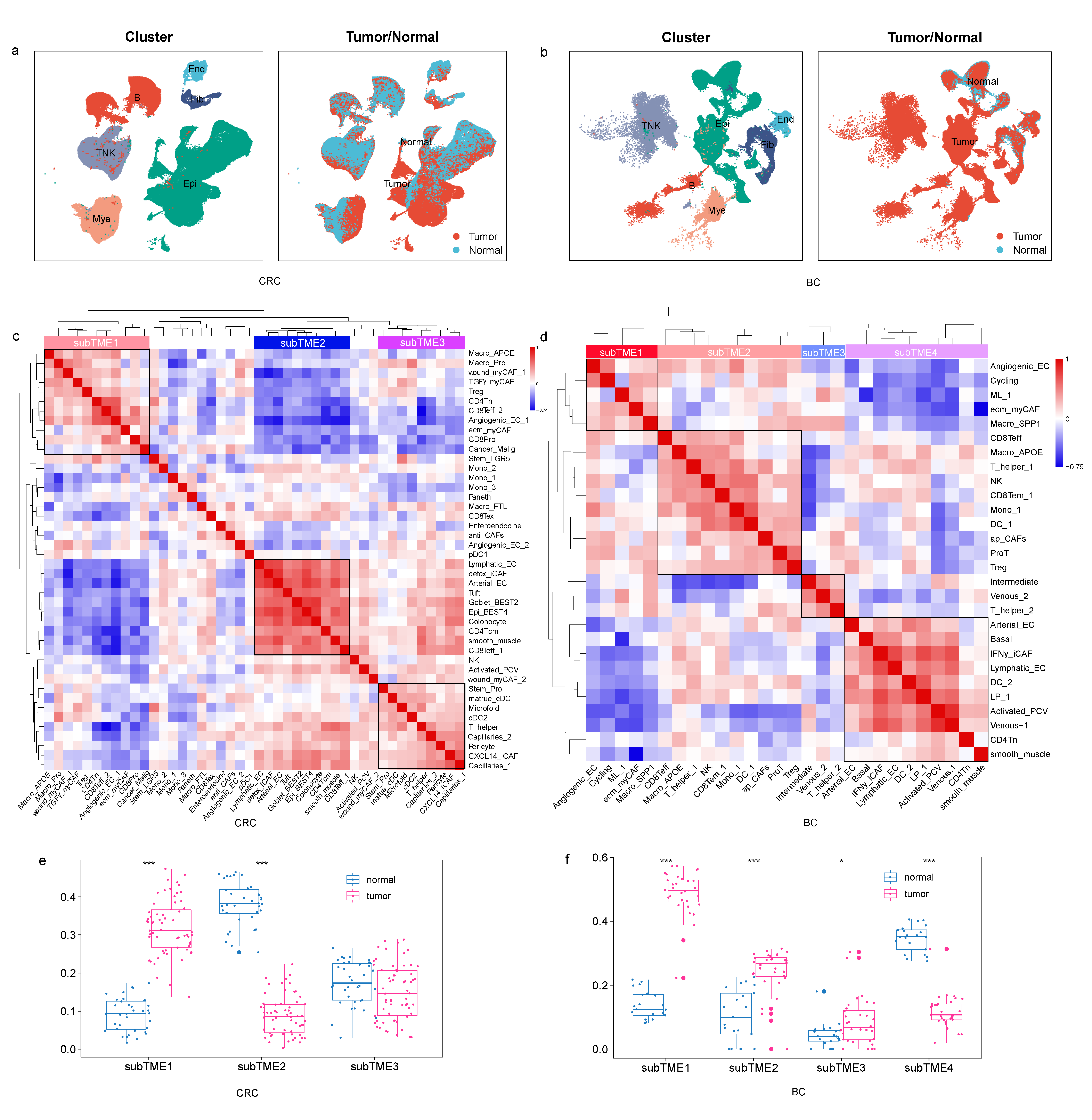
3.1. Cellular Co-Infiltration Pattern Identifies TME Subtypes across Four Cancer Types
3.2. Different Molecular Characteristics between TME Subtypes
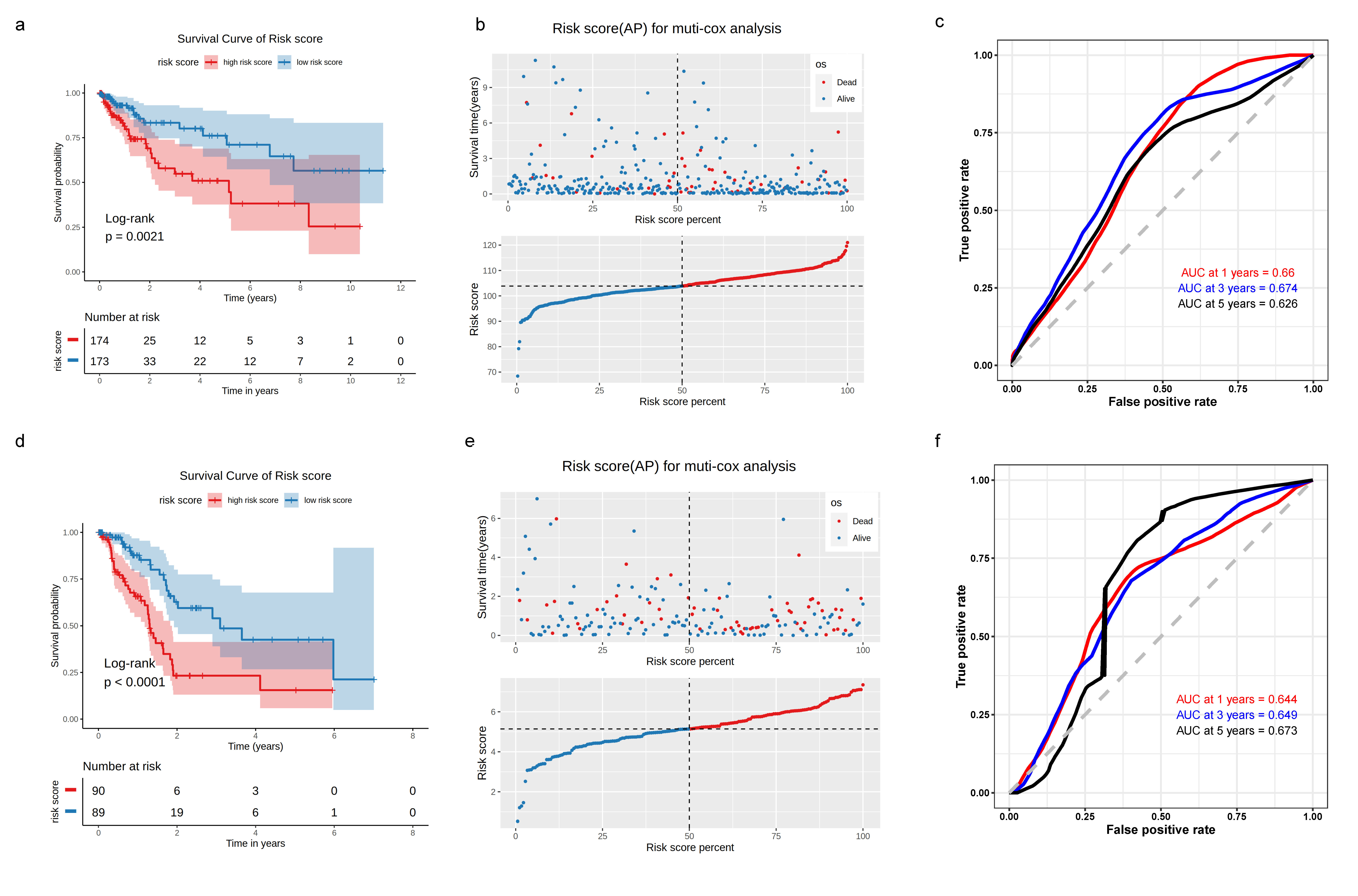
3.3. Construction of TME-Subtype-Based Risk Model
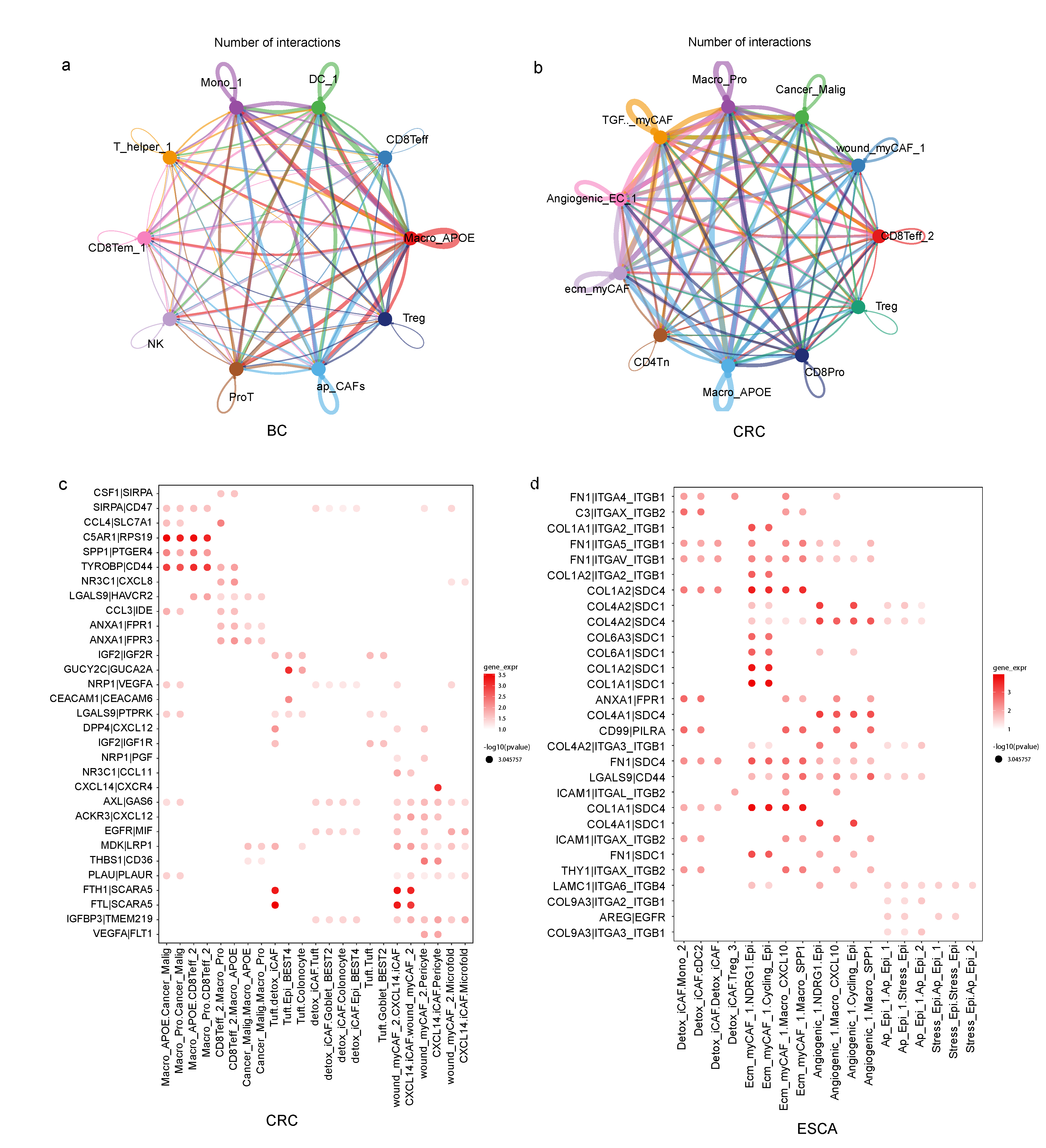
3.4. TME Subtypes Exhibit Distinct Features in Cell-Cell Interaction Pairs
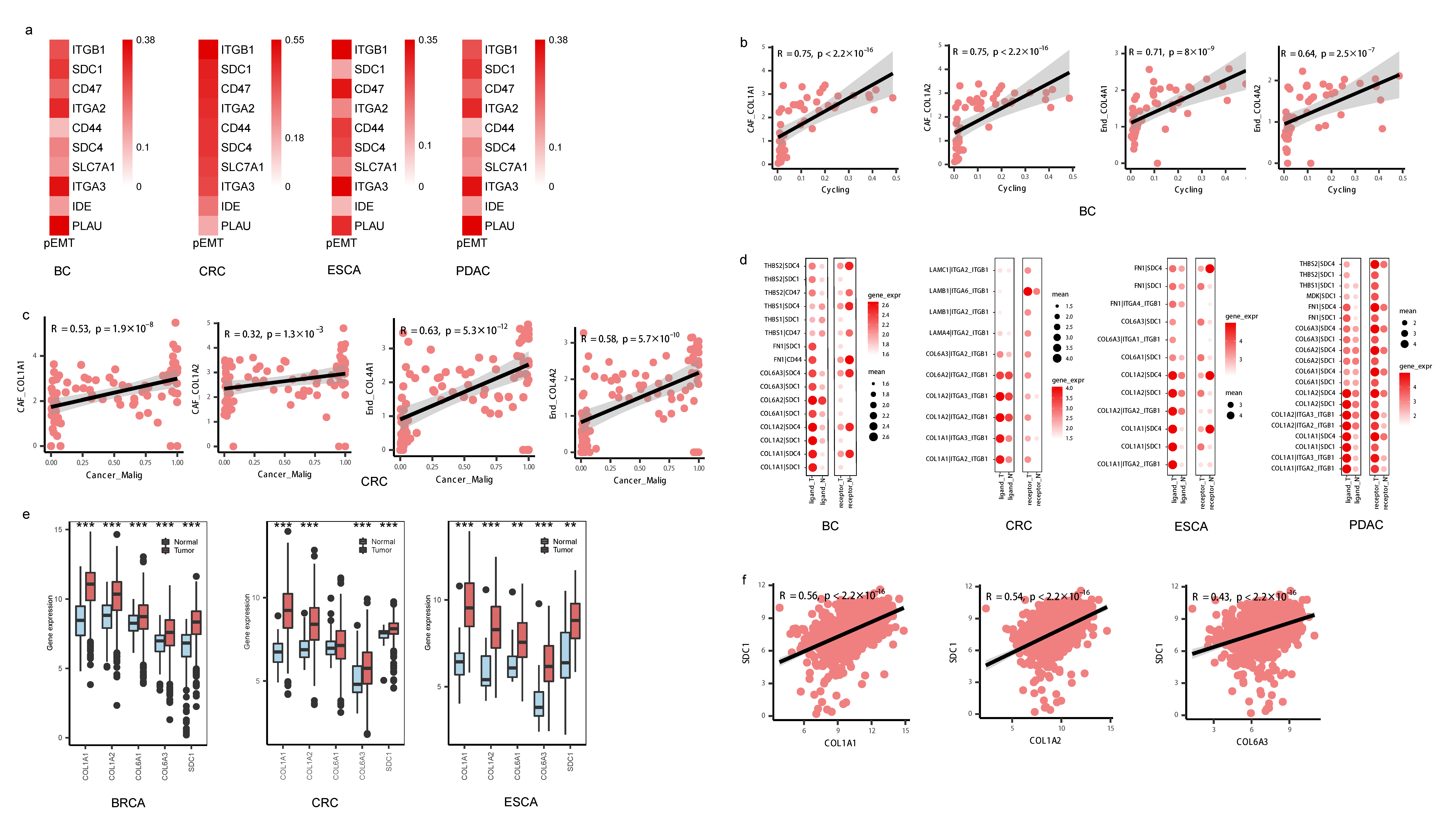
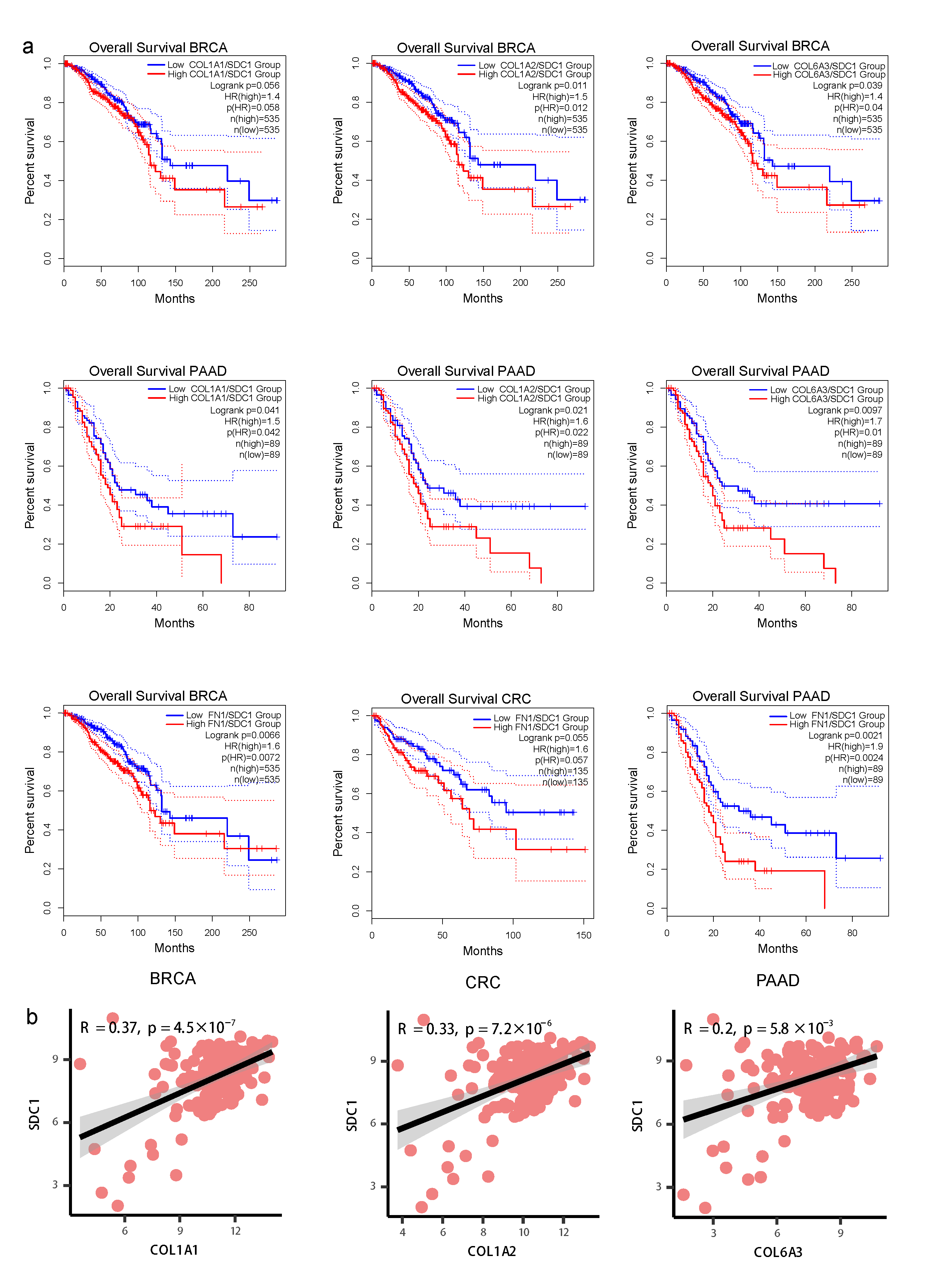
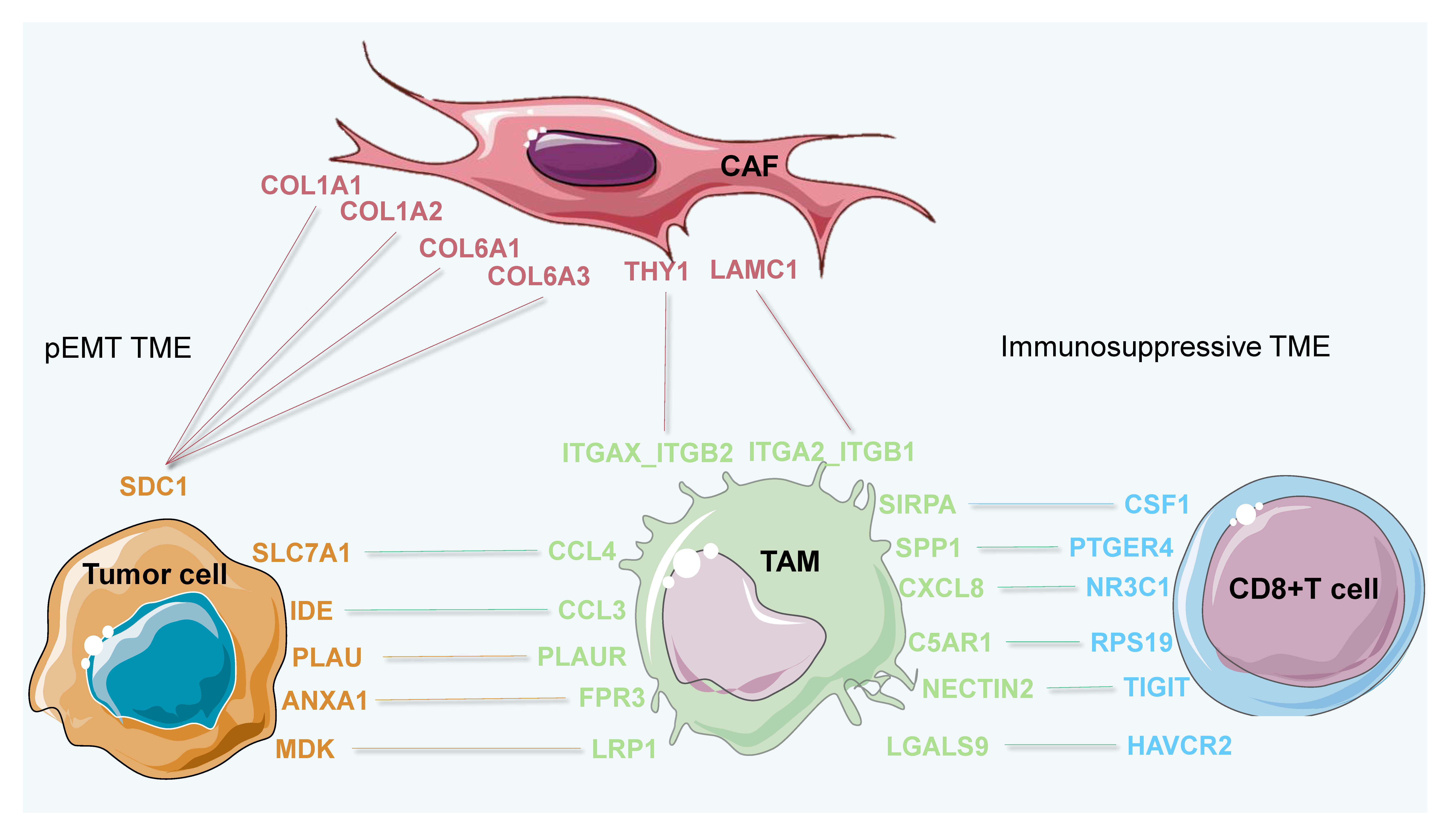
3.5. Invasive States of Tumor Cells Related to Cell-Cell Interaction Patterns in subTME-MRM
3.6. Immunosuppression States and Immune Dysfunction Caused by TAMs Related to Cell-Cell Interaction Patterns in subTME-IS
3.7. Conserved Cell-Cell Interaction Patterns and subTME Landscape in Patients in Pan-Cancer Reveal Stratification Patterns
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AUC | area under the curve |
| BC | breast cancer |
| CAF | cancer-associated fibroblast |
| Cancer_Malig | cancer malignant cell |
| CCI | Cell-cell interaction |
| CD4+T | CD4+ T lymphocytes |
| CD8+T | CD8+ T lymphocytes |
| cDC | conventional DC |
| CRC | colorectal cancer |
| DC | dendritic cell |
| DEGs | differentially expressed genes |
| ECM | extracellular matrix |
| EMT | epithelial-to-mesenchymal transition |
| End | endothelial cells |
| ESCA | esophageal cancer |
| GIST | gastrointestinal stromal tumor |
| KM | Kaplan–Meier |
| LC | lung cancer |
| L-R | ligand–receptor |
| Macro | macrophage |
| myCAF | myofibroblast |
| NK | natural killer |
| OV | ovarian cancer |
| PC | principal component |
| PDAC | pancreatic ductal adenocarcinoma |
| pEMT | partial epithelial-to-mesenchymal transition |
| RCC | renal cell carcinoma |
| ROC | receiver operating characteristic |
| scRNA | single-cell RNA |
| ssGSEA | single sample gene set enrichment analysis |
| ST | spatial transcriptome |
| subTME | tumor microenvironment subtype |
| subTME-ICI | immune cell infiltration tumor microenvironment subtype |
| subTME-IS | immune-suppressive tumor microenvironment subtype |
| subTME-MRM | matrix remodeling with malignant cells tumor microenvironment subtype |
| subTME-PSE | precancerous state of epithelial cells tumor microenvironment subtype |
| TAM | tumor-associated macrophage |
| Teff | T lymphocyte effector |
| Tex | T lymphocyte exhaustion |
| TME | tumor microenvironment |
| Treg | T regulatory |
References
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef]
- Barkley, D.; Moncada, R.; Pour, M.; Liberman, D.A.; Dryg, I.; Werba, G.; Wang, W.; Baron, M.; Rao, A.; Xia, B.; et al. Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment. Nat. Genet. 2022, 54, 1192–1201. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. A timeline of tumour-associated macrophage biology. Nat. Rev. Cancer 2023, 23, 238–257. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, X.; Jiang, L.; Li, Y.; Zheng, Q. Tumor endothelial cell-derived extracellular vesicles contribute to tumor microenvironment remodeling. Cell Commun. Signal. 2022, 20, 97. [Google Scholar] [CrossRef]
- Li, C.; Sun, Y.D.; Yu, G.Y.; Cui, J.R.; Lou, Z.; Zhang, H.; Huang, Y.; Bai, C.G.; Deng, L.L.; Liu, P.; et al. Integrated Omics of Metastatic Colorectal Cancer. Cancer Cell 2020, 38, 734–747.e9. [Google Scholar] [CrossRef]
- Orouji, E.; Raman, A.T.; Singh, A.K.; Sorokin, A.; Arslan, E.; Ghosh, A.K.; Schulz, J.; Terranova, C.; Jiang, S.; Tang, M.; et al. Chromatin state dynamics confers specific therapeutic strategies in enhancer subtypes of colorectal cancer. Gut 2022, 71, 938–949. [Google Scholar] [CrossRef]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef]
- Luo, H.; Xia, X.; Huang, L.B.; An, H.; Cao, M.; Kim, G.D.; Chen, H.N.; Zhang, W.H.; Shu, Y.; Kong, X.; et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat. Commun. 2022, 13, 6619. [Google Scholar] [CrossRef]
- Cheng, S.; Li, Z.; Gao, R.; Xing, B.; Gao, Y.; Yang, Y.; Qin, S.; Zhang, L.; Ouyang, H.; Du, P.; et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 2021, 184, 792–809.e23. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Qin, S.; Si, W.; Wang, A.; Xing, B.; Gao, R.; Ren, X.; Wang, L.; Wu, X.; Zhang, J.; et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021, 374, abe6474. [Google Scholar] [CrossRef]
- Gong, Z.; Li, Q.; Shi, J.; Wei, J.; Li, P.; Chang, C.H.; Shultz, L.D.; Ren, G. Lung fibroblasts facilitate pre-metastatic niche formation by remodeling the local immune microenvironment. Immunity 2022, 55, 1483–1500.e9. [Google Scholar] [CrossRef] [PubMed]
- Verginadis, I.I.; Avgousti, H.; Monslow, J.; Skoufos, G.; Chinga, F.; Kim, K.; Leli, N.M.; Karagounis, I.V.; Bell, B.I.; Velalopoulou, A.; et al. A stromal Integrated Stress Response activates perivascular cancer-associated fibroblasts to drive angiogenesis and tumour progression. Nat. Cell Biol. 2022, 24, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurty, A.T.; Shyer, J.A.; Thai, M.; Gandham, V.; Buechler, M.B.; Yang, Y.A.; Pradhan, R.N.; Wang, A.W.; Sanchez, P.L.; Qu, Y.; et al. LRRC15+ myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature 2022, 611, 148–154. [Google Scholar] [CrossRef]
- Timperi, E.; Gueguen, P.; Molgora, M.; Magagna, I.; Kieffer, Y.; Lopez-Lastra, S.; Sirven, P.; Baudrin, L.G.; Baulande, S.; Nicolas, A.; et al. Lipid-Associated Macrophages Are Induced by Cancer-Associated Fibroblasts and Mediate Immune Suppression in Breast Cancer. Cancer Res. 2022, 82, 3291–3306. [Google Scholar] [CrossRef]
- Tkach, M.; Thalmensi, J.; Timperi, E.; Gueguen, P.; Névo, N.; Grisard, E.; Sirven, P.; Cocozza, F.; Gouronnec, A.; Martin-Jaular, L.; et al. Extracellular vesicles from triple negative breast cancer promote pro-inflammatory macrophages associated with better clinical outcome. Proc. Natl. Acad. Sci. USA 2022, 119, e2107394119. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, J.H.; Kim, H.C.; Kim, C.W.; Kang, I.; Lee, H.K. Blood monocyte-derived CD169+ macrophages contribute to antitumor immunity against glioblastoma. Nat. Commun. 2022, 13, 6211. [Google Scholar] [CrossRef]
- Binnewies, M.; Pollack, J.L.; Rudolph, J.; Dash, S.; Abushawish, M.; Lee, T.; Jahchan, N.S.; Canaday, P.; Lu, E.; Norng, M.; et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 2021, 37, 109844. [Google Scholar] [CrossRef]
- Wei, J.; Chen, Z.; Hu, M.; He, Z.; Jiang, D.; Long, J.; Du, H. Characterizing Intercellular Communication of Pan-Cancer Reveals SPP1+ Tumor-Associated Macrophage Expanded in Hypoxia and Promoting Cancer Malignancy Through Single-Cell RNA-Seq Data. Front. Cell Dev. Biol. 2021, 9, 749210. [Google Scholar] [CrossRef]
- Wei, J.; Yu, W.; Chen, J.; Huang, G.; Zhang, L.; Chen, Z.; Hu, M.; Gong, X.; Du, H. Single-cell and spatial analyses reveal the association between gene expression of glutamine synthetase with the immunosuppressive phenotype of APOE+CTSZ+TAM in cancers. Mol. Oncol. 2023, 17, 611–628. [Google Scholar] [CrossRef] [PubMed]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; Ge, W.H.; Xu, K.H.; et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 2021, 184, 4734–4752.e20. [Google Scholar] [CrossRef] [PubMed]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Peng, L.; Luo, Y.; Zhang, S.; Pu, Y.; Chen, Y.; Guo, W.; Yao, J.; Shao, M.; Fan, W.; et al. Dissecting esophageal squamous-cell carcinoma ecosystem by single-cell transcriptomic analysis. Nat. Commun. 2021, 12, 5291. [Google Scholar] [CrossRef]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; Gupta, S.; Vanderbilt, C.; Purohit, T.A.; Liu, M.; et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 2021, 39, 662–677.e6. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.I.; Suh, Y.L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Xue, R.; Zhang, Q.; Cao, Q.; Kong, R.; Xiang, X.; Liu, H.; Feng, M.; Wang, F.; Cheng, J.; Li, Z.; et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature 2022, 612, 141–147. [Google Scholar] [CrossRef]
- Chu, Y.; Dai, E.; Li, Y.; Han, G.; Pei, G.; Ingram, D.R.; Thakkar, K.; Qin, J.J.; Dang, M.; Le, X.; et al. Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance. Nat. Med. 2023, 29, 1550–1562. [Google Scholar] [CrossRef]
- Qi, J.; Sun, H.; Zhang, Y.; Wang, Z.; Xun, Z.; Li, Z.; Ding, X.; Bao, R.; Hong, L.; Jia, W.; et al. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat. Commun. 2022, 13, 1742. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Tietscher, S.; Wagner, J.; Anzeneder, T.; Langwieder, C.; Rees, M.; Sobottka, B.; de Souza, N.; Bodenmiller, B. A comprehensive single-cell map of T cell exhaustion-associated immune environments in human breast cancer. Nat. Commun. 2023, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef] [PubMed]
- Park, B.V.; Freeman, Z.T.; Ghasemzadeh, A.; Chattergoon, M.A.; Rutebemberwa, A.; Steigner, J.; Winter, M.E.; Huynh, T.V.; Sebald, S.M.; Lee, S.J.; et al. TGFβ1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016, 6, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Ullman, N.A.; Burchard, P.R.; Dunne, R.F.; Linehan, D.C. Immunologic Strategies in Pancreatic Cancer: Making Cold Tumors Hot. J. Clin. Oncol. 2022, 40, 2789–2805. [Google Scholar] [CrossRef]
- Waks, A.G.; Stover, D.G.; Guerriero, J.L.; Dillon, D.; Barry, W.T.; Gjini, E.; Hartl, C.; Lo, W.; Savoie, J.; Brock, J.; et al. The Immune Microenvironment in Hormone Receptor-Positive Breast Cancer Before and After Preoperative Chemotherapy. Clin. Cancer Res. 2019, 25, 4644–4655. [Google Scholar] [CrossRef]
- Wang, M.; Wu, M.; Liu, X.; Shao, S.; Huang, J.; Liu, B.; Liang, T. Pyroptosis Remodeling Tumor Microenvironment to Enhance Pancreatic Cancer Immunotherapy Driven by Membrane Anchoring Photosensitizer. Adv. Sci. 2022, 9, e2202914. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, W.; Dong, B.; Xin, Z.; Ji, Y.; Su, R.; Shen, K.; Pan, J.; Wang, Q.; Xue, W. Docetaxel remodels prostate cancer immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. Theranostics 2022, 12, 4965–4979. [Google Scholar] [CrossRef]
- Jenkins, L.; Jungwirth, U.; Avgustinova, A.; Iravani, M.; Mills, A.; Haider, S.; Harper, J.; Isacke, C.M. Cancer-Associated Fibroblasts Suppress CD8+ T-cell Infiltration and Confer Resistance to Immune-Checkpoint Blockade. Cancer Res. 2022, 82, 2904–2917. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Cameron, S.; Haller, F.; Dudas, J.; Moriconi, F.; Gunawan, B.; Armbrust, T.; Langer, C.; Füzesi, L.; Ramadori, G. Immune cells in primary gastrointestinal stromal tumors. Eur. J. Gastroenterol. Hepatol. 2008, 20, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Trent, J.C.; Wilky, B.A.; Kerr, D.A.; Rosenberg, A.E. Current status of immunotherapy for gastrointestinal stromal tumor. Cancer Gene Ther. 2017, 24, 130–133. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Tarantino, G.; Agostinelli, C.; Urbini, M.; Nannini, M.; Saponara, M.; Castelli, C.; Stacchiotti, S.; Fumagalli, E.; Gatto, L.; et al. Immune microenvironment profiling of gastrointestinal stromal tumors (GIST) shows gene expression patterns associated to immune checkpoint inhibitors response. Oncoimmunology 2019, 8, e1617588. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Yang, X.; Chen, X.; Yu, S.; Yu, S.; Zhang, B.; Ji, Y.; Chen, Y.; Ouyang, Y.; Luo, W. Single-cell transcriptome analysis revealed the heterogeneity and microenvironment of gastrointestinal stromal tumors. Cancer Sci. 2021, 112, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Gatti-Mays, M.E.; Balko, J.M.; Gameiro, S.R.; Bear, H.D.; Prabhakaran, S.; Fukui, J.; Disis, M.L.; Nanda, R.; Gulley, J.L.; Kalinsky, K.; et al. If we build it they will come: Targeting the immune response to breast cancer. NPJ Breast Cancer 2019, 5, 37. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Zhang, J.; Zhong, J.; Yang, R. COL1A1 promotes metastasis in colorectal cancer by regulating the WNT/PCP pathway. Mol. Med. Rep. 2018, 17, 5037–5042. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, J.; Yu, W.; Wu, L.; Chen, Z.; Huang, G.; Hu, M.; Du, H. Intercellular Molecular Crosstalk Networks within Invasive and Immunosuppressive Tumor Microenvironment Subtypes Associated with Clinical Outcomes in Four Cancer Types. Biomedicines 2023, 11, 3057. https://doi.org/10.3390/biomedicines11113057
Wei J, Yu W, Wu L, Chen Z, Huang G, Hu M, Du H. Intercellular Molecular Crosstalk Networks within Invasive and Immunosuppressive Tumor Microenvironment Subtypes Associated with Clinical Outcomes in Four Cancer Types. Biomedicines. 2023; 11(11):3057. https://doi.org/10.3390/biomedicines11113057
Chicago/Turabian StyleWei, Jinfen, Wenqi Yu, Lei Wu, Zixi Chen, Guanda Huang, Meiling Hu, and Hongli Du. 2023. "Intercellular Molecular Crosstalk Networks within Invasive and Immunosuppressive Tumor Microenvironment Subtypes Associated with Clinical Outcomes in Four Cancer Types" Biomedicines 11, no. 11: 3057. https://doi.org/10.3390/biomedicines11113057
APA StyleWei, J., Yu, W., Wu, L., Chen, Z., Huang, G., Hu, M., & Du, H. (2023). Intercellular Molecular Crosstalk Networks within Invasive and Immunosuppressive Tumor Microenvironment Subtypes Associated with Clinical Outcomes in Four Cancer Types. Biomedicines, 11(11), 3057. https://doi.org/10.3390/biomedicines11113057