
Hepatic Macrophages as Targets for the MSC-Based Cell Therapy in Non-Alcoholic Steatohepatitis



Abstract
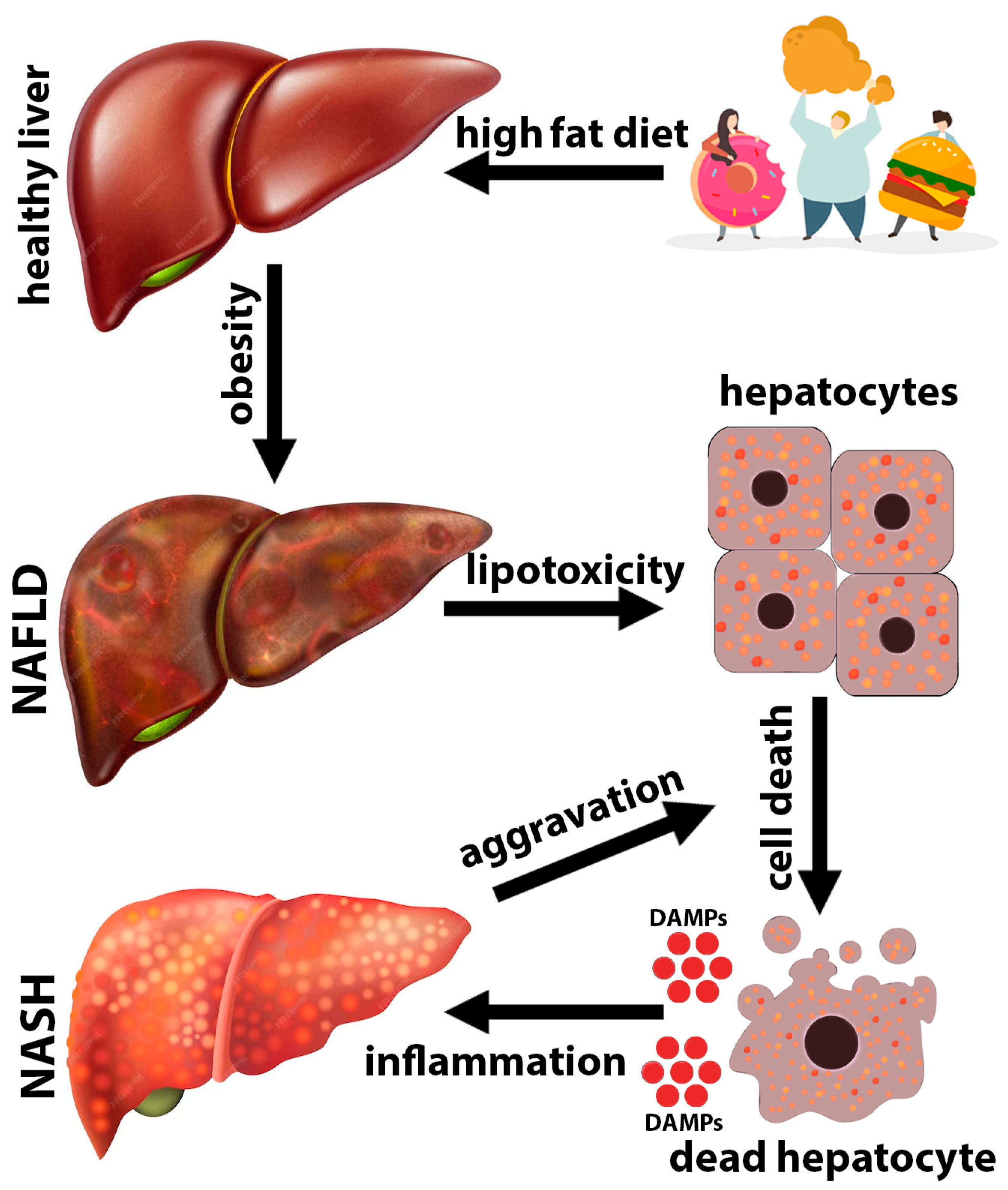
:1. Introduction
2. NAFLD/NASH Pathogenesis
3. Role of Kupffer Cells in the Pathogenesis of NASH
4. Cell Therapy of NASH Based on the Cross-Talk between Transplanted MSCs and Liver Macrophages

{kind=link}
{kind=link}
| MSCs/MSC Derivatives | Effect on Kupffer Cells/Macrophages | References |
|---|---|---|
| PGE2 | Inhibition of apoptosis via TLR4-ERK1/2-caspase 3 pathway regulation | [167] |
| TGF-β | Induction of M2 polarization | [168] |
| Indoleamine 2,3-dioxygenase | Induction of M2 polarization | [169] |
| Mitochondria | Enhancement of swallowing ability | [170] |
| Phagocytosis promotion and pro-inflammatory cytokines secretion suppression | [171] | |
| MSCs | Inhibition of Drp-1 dependent mitochondrial fission and promotion of M2 polarization | [15] |
| MSC-derived extracellular vesicles containing IL-10 | Induction of the expression of the protein tyrosine phosphatase non receptor 22 (PTPN22) and inhibition of the pro-inflammatory macrophage activation | [172] |
| Dead MSCs | Induction of M2 polarization | [175] |
| Direct cell-to-cell contacts via CD47 on MSCs and SIRPα on Kupffer cells | Inhibition of inflammosomes in Kupffer cells | [176] |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipose tissue, obesity and non-alcoholic fatty liver disease. Minerva Endocrinol. 2017, 42, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Targher, G. Global epidemiology of nonalcoholic fatty liver disease: Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 1388–1389. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P. Pathology of non-alcoholic fatty liver disease. Liver Int. 2017, 37 (Suppl. S1), 85–89. [Google Scholar] [CrossRef]
- Xu, X.; Poulsen, K.L.; Wu, L.; Liu, S.; Miyata, T.; Song, Q.; Wei, Q.; Zhao, C.; Lin, C.; Yang, J. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct. Target. Ther. 2022, 7, 287. [Google Scholar] [CrossRef]
- Day, C.; Saksena, S. Non-alcoholic steatohepatitis: Definitions and pathogenesis. J. Gastroenterol. Hepatol. 2002, 17, S377–S384. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, L. Current, emerging, and potential therapies for non-alcoholic steatohepatitis. Front. Pharmacol. 2023, 14, 1152042. [Google Scholar] [CrossRef]
- Hu, C.; Wu, Z.; Li, L. Mesenchymal stromal cells promote liver regeneration through regulation of immune cells. Int. J. Biol. Sci. 2020, 16, 893–903. [Google Scholar] [CrossRef]
- Eguchi, A.; Iwasa, M.; Nakagawa, H. Extracellular vesicles in fatty liver disease and steatohepatitis: Role as biomarkers and therapeutic targets. Liver Int. 2023, 43, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Sui, B.; Zhang, X.; Hu, J.; Chen, J.; Liu, J.; Wu, D.; Ye, Q.; Xiang, L.; Qiu, X.; et al. Apoptotic vesicles restore liver macrophage homeostasis to counteract type 2 diabetes. J. Extracell. Vesicles 2021, 10, e12109. [Google Scholar] [CrossRef] [PubMed]
- Yarygin, K.N.; Lupatov, A.Y.; Sukhikh, G.T. Modulation of Immune Responses by Mesenchymal Stromal Cells. Bull. Exp. Biol. Med. 2016, 161, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Xu, Y.; Liu, Q.; Zhang, Q. Mesenchymal Stem Cell-Macrophage Crosstalk and Maintenance of Inflammatory Microenvironment Homeostasis. Front. Cell Dev. Biol. 2021, 9, 681171. [Google Scholar] [CrossRef]
- Shang, L.C.; Wang, M.; Liu, Y.; Zhu, X.; Wang, S. MSCs Ameliorate Hepatic IR Injury by Modulating Phenotypic Transformation of Kupffer Cells Through Drp-1 Dependent Mitochondrial Dynamics. Stem Cell Rev. Rep. 2023, 19, 1965–1980. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef]
- Amarapurka, D.N.; Amarapurkar, A.D.; Patel, N.D.; Agal, S.; Baigal, R.; Gupte, P.; Pramanik, S. Nonalcoholic steatohepatitis (NASH) with diabetes: Predictors of liver fibrosis. Ann. Hepatol. 2006, 5, 30–33. [Google Scholar] [CrossRef]
- Angulo, P.; Keach, J.C.; Batts, K.P.; Lindor, K.D. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 1999, 30, 1356–1362. [Google Scholar] [CrossRef]
- Lee, T.H.; Kim, W.R.; Benson, J.T.; Therneau, T.M.; Melton, L.J., 3rd. Serum aminotransferase activity and mortality risk in a United States community. Hepatology 2008, 47, 880–887. [Google Scholar] [CrossRef]
- Kim, H.C.; Nam, C.M.; Jee, S.H.; Han, K.H.; Oh, D.K.; Suh, I. Normal serum aminotransferase concentration and risk of mortality from liver diseases: Prospective cohort study. BMJ 2004, 328, 983. [Google Scholar] [CrossRef]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 Pathway Links Endoplasmic Reticulum Stress with Hepatocyte Apoptosis in Early Alcoholic Liver Disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ni, H.M.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Nawabi, A.; Komatsu, M.; Huang, H.; Ding, W.X. Increased Hepatic Receptor Interacting Protein Kinase 3 Expression Due to Impaired Proteasomal Functions Contributes to Alcohol-Induced Steatosis and Liver Injury. Oncotarget 2016, 7, 17681–17698. [Google Scholar] [CrossRef]
- Khambu, B.; Wang, L.; Zhang, H.; Yin, X.-M. The Activation and Function of Autophagy in Alcoholic Liver Disease. Curr. Mol. Pharmacol. 2017, 10, 165–171. [Google Scholar] [CrossRef]
- Heo, M.J.; Kim, T.H.; You, J.S.; Blaya, D.; Sancho-Bru, P.; Kim, S.G. Alcohol Dysregulates MiR-148a in Hepatocytes through FoxO1, Facilitating Pyroptosis via TXNIP Overexpression. Gut 2019, 68, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ye, T.J.; DeCaro, E.; Buehler, B.; Stahl, Z.; Bonavita, G.; Daniels, M.; You, M. Intestinal SIRT1 Deficiency Protects Mice from Ethanol-Induced Liver Injury by Mitigating Ferroptosis. Am. J. Pathol. 2020, 190, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Shojaie, L.; Iorga, A.; Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Ramachandran, A.; Chao, X.; Ding, W.X. Emerging and Established Modes of Cell Death during Acetaminophen-Induced Liver Injury. Arch. Toxicol. 2019, 93, 3491–3502. [Google Scholar] [CrossRef] [PubMed]
- Cazanave, S.C.; Gores, G.J. Mechanisms and clinical implications of hepatocyte lipoapoptosis. Clin. Lipidol. 2010, 5, 71–85. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef]
- Ratziu, V.; Sheikh, M.Y.; Sanyal, A.J.; Lim, J.K.; Conjeevaram, H.; Chalasani, N.; Abdelmalek, M.; Bakken, A.; Renou, C.; Palmer, M.; et al. A phase 2, randomized, double-blind, placebo-controlled study of GS-9450 in subjects with nonalcoholic steatohepatitis. Hepatology 2012, 55, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Shiffman, M.; Freilich, B.; Vuppalanchi, R.; Watt, K.; Chan, J.L.; Spada, A.; Hagerty, D.T.; Schiff, E. Randomised clinical trial: Emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2019, 49, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Goodman, Z.; Jabbar, A.; Vemulapalli, R.; Younes, Z.H.; Freilich, B.; Sheikh, M.Y.; Schattenberg, J.M.; Kayali, Z.; Zivony, A.; et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J. Hepatol. 2020, 72, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V. Nonalcoholic Steatohepatitis Clinical Research Network. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef]
- Caballero, F.; Fernandez, A.; De Lacy, A.M.; Fernandez-Checa, J.C.; Caballeria, J.; Garcia-Ruiz, C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef]
- Eguchi, A.; Wree, A.; Feldstein, A.E. Biomarkers of liver cell death. J. Hepatol. 2014, 60, 1063–1074. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lavine, K.J.; Epelman, S.; Evans, S.A.; Weinheimer, C.J.; Barger, P.M.; Mann, D.L. Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J. Am. Heart Assoc. 2015, 4, e001993. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sursal, T.; Adibnia, Y.; Zhao, C.; Zheng, Y.; Li, H.; Otterbein, L.E.; Hauser, C.J.; Itagaki, K. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS ONE 2013, 8, e59989. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Mechanisms of Inflammatory Liver Injury and Drug-Induced Hepatotoxicity. Curr. Pharmacol. Rep. 2018, 4, 346–357. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Xiao, F.; Waldrop, S.L.; Khimji, A.K.; Kilic, G. Pannexin1 contributes to pathophysiological ATP release in lipoapoptosis induced by saturated free fatty acids in liver cells. Am. J. Physiol. Cell Physiol. 2012, 303, C1034–C1044. [Google Scholar] [CrossRef]
- Xiao, F.; Waldrop, S.L.; Bronk, S.F.; Gores, G.J.; Davis, L.S.; Kilic, G. Lipoapoptosis induced by saturated free fatty acids stimulates monocyte migration: A novel role for Pannexin1 in liver cells. Purinergic Signal. 2015, 11, 347–359. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366, Erratum in Science 2011, 331, 1517. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef]
- Handa, P.; Vemulakonda, A.; Kowdley, K.V.; Uribe, M.; Méndez-Sánchez, N. Mitochondrial DNA from hepatocytes as a ligand for TLR9: Drivers of nonalcoholic steatohepatitis? World J. Gastroenterol. 2016, 22, 6965–6971. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Coulombe, P.A.; McLean, W.H. Intermediate filament proteins and their associated diseases. N. Engl. J. Med. 2004, 351, 2087–2100. [Google Scholar] [CrossRef]
- Omary, M.B.; Ku, N.O.; Strnad, P.; Hanada, S. Toward unraveling the complexity of simple epithelial keratins in human disease. J. Clin. Investig. 2009, 119, 1794–1805. [Google Scholar] [CrossRef]
- MacFarlane, M.; Merrison, W.; Dinsdale, D.; Cohen, G.M. Active caspases and cleaved cytokeratins are sequestered into cytoplasmic inclusions in TRAIL-induced apoptosis. J. Cell Biol. 2000, 148, 1239–1254. [Google Scholar] [CrossRef]
- Weerasinghe, S.V.; Ku, N.O.; Altshuler, P.J.; Kwan, R.; Omary, M.B. Mutation of caspase-digestion sites in keratin 18 interferes with filament reorganization, and predisposes to hepatocyte necrosis and loss of membrane integrity. J. Cell Sci. 2014, 127, 1464–1475. [Google Scholar] [CrossRef]
- Ku, N.O.; Strnad, P.; Bantel, H.; Omary, M.B. Keratins: Biomarkers and modulators of apoptotic and necrotic cell death in the liver. Hepatology 2016, 64, 966–976. [Google Scholar] [CrossRef]
- Li, L.; Chen, L.; Hu, L.; Liu, Y.; Sun, H.Y.; Tang, J.; Hou, Y.J.; Chang, Y.X.; Tu, Q.Q.; Feng, G.S.; et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 2011, 54, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Hirsova, P.; Gores, G.J. Non-alcoholic steatohepatitis pathogenesis: Sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018, 67, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Nobili, V.; Ceccarelli, S.; Panera, N.; De Stefanis, C.; De Vito, R.; Vitali, R.; Bedogni, G.; Balsano, C.; Cucchiara, S.; et al. Plasma high mobility group box 1 protein reflects fibrosis in pediatric nonalcoholic fatty liver disease. Expert Rev. Mol. Diagn. 2014, 14, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195, Erratum in Nature 2010, 467, 622. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ochani, M.; Li, J.; Qiang, X.; Tanovic, M.; Harris, H.E.; Susarla, S.M.; Ulloa, L.; Wang, H.; DiRaimo, R.; et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc. Natl. Acad. Sci. USA 2004, 101, 296–301. [Google Scholar] [CrossRef]
- Ge, X.; Arriazu, E.; Magdaleno, F.; Antoine, D.J.; Cruz, R.D.; Theise, N.; Nieto, N. High Mobility Group Box-1 Drives Fibrosis Progression Signaling via the Receptor for Advanced Glycation End Products in Mice. Hepatology 2018, 68, 2380–2404. [Google Scholar] [CrossRef]
- Arriazu, E.; Ge, X.; Leung, T.M.; Magdaleno, F.; Lopategi, A.; Lu, Y.; Kitamura, N.; Urtasun, R.; Theise, N.; Antoine, D.J.; et al. Signalling via the osteopontin and high mobility group box-1 axis drives the fibrogenic response to liver injury. Gut 2017, 66, 1123–1137, Erratum in Gut 2020, 69, e2. [Google Scholar] [CrossRef]
- Vonghia, L.; Michielsen, P.; Francque, S. Immunological mechanisms in the pathophysiology of non-alcoholic steatohepatitis. Int. J. Mol. Sci. 2013, 14, 19867–19890. [Google Scholar] [CrossRef]
- Ramadori, G.; Christ, B. Cytokines and the hepatic acute-phase response. Semin. Liver Dis. 1999, 19, 141–155. [Google Scholar] [CrossRef]
- Mollica, M.P.; Lionetti, L.; Putti, R.; Cavaliere, G.; Gaita, M.; Barletta, A. From chronic overfeeding to hepatic injury: Role of endoplasmic reticulum stress and inflammation. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 222–230. [Google Scholar] [CrossRef]
- Winkler, S.; Borkham-Kamphorst, E.; Stock, P.; Brückner, S.; Dollinger, M.; Weiskirchen, R.; Christ, B. Human mesenchymal stem cells towards non-alcoholic steatohepatitis in an immunodeficient mouse model. Exp. Cell Res. 2014, 326, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Pelz, S.; Stock, P.; Brückner, S.; Christ, B. A methionine-choline-deficient diet elicits NASH in the immunodeficient mouse featuring a model for hepatic cell transplantation. Exp. Cell Res. 2012, 318, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Bouwens, L.; Baekeland, M.; De Zanger, R.; Wisse, E. Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology 1986, 6, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Deppermann, C.; Kratofil, R.M.; Peiseler, M.; David, B.A.; Zindel, J.; Castanheira, F.V.E.S.; van der Wal, F.; Carestia, A.; Jenne, C.N.; Marth, J.D.; et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J. Exp. Med. 2020, 217, e20190723. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, W.D.; Crane, A.; Johansson, J.U.; Yen, K.; Garfinkel, K.; Mastroeni, D.; Asok, P.; Bradt, B.; Sabbagh, M.; Wallace, T.L.; et al. Peripheral complement interactions with amyloid β peptide: Erythrocyte clearance mechanisms. Alzheimers Dement. 2017, 13, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Willekens, F.L.; Were, J.M.; Kruijt, J.K.; Roerdinkholder-Stoelwinder, B.; Groenen-Döpp, Y.A.; van den Bos, A.G.; Bosman, G.J.; van Berkel, T.J. Liver Kupffer cells rapidly remove red blood cell-derived vesicles from the circulation by scavenger receptors. Blood 2005, 105, 2141–2145. [Google Scholar] [CrossRef]
- Terpstra, V.; van Berkel, T.J. Scavenger receptors on liver Kupffer cells mediate the in vivo uptake of oxidatively damaged red blood cells in mice. Blood 2000, 95, 2157–2163. [Google Scholar] [CrossRef]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.; Holderried, T.A.; Seifert, M.; et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 2016, 22, 945–951. [Google Scholar] [CrossRef]
- Scott, C.L.; Guilliams, M. The role of Kupffer cells in hepatic iron and lipid metabolism. J. Hepatol. 2018, 69, 1197–1199. [Google Scholar] [CrossRef]
- Wang, Y.; van der Tuin, S.; Tjeerdema, N.; van Dam, A.D.; Rensen, S.S.; Hendrikx, T.; Berbée, J.F.; Atanasovska, B.; Fu, J.; Hoekstra, M.; et al. Plasma cholesteryl ester transfer protein is predominantly derived from Kupffer cells. Hepatology 2015, 62, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Helmy, K.Y.; Katschke, K.J., Jr.; Gorgani, N.N.; Kljavin, N.M.; Elliott, J.M.; Diehl, L.; Scales, S.J.; Ghilardi, N.; van Lookeren Campagne, M. CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 2006, 124, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Surewaard, B.G.; Wong, C.H.; Geoghegan, J.A.; Jenne, C.N.; Kubes, P. CRIg Functions as a Macrophage Pattern Recognition Receptor to Directly Bind and Capture Blood-Borne Gram-Positive Bacteria. Cell Host Microbe 2016, 20, 99–106. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Cheng, L.; Kedl, R.M.; Ju, C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology 2008, 48, 978–990. [Google Scholar] [CrossRef]
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; van Rooijen, N.; Ochando, J.C.; Randolph, G.J.; et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 2015, 62, 279–291. [Google Scholar] [CrossRef]
- Li, W.; Chang, N.; Li, L. Heterogeneity and Function of Kupffer Cells in Liver Injury. Front. Immunol. 2022, 13, 940867. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Bertrand, J.Y.; Jalil, A.; Klaine, M.; Jung, S.; Cumano, A.; Godin, I. Three pathways to mature macrophages in the early mouse yolk sac. Blood 2005, 106, 3004–3011. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef]
- MacParland, S.A.; Liu, J.C.; Ma, X.Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef]
- Aizarani, N.; Saviano, A.; Sagar Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Hollingshead, N.; Roberto, J.; Knupp, A.; Kenerson, H.; Chen, A.; Strickland, I.; Horton, H.; Yeung, R.; Soysa, R.; et al. Human Liver Macrophage Subsets Defined by CD32. Front. Immunol. 2020, 11, 2108. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thoné, T.; Browaeys, R.; et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022, 185, 379–396.e38. [Google Scholar] [CrossRef] [PubMed]
- Blériot, C.; Barreby, E.; Dunsmore, G.; Ballaire, R.; Chakarov, S.; Ficht, X.; De Simone, G.; Andreata, F.; Fumagalli, V.; Guo, W.; et al. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity 2021, 54, 2101–2116.e6. [Google Scholar] [CrossRef]
- Martrus, G.; Goebels, H.; Langeneckert, A.E.; Kah, J.; Flomm, F.; Ziegler, A.E.; Niehrs, A.; Löbl, S.M.; Russu, K.; Hess, L.U.; et al. CD49a Expression Identifies a Subset of Intrahepatic Macrophages in Humans. Front. Immunol. 2019, 10, 1247. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Stutchfield, B.M.; Antoine, D.J.; Mackinnon, A.C.; Gow, D.J.; Bain, C.C.; Hawley, C.A.; Hughes, M.J.; Francis, B.; Wojtacha, D.; Man, T.Y.; et al. CSF1 Restores Innate Immunity After Liver Injury in Mice and Serum Levels Indicate Outcomes of Patients With Acute Liver Failure. Gastroenterology 2015, 149, 1896–1909.e14. [Google Scholar] [CrossRef]
- Nascimento, M.; Huang, S.C.; Smith, A.; Everts, B.; Lam, W.; Bassity, E.; Gautier, E.L.; Randolph, G.J.; Pearce, E.J. Ly6Chi monocyte recruitment is responsible for Th2 associated host-protective macrophage accumulation in liver inflammation due to schistosomiasis. PLoS Pathog. 2014, 10, e1004282. [Google Scholar] [CrossRef]
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z.; Varol, C. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J. Immunol. 2014, 193, 344–353. [Google Scholar] [CrossRef]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef] [PubMed]
- Fogg, D.K.; Sibon, C.; Miled, C.; Jung, S.; Aucouturier, P.; Littman, D.R.; Cumano, A.; Geissmann, F. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 2006, 311, 83–87, Erratum in Science 2006, 311, 1242. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Hammerich, L.; Storch, D.; Bartneck, M.; Huss, S.; Rüsseler, V.; Gassler, N.; Lira, S.A.; Luedde, T.; Trautwein, C.; et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology 2012, 55, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Nakamoto, N.; Ebinuma, H.; Kanai, T.; Chu, P.S.; Ono, Y.; Mikami, Y.; Ojiro, K.; Lipp, M.; Love, P.E.; Saito, H.; et al. CCR9+ macrophages are required for acute liver inflammation in mouse models of hepatitis. Gastroenterology 2012, 142, 366–376. [Google Scholar] [CrossRef]
- Chu, P.S.; Nakamoto, N.; Ebinuma, H.; Usui, S.; Saeki, K.; Matsumoto, A.; Mikami, Y.; Sugiyama, K.; Tomita, K.; Kanai, T.; et al. C-C motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology 2013, 58, 337–350. [Google Scholar] [CrossRef]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef]
- McGettigan, B.; McMahan, R.; Orlicky, D.; Burchill, M.; Danhorn, T.; Francis, P.; Cheng, L.L.; Golden-Mason, L.; Jakubzick, C.V.; Rosen, H.R. Dietary Lipids Differentially Shape Nonalcoholic Steatohepatitis Progression and the Transcriptome of Kupffer Cells and Infiltrating Macrophages. Hepatology 2019, 70, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2019, 129, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Tacke, F. Immunology in the liver—from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594.e1. [Google Scholar] [CrossRef] [PubMed]
- Dambach, D.M.; Watson, L.M.; Gray, K.R.; Durham, S.K.; Laskin, D.L. Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology 2002, 35, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tohme, S.; Al-Khafaji, A.B.; Tai, S.; Loughran, P.; Chen, L.; Wang, S.; Kim, J.; Billiar, T.; Wang, Y.; et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 2015, 62, 600–614. [Google Scholar] [CrossRef]
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J. Immunol. 2013, 190, 5226–5236. [Google Scholar] [CrossRef]
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682. [Google Scholar] [CrossRef]
- Xia, C.; Razavi, M.; Rao, X.; Braunstein, Z.; Mao, H.; Toomey, A.C.; Wang, Y.; Simon, D.I.; Zhao, S.; Rajagopalan, S.; et al. MRP14 enhances the ability of macrophage to recruit T cells and promotes obesity-induced insulin resistance. Int. J. Obes. 2019, 43, 2434–2447. [Google Scholar] [CrossRef]
- Liaskou, E.; Zimmermann, H.W.; Li, K.K.; Oo, Y.H.; Suresh, S.; Stamataki, Z.; Qureshi, O.; Lalor, P.F.; Shaw, J.; Syn, W.K.; et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology 2013, 57, 385–398. [Google Scholar] [CrossRef]
- Ogura, Y.; Sutterwala, F.S.; Flavell, R.A. The inflammasome: First line of the immune response to cell stress. Cell 2006, 126, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lan, P.; Hou, X.; Han, Q.; Lu, N.; Li, T.; Jiao, C.; Zhang, J.; Zhang, C.; Tian, Z. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production. J. Hepatol. 2017, 66, 693–702. [Google Scholar] [CrossRef]
- Petrasek, J.; Iracheta-Vellve, A.; Saha, B.; Satishchandran, A.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J. Leukoc. Biol. 2015, 98, 249–256. [Google Scholar] [CrossRef]
- Ganz, M.; Bukong, T.N.; Csak, T.; Saha, B.; Park, J.K.; Ambade, A.; Kodys, K.; Szabo, G. Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice. J. Transl. Med. 2015, 13, 193. [Google Scholar] [CrossRef]
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306. [Google Scholar] [CrossRef]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Martínez-Micaelo, N.; González-Abuín, N.; Pinent, M.; Ardévol, A.; Blay, M. Dietary fatty acid composition is sensed by the NLRP3 inflammasome: Omega-3 fatty acid (DHA) prevents NLRP3 activation in human macrophages. Food Funct. 2016, 7, 3480–3487. [Google Scholar] [CrossRef]
- Pan, J.; Ou, Z.; Cai, C.; Li, P.; Gong, J.; Ruan, X.Z.; He, K. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell. Immunol. 2018, 332, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Liu, Y.; Shi, Y.; Zhang, H.; Sun, Y.; Zhangyuan, G.; Wang, F.; Yu, W.; Wang, J.; Tao, X.; et al. PTPROt aggravates inflammation by enhancing NF-κB activation in liver macrophages during nonalcoholic steatohepatitis. Theranostics 2020, 10, 5290–5304, Erratum in Theranostics 2022, 12, 3604–3606. [Google Scholar] [CrossRef]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef]
- Wree, A.; McGeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Devisscher, L.; Scott, C.L.; Lefere, S.; Raevens, S.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Geerts, A.; Guilliams, M.; Van Vlierberghe, H. Non-alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell. Immunol. 2017, 322, 74–83. [Google Scholar] [CrossRef]
- Daemen, S.; Gainullina, A.; Kalugotla, G.; He, L.; Chan, M.M.; Beals, J.W.; Liss, K.H.; Klein, S.; Feldstein, A.E.; Finck, B.N.; et al. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep. 2021, 34, 108626, Erratum in Cell Rep. 2022, 41, 111660. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Degroote, H.; Van Vlierberghe, H.; Devisscher, L. Unveiling the depletion of Kupffer cells in experimental hepatocarcinogenesis through liver macrophage subtype-specific markers. J. Hepatol. 2019, 71, 631–633. [Google Scholar] [CrossRef]
- Tran, S.; Baba, I.; Poupel, L.; Dussaud, S.; Moreau, M.; Gélineau, A.; Marcelin, G.; Magréau-Davy, E.; Ouhachi, M.; Lesnik, P.; et al. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity 2020, 53, 627–640.e5. [Google Scholar] [CrossRef]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020, 52, 1057–1074.e7. [Google Scholar] [CrossRef]
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9. [Google Scholar] [CrossRef]
- Sakai, M.; Troutman, T.D.; Seidman, J.S.; Ouyang, Z.; Spann, N.J.; Abe, Y.; Ego, K.M.; Bruni, C.M.; Deng, Z.; Schlachetzki, J.C.M.; et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity 2019, 51, 655–670.e8. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Duarte, N.; Coelho, I.C.; Patarrão, R.S.; Almeida, J.I.; Penha-Gonçalves, C.; Macedo, M.P. How Inflammation Impinges on NAFLD: A Role for Kupffer Cells. Biomed. Res. Int. 2015, 2015, 984578. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Baker, R.D.; Bhatia, T.; Zhu, L.; Baker, S.S. Pathogenesis of nonalcoholic steatohepatitis. Cell. Mol. Life Sci. 2016, 73, 1969–1987. [Google Scholar] [CrossRef]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, H.J.; Na, H.; Nam, M.W.; Kim, J.Y.; Kim, J.S.; Koo, S.H.; Lee, M.O. RORα Induces KLF4-Mediated M2 Polarization in the Liver Macrophages that Protect against Nonalcoholic Steatohepatitis. Cell Rep. 2017, 20, 124–135. [Google Scholar] [CrossRef]
- Park, J.W.; Jeong, G.; Kim, S.J.; Kim, M.K.; Park, S.M. Predictors reflecting the pathological severity of non-alcoholic fatty liver disease: Comprehensive study of clinical and immunohistochemical findings in younger Asian patients. J. Gastroenterol. Hepatol. 2007, 22, 491–497. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Lebensztejn, D.M. The role of Kupffer cells in the morphogenesis of nonalcoholic steatohepatitis—Ultrastructural findings. The first report in pediatric patients. Scand. J. Gastroenterol. 2013, 48, 352–357. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Macrophages in Nonalcoholic Fatty Liver Disease: A Role Model of Pathogenic Immunometabolism. Semin. Liver Dis. 2017, 37, 189–197. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Tawfeek, G.A.; Kasem, H.A. Curcumin preconditioned mesenchymal stem cells derived exosomes transplantation ameliorate and protect against non- alcoholic steatohepatitis by regulation the expression of key genes of inflammation and oxidative stress. Transpl. Immunol. 2023, 78, 101837. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wu, Y.; Chen, Y.; Xi, J.; Chu, Y.; Jin, J.; Yan, Y. Human umbilical cord mesenchymal stem cell-derived exosomes ameliorate liver steatosis by promoting fatty acid oxidation and reducing fatty acid synthesis. JHEP Rep. 2023, 5, 100746. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Hsiao, W.T.; Lee, O.K. Mesenchymal stromal cell-based therapies reduce obesity and metabolic syndromes induced by a high-fat diet. Transl. Res. 2017, 182, 61–74.e8. [Google Scholar] [CrossRef]
- Kholodenko, I.V.; Kholodenko, R.V.; Yarygin, K.N. The Crosstalk between Mesenchymal Stromal/Stem Cells and Hepatocytes in Homeostasis and under Stress. Int. J. Mol. Sci. 2023, 24, 15212. [Google Scholar] [CrossRef]
- Ezquer, M.; Ezquer, F.; Ricca, M.; Allers, C.; Conget, P. Intravenous administration of multipotent stromal cells prevents the onset of non-alcoholic steatohepatitis in obese mice with metabolic syndrome. J. Hepatol. 2011, 55, 1112–1120. [Google Scholar] [CrossRef]
- Shi, Y.; Yang, X.; Wang, S.; Wu, Y.; Zheng, L.; Tang, Y.; Gao, Y.; Niu, J. Human umbilical cord mesenchymal stromal cell-derived exosomes protect against MCD-induced NASH in a mouse model. Stem Cell Res. Ther. 2022, 13, 517. [Google Scholar] [CrossRef]
- Muto, H.; Ito, T.; Tanaka, T.; Yokoyama, S.; Yamamoto, K.; Imai, N.; Ishizu, Y.; Maeda, K.; Honda, T.; Ishikawa, T.; et al. Conditioned medium from stem cells derived from human exfoliated deciduous teeth ameliorates NASH via the Gut-Liver axis. Sci. Rep. 2021, 11, 18778. [Google Scholar] [CrossRef]
- Watanabe, T.; Tsuchiya, A.; Takeuchi, S.; Nojiri, S.; Yoshida, T.; Ogawa, M.; Itoh, M.; Takamura, M.; Suganami, T.; Ogawa, Y.; et al. Development of a non-alcoholic steatohepatitis model with rapid accumulation of fibrosis, and its treatment using mesenchymal stem cells and their small extracellular vesicles. Regen. Ther. 2020, 14, 252–261. [Google Scholar] [CrossRef]
- Grunhut, J.; Wang, W.; Aykut, B.; Gakhal, I.; Torres−Hernandez, A.; Miller, G. Macrophages in Nonalcoholic Steatohepatitis: Friend or Foe? Eur. Med. J. Hepatol. 2018, 6, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Trautwein, C.; Tacke, F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm. Allergy Drug Targets 2009, 8, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, B.; Lai, R.C.; Sim, W.K.; Lam, K.P.; Lim, S.K. MSC-sEV Treatment Polarizes Pro-Fibrotic M2 Macrophages without Exacerbating Liver Fibrosis in NASH. Int. J. Mol. Sci. 2023, 24, 8092. [Google Scholar] [CrossRef] [PubMed]
- Ohara, M.; Ohnishi, S.; Hosono, H.; Yamamoto, K.; Yuyama, K.; Nakamura, H.; Fu, Q.; Maehara, O.; Suda, G.; Sakamoto, N. Extracellular Vesicles from Amnion-Derived Mesenchymal Stem Cells Ameliorate Hepatic Inflammation and Fibrosis in Rats. Stem Cells Int. 2018, 2018, 3212643. [Google Scholar] [CrossRef]
- Liang, X.; Li, T.; Zhou, Q.; Pi, S.; Li, Y.; Chen, X.; Weng, Z.; Li, H.; Zhao, Y.; Wang, H.; et al. Mesenchymal stem cells attenuate sepsis-induced liver injury via inhibiting M1 polarization of Kupffer cells. Mol. Cell. Biochem. 2019, 452, 187–197. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Ding, H.; Shi, X.; Ren, H. Mesenchymal stem cell-secreted prostaglandin E2 ameliorates acute liver failure via attenuation of cell death and regulation of macrophage polarization. Stem Cell Res. Ther. 2021, 12, 15. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, J.; Wang, W.; Ding, Y.; Sun, Z.; Zhang, Q.; Wang, Y.; Xie, H.; Yan, S.; Zheng, S. Mesenchymal stem cells improve mouse non-heart-beating liver graft survival by inhibiting Kupffer cell apoptosis via TLR4-ERK1/2-Fas/FasL-caspase3 pathway regulation. Stem Cell Res. Ther. 2016, 7, 157. [Google Scholar] [CrossRef]
- Liu, F.; Qiu, H.; Xue, M.; Zhang, S.; Zhang, X.; Xu, J.; Chen, J.; Yang, Y.; Xie, J. MSC-secreted TGF-β regulates lipopolysaccharide-stimulated macrophage M2-like polarization via the Akt/FoxO1 pathway. Stem Cell Res. Ther. 2019, 10, 345. [Google Scholar] [CrossRef]
- François, M.; Romieu-Mourez, R.; Li, M.; Galipeau, J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol. Ther. 2012, 20, 187–195. [Google Scholar] [CrossRef]
- Jackson, M.V.; Krasnodembskaya, A.D. Analysis of Mitochondrial Transfer in Direct Co-cultures of Human Monocyte-derived Macrophages (MDM) and Mesenchymal Stem Cells (MSC). Bio-Protocol 2017, 7, e2255. [Google Scholar] [CrossRef]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Zhang, H.; Song, P.; Pan, W.; Xu, P.; Wang, G.; Hu, P.; Wang, Z.; Huang, K.; et al. Mesenchymal Stem Cells Derived Extracellular Vesicles Alleviate Traumatic Hemorrhagic Shock Induced Hepatic Injury via IL-10/PTPN22-Mediated M2 Kupffer Cell Polarization. Front. Immunol. 2022, 12, 811164. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Miaw, S.C.; Tseng, W.; Sun, Y.W.; Liu, C.C.; Tsao, H.W.; Ho, I.C. PTPN22 modulates macrophage polarization and susceptibility to dextran sulfate sodium-induced colitis. J. Immunol. 2013, 191, 2134–2143. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, I.V.; Kholodenko, R.V.; Majouga, A.G.; Yarygin, K.N. Apoptotic MSCs and MSC-Derived Apoptotic Bodies as New Therapeutic Tools. Curr. Issues Mol. Biol. 2022, 44, 5153–5172. [Google Scholar] [CrossRef]
- de Witte, S.F.H.; Luk, F.; Sierra Parraga, J.M.; Gargesha, M.; Merino, A.; Korevaar, S.S.; Shankar, A.S.; O’Flynn, L.; Elliman, S.J.; Roy, D.; et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells 2018, 36, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Lin, Y.; Xu, D.; Tian, Y.; Zhan, Y.; Li, C.; Farmer, D.G.; Kupiec-Weglinski, J.W.; Ke, B. CD47-Mediated Hedgehog/SMO/GLI1 Signaling Promotes Mesenchymal Stem Cell Immunomodulation in Mouse Liver Inflammation. Hepatology 2021, 74, 1560–1577. [Google Scholar] [CrossRef]
- Francque, S.; Moreno, C.; Saliba, F.; Vargas, V.; Ibañez, L.; Soriano, G.; Genov, J.; Katzarov, K.; Janczewska, E.; Iliescu, E.L.; et al. TOP-077. Safety, tolerability, and preliminary efficacy of ascending doses of Human Allogeneic Liver-derived Progenitor Cells (HepaStem®) in patients with cirrhotic and pre-cirrhotic non-alcoholic steatohepatitis (NASH). J. Hepatol. 2023, 78 (Suppl. S1), S807–S808. [Google Scholar] [CrossRef]
- Najimi, M.; Michel, S.; Binda, M.M.; Gellynck, K.; Belmonte, N.; Mazza, G.; Gordillo, N.; Vainilovich, Y.; Sokal, E. Human Allogeneic Liver-Derived Progenitor Cells Significantly Improve NAFLD Activity Score and Fibrosis in Late-Stage NASH Animal Model. Cells 2022, 11, 2854. [Google Scholar] [CrossRef]
- Sakai, Y.; Fukunishi, S.; Takamura, M.; Kawaguchi, K.; Inoue, O.; Usui, S.; Takashima, S.; Seki, A.; Asai, A.; Tsuchimoto, Y.; et al. Clinical trial of autologous adipose tissue-derived regenerative (stem) cells therapy for exploration of its safety and efficacy. Regen. Ther. 2021, 18, 97–101. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kholodenko, I.V.; Yarygin, K.N. Hepatic Macrophages as Targets for the MSC-Based Cell Therapy in Non-Alcoholic Steatohepatitis. Biomedicines 2023, 11, 3056. https://doi.org/10.3390/biomedicines11113056
Kholodenko IV, Yarygin KN. Hepatic Macrophages as Targets for the MSC-Based Cell Therapy in Non-Alcoholic Steatohepatitis. Biomedicines. 2023; 11(11):3056. https://doi.org/10.3390/biomedicines11113056
Chicago/Turabian StyleKholodenko, Irina V., and Konstantin N. Yarygin. 2023. "Hepatic Macrophages as Targets for the MSC-Based Cell Therapy in Non-Alcoholic Steatohepatitis" Biomedicines 11, no. 11: 3056. https://doi.org/10.3390/biomedicines11113056
APA StyleKholodenko, I. V., & Yarygin, K. N. (2023). Hepatic Macrophages as Targets for the MSC-Based Cell Therapy in Non-Alcoholic Steatohepatitis. Biomedicines, 11(11), 3056. https://doi.org/10.3390/biomedicines11113056