
ADMET and Solubility Analysis of New 5-Nitroisatine-Based Inhibitors of CDK2 Enzymes

 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. ADMET Analysis
2.2. Materials
2.3. Synthesis
2.4. Characterizations of the New Compounds
2.5. NMR Measurements
2.6. Elemental Analysis Measurements
2.7. Calibration Curves
2.8. Solubility Determination
2.9. FTIR Analysis of the Samples
3. Results and Discussion
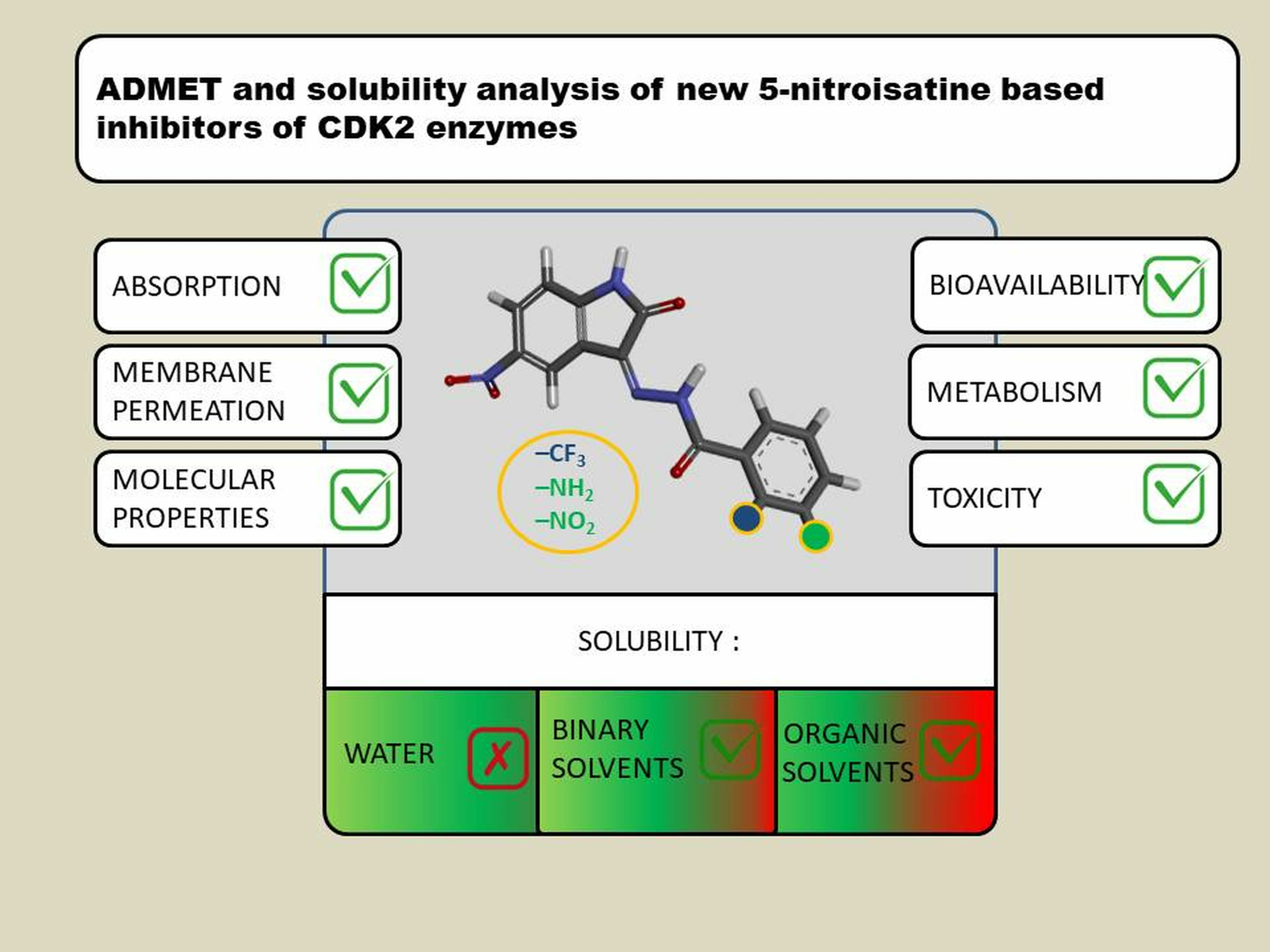
3.1. ADMET Analysis
3.2. Solubility Determination
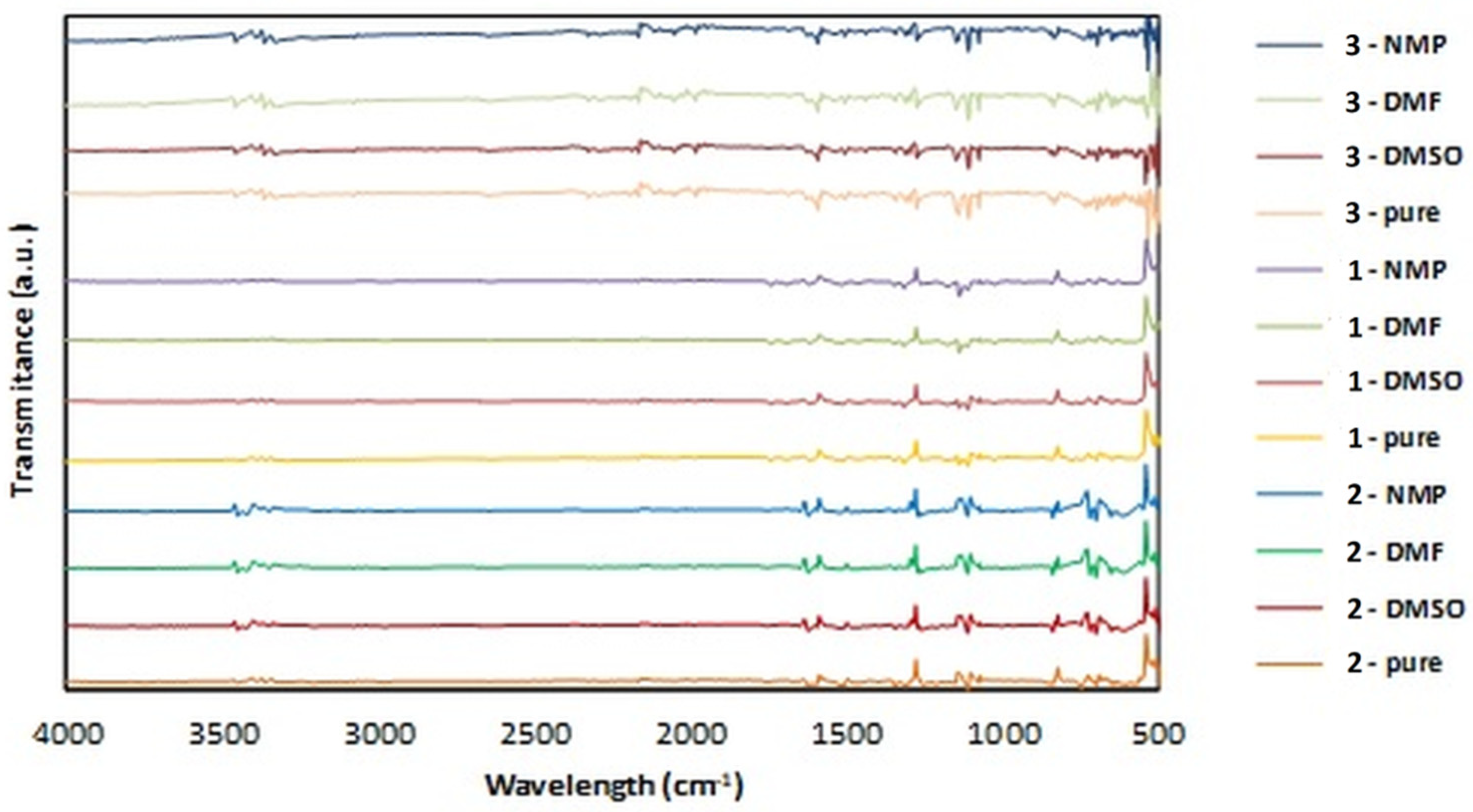
3.3. FTIR Analysis of the Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Hancock, B.C.; York, P.; Rowe, R.C. The use of solubility parameters in pharmaceutical dosage form design. Int. J. Pharm. 1997, 148, 1–21. [Google Scholar] [CrossRef]
- Constable, D.J.C.; Jimenez-Gonzalez, C.; Henderson, R.K. Perspective on solvent use in the pharmaceutical industry. Org. Process Res. Dev. 2007, 11, 133–137. [Google Scholar] [CrossRef]
- Wang, N.-N.; Dong, J.; Deng, Y.-H.; Zhu, M.-F.; Wen, M.; Yao, Z.-J.; Lu, A.-P.; Wang, J.-B.; Cao, D.-S. ADME Properties Evaluation in Drug Discovery: Prediction of Caco-2 Cell Permeability Using a Combination of NSGA-II and Boosting. J. Chem. Inf. Model. 2016, 56, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fu, L.; Zhang, J.-W.; Wei, H.; Ye, W.-L.; Deng, Z.-K.; Zhang, L.; Cheng, Y.; Ouyang, D.; Cao, Q.; et al. Three-Level Hepatotoxicity Prediction System Based on Adverse Hepatic Effects. Mol. Pharm. 2019, 16, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Li, Y.; Song, Y.; Li, D.; Sun, H.; Hou, T. ADMET evaluation in drug discovery: 15. Accurate prediction of rat oral acute toxicity using relevance vector machine and consensus modeling. J. Cheminform. 2016, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Sun, H.; Kang, Y.; Zhu, F.; Liu, H.; Zhou, W.; Wang, Z.; Li, D.; Li, Y.; Hou, T. ADMET Evaluation in Drug Discovery. 18. Reliable Prediction of Chemical-Induced Urinary Tract Toxicity by Boosting Machine Learning Approaches. Mol. Pharm. 2017, 14, 3935–3953. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, H.; Liu, H.; Li, D.; Li, Y.; Hou, T. ADMET Evaluation in Drug Discovery. 16. Predicting hERG Blockers by Combining Multiple Pharmacophores and Machine Learning Approaches. Mol. Pharm. 2016, 13, 2855–2866. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Czeleń, P.; Skotnicka, A.; Szefler, B. Designing and Synthesis of New Isatin Derivatives as Potential CDK2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 8046. [Google Scholar] [CrossRef]
- Martínez, F.; Jouyban, A.; Acree, W.E. Pharmaceuticals Solubility is Still Nowadays Widely Studied Everywhere. Pharm. Sci. 2017, 23, 1–2. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.C.; Kim, D.H.; Lee, S.E.; Kim, J.K.; Park, J.S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Grossmann, L.; McClements, D.J. Current insights into protein solubility: A review of its importance for alternative proteins. Food Hydrocoll. 2023, 137, 108416. [Google Scholar] [CrossRef]
- Sou, T.; Bergström, C.A.S. Automated assays for thermodynamic (equilibrium) solubility determination. Drug Discov. Today Technol. 2018, 27, 11–19. [Google Scholar] [CrossRef]
- Lu, W.; Chen, H. Application of deep eutectic solvents (DESs) as trace level drug extractants and drug solubility enhancers: State-of-the-art, prospects and challenges. J. Mol. Liq. 2022, 349, 118105. [Google Scholar] [CrossRef]
- Suwanwong, Y.; Boonpangrak, S. Molecularly imprinted polymers for the extraction and determination of water-soluble vitamins: A review from 2001 to 2020. Eur. Polym. J. 2021, 161, 110835. [Google Scholar] [CrossRef]
- Smith, P.E.; Mazo, R.M. On the theory of solute solubility in mixed solvents. J. Phys. Chem. B 2008, 112, 7875–7884. [Google Scholar] [CrossRef]
- Seedher, N.; Kanojia, M. Co-solvent solubilization of some poorly-soluble antidiabetic drugs. Pharm. Dev. Technol. 2009, 14, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Cysewski, P.; Jeliński, T.; Przybyłek, M.; Nowak, W.; Olczak, M. Solubility Characteristics of Acetaminophen and Phenacetin in Binary Mixtures of Aqueous Organic Solvents: Experimental and Deep Machine Learning Screening of Green Dissolution Media. Pharmaceutics 2022, 14, 2828. [Google Scholar] [CrossRef]
- Cysewski, P.; Jeliński, T.; Cymerman, P.; Przybyłek, M. Solvent Screening for Solubility Enhancement of Theophylline in Neat, Binary and Ternary NADES Solvents: New Measurements and Ensemble Machine Learning. Int. J. Mol. Sci. 2021, 22, 7347. [Google Scholar] [CrossRef] [PubMed]
- DeSimone, J.M. Practical approaches to green solvents. Science 2002, 297, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Jessop, P.G. Searching for green solvents. Green Chem. 2011, 13, 1391–1398. [Google Scholar] [CrossRef]
- Cvjetko Bubalo, M.; Vidović, S.; Radojčić Redovniković, I.; Jokić, S. Green solvents for green technologies. J. Chem. Technol. Biotechnol. 2015, 90, 1631–1639. [Google Scholar] [CrossRef]
- Häckl, K.; Kunz, W. Some aspects of green solvents. Comptes Rendus Chim. 2018, 21, 572–580. [Google Scholar] [CrossRef]
- Xie, W.; Li, T.; Chen, C.; Wu, H.; Liang, S.; Chang, H.; Liu, B.; Drioli, E.; Wang, Q.; Crittenden, J.C. Using the Green Solvent Dimethyl Sulfoxide to Replace Traditional Solvents Partly and Fabricating PVC/PVC- g-PEGMA Blended Ultrafiltration Membranes with High Permeability and Rejection. Ind. Eng. Chem. Res. 2019, 58, 6413–6423. [Google Scholar] [CrossRef]
- Kumar, A.; Jad, Y.E.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Green solid-phase peptide synthesis 4. γ-Valerolactone and N-formylmorpholine as green solvents for solid phase peptide synthesis. Tetrahedron Lett. 2017, 58, 2986–2988. [Google Scholar] [CrossRef]
- Tang, S.; Zhao, H. Glymes as versatile solvents for chemical reactions and processes: From the laboratory to industry. RSC Adv. 2014, 4, 11251–11287. [Google Scholar] [CrossRef]
- Cavuoto, D.; Gervasini, A.; Zaccheria, F.; Scotti, N.; Marelli, M.; Bisio, C.; Begni, F.; Ravasio, N. Synthesis of green solvents from bio-based lactones using heterogeneous copper catalysts. Catal. Today 2023, 418, 114104. [Google Scholar] [CrossRef]
- Marino, T.; Galiano, F.; Molino, A.; Figoli, A. New frontiers in sustainable membrane preparation: CyreneTM as green bioderived solvent. J. Memb. Sci. 2019, 580, 224–234. [Google Scholar] [CrossRef]
- Aparicio, S.; Alcalde, R. The green solvent ethyl lactate: An experimental and theoretical characterization. Green Chem. 2009, 11, 65–78. [Google Scholar] [CrossRef]
- Davies, T.G.; Tunnah, P.; Meijer, L.; Marko, D.; Eisenbrand, G.; Endicott, J.A.; Noble, M.E. Inhibitor binding to active and inactive CDK2: The crystal structure of CDK2-cyclin A/indirubin-5-sulphonate. Structure 2001, 9, 389–397. [Google Scholar] [CrossRef]
- Harten, P.; Martin, T.; Gonzalez, M.; Young, D. The software tool to find greener solvent replacements, PARIS III. Environ. Prog. Sustain. Energy 2020, 39, 13331. [Google Scholar] [CrossRef]
- Harten, P.; Martin, T.; Chang, D.; Young, D. Finding Potential Replacements for TRI Solvents Using the Environmental Impact of the Average Solvent. J. Solut. Chem. 2022, 51, 838–849. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef]
- Locher, K.P. Review. Structure and mechanism of ATP-binding cassette transporters. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, O.; Turpeinen, M.; Hakkola, J.; Honkakoski, P.; Hukkanen, J.; Raunio, H. Inhibition and induction of human cytochrome P450 enzymes: Current status. Arch. Toxicol. 2008, 82, 667–715. [Google Scholar] [CrossRef]
- Du, L.; Li, M.; You, Q. The interactions between hERG potassium channel and blockers. Curr. Top. Med. Chem. 2009, 9, 330–338. [Google Scholar] [CrossRef]
- Lee, W.M. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003, 349, 474–485. [Google Scholar] [CrossRef]
- Goda, K.; Kobayashi, A.; Takahashi, A.; Takahashi, T.; Saito, K.; Maekawa, K.; Saito, Y.; Sugai, S. Evaluation of the Potential Risk of Drugs to Induce Hepatotoxicity in Human—Relationships between Hepatic Steatosis Observed in Non-Clinical Toxicity Study and Hepatotoxicity in Humans. Int. J. Mol. Sci. 2017, 18, 810. [Google Scholar] [CrossRef]
- Brawley, O.W.; Boyle, P. Avoiding population exposure to carcinogens from chemotherapy. J. Clin. Oncol. 2015, 33, e12582. [Google Scholar] [CrossRef]
- Weisburger, J.H.; Griswold, D.P.; Prejean, J.D.; Casey, A.E.; Wood, H.B.; Weisburger, E.K. The carcinogenic properties of some of the principal drugs used in clinical cancer chemotherapy. In The Ambivalence of Cytostatic Therapy; Springer: Berlin/Heidelberg, Germany, 1975; Volume 52, pp. 1–17. [Google Scholar]
- Shortt, J.; Hsu, A.K.; Martin, B.P.; Doggett, K.; Matthews, G.M.; Doyle, M.A.; Ellul, J.; Jockel, T.E.; Andrews, D.M.; Hogg, S.J.; et al. The Drug Vehicle and Solvent N-Methylpyrrolidone Is an Immunomodulator and Antimyeloma Compound. Cell Rep. 2014, 7, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Moshikur, R.M.; Chowdhury, M.R.; Wakabayashi, R.; Tahara, Y.; Kamiya, N.; Moniruzzaman, M.; Goto, M. Ionic liquids with N-methyl-2-pyrrolidonium cation as an enhancer for topical drug delivery: Synthesis, characterization, and skin-penetration evaluation. J. Mol. Liq. 2020, 299, 112166. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | UNIT | 1 | 2 | 3 |
|---|---|---|---|---|
| LogS | log mol/dm3 | −4.831 | −4.83 | −4.806 |
| LogD | log mol/dm3 | 2.661 | 2.021 | 2.122 |
| LogP | log mol/dm3 | 3.15 | 2.29 | 2.635 |
| TPSA | Å2 | 113.7 | 139.72 | 156.84 |
| MW | g/mol | 378.06 | 325.08 | 355.06 |
| Pgp-inh | - | 0.007 | 0.002 | 0.007 |
| Pgp-sub | - | 0.016 | 0.635 | 0.01 |
| HIA | - | 0.012 | 0.027 | 0.043 |
| F(30%) | - | 0.001 | 0.002 | 0 |
| Caco-2 | log cm/s | −5.011 | −5.585 | −5.349 |
| MDCK | cm/s | 4.80 × 10−5 | 2.27 × 10−5 | 0.000279072 |
| Enzyme | Probability | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| CYP1A2-inh | 0.831 | 0.739 | 0.555 |
| CYP1A2-sub | 0.684 | 0.1 | 0.104 |
| CYP2C19-inh | 0.489 | 0.211 | 0.265 |
| CYP2C19-sub | 0.096 | 0.062 | 0.062 |
| CYP2C9-inh | 0.566 | 0.403 | 0.338 |
| CYP2C9-sub | 0.691 | 0.278 | 0.501 |
| CYP2D6-inh | 0.02 | 0.002 | 0.011 |
| CYP2D6-sub | 0.136 | 0.15 | 0.133 |
| CYP3A4-inh | 0.392 | 0.708 | 0.74 |
| CYP3A4-sub | 0.154 | 0.096 | 0.069 |
| Property | Probability | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| hERG | 0.109 | 0.205 | 0.514 |
| H-HT | 0.611 | 0.328 | 0.561 |
| ROA | 0.28 | 0.117 | 0.105 |
| SkinSen | 0.23 | 0.508 | 0.544 |
| Carcinogenicity | 0.584 | 0.767 | 0.793 |
| EC | 0.003 | 0.003 | 0.003 |
| EI | 0.033 | 0.217 | 0.183 |
| Solvent | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| x | c (mg/mL) | x | c (mg/mL) | x | c (mg/mL) | |
| diglyme | 9.43 (0.90) × 10−4 | 2.52 (0.24) | 5.22 (0.10) × 10−4 | 1.19 (0.02) | 2.08 (0.07) × 10−4 | 0.52 (0.02) |
| triglyme | 4.48 (0.06) × 10−4 | 0.95 (0.01) | 2.34 (0.06) × 10−4 | 0.42 (0.01) | 1.17 (0.05) × 10−4 | 0.23 (0.01) |
| tetraglyme | 2.60 (0.17) × 10−4 | 4.62 (0.30) | 1.20 (0.02) × 10−3 | 1.79 (0.03) | 2.67 (0.09) × 10−4 | 0.43 (0.01) |
| DMP | 2.96 (0.11) × 10−4 | 0.94 (0.03) | 1.05 (0.03) × 10−4 | 0.29 (0.01) | 0.27 (0.01) × 10−4 | 0.08 (<0.01) |
| NMP | 8.06 (0.25) × 10−2 | 2.65 (0.07) × 102 | 9.80 (0.13) × 10−3 | 3.27 (0.05) × 101 | 2.44 (0.07) × 10−3 | 8.99 (0.25) |
| 4FM | 1.88 (0.04) × 10−3 | 7.22 (0.16) | 1.06 (0.04) × 10−3 | 3.47 (0.12) | 2.65 (0.11) × 10−4 | 0.94 (0.04) |
| γ-heptalactone | 5.92 (0.08) × 10−4 | 1.78 (0.02) | 4.25 (0.06) × 10−4 | 1.09 (0.02) | 1.59 (0.04) × 10−4 | 0.44 (0.01) |
| γ-nonanoic lactone | 4.81 (0.06) × 10−4 | 1.16 (0.01) | 3.46 (0.07) × 10−4 | 0.71 (0.01) | 1.26 (0.05) × 10−4 | 0.28 (0.01) |
| ε-caprolactone | 4.42 (0.09) × 10−4 | 1.60 (0.03) | 3.25 (0.09) × 10−4 | 1.01 (0.03) | 1.02 (0.02) × 10−4 | 0.34 (0.01) |
| cyrene | 4.37 (0.09) × 10−4 | 1.62 (0.03) | 2.36 (0.04) × 10−4 | 0.75 (0.01) | 0.83 (0.01) × 10−4 | 0.29 (<0.01) |
| ethyl lactate | 3.75 (0.10) × 10−4 | 1.25 (0.03) | 1.95 (0.04) × 10−4 | 0.56 (0.01) | 0.57 (0.01) × 10−4 | 0.18 (<0.01) |
| DMSO | 7.70 (0.11) × 10−2 | 3.27 (0.04) × 102 | 4.63 (0.14) × 10−3 | 2.14 (0.07) × 101 | 6.12 (0.10) × 10−4 | 3.09 (0.05) |
| DMF | 2.66 (0.55) × 10−2 | 1.19 (0.02) × 102 | 3.66 (0.06) × 10−3 | 1.53 (0.02) × 101 | 3.69 (0.05) × 10−4 | 1.69 (0.02) |
| dioxane | 9.21 (0.21) × 10−4 | 4.17 (0.09) | 2.49 (0.05) × 10−4 | 0.96 (0.02) | 0.74 (0.01) × 10−4 | 0.31 (0.01) |
| acetone | 8.38 (0.16) × 10−4 | 4.39 (0.08) | 2.00 (0.09 × 10−4 | 0.90 (0.04) | 0.43 (0.01) × 10−4 | 0.21 (0.01) |
| acetonitrile | 2.76 (0.12) × 10−4 | 2.02 (0.09) | 0.96 (0.03) × 10−4 | 0.61 (0.02) | 0.25 (<0.01) × 10−4 | 0.17 (<0.01) |
| methanol | 1.60 (0.05) × 10−4 | 1.51 (0.05) | 0.39 (0.01) × 10−4 | 0.31 (0.01) | 0.07 (<0.01) × 10−4 | 0.06 (<0.01) |
| ethanol | 1.26 (0.04) × 10−4 | 0.82 (0.03) | 0.26 (0.01) × 10−4 | 0.15 (0.01) | 0.05 (<0.01) × 10−4 | 0.03 (<0.01) |
| 1-propanol | 1.19 (0.05) × 10−4 | 0.61 (0.03) | 0.23 (0.01) × 10−4 | 0.10 (<0.01) | 0.04 (<0.01) × 10−4 | 0.02 (<0.01) |
| 1-butanol | 1.11 (0.04) × 10−4 | 0.47 (0.02) | 0.21 (0.01) × 10−4 | 0.07 (<0.01) | 0.04 (<0.01) × 10−4 | 0.02 (<0.01) |
| x*DMSO | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| x | c (mg/mL) | x | c (mg/mL) | x | c (mg/mL) | |
| 0.1 | 5.27 (0.10) × 10−3 | 8.80 (0.16) × 101 | 2.11 (0.02) × 10−4 | 3.22 (0.02) | 0.52 (<0.01) × 10−4 | 0.86 (0.01) |
| 0.2 | 1.47 (0.02) × 10−2 | 1.79 (0.02) × 102 | 5.76 (0.06) × 10−4 | 6.95 (0.07) | 1.21 (0.01) × 10−4 | 1.58 (0.01) |
| 0.3 | 2.25 (0.03) × 10−2 | 2.19 (0.02) × 102 | 1.08 (0.01) × 10−3 | 1.08 (0.01) × 101 | 2.29 (0.01) × 10−4 | 2.48 (0.01) |
| 0.4 | 3.25 (0.05) × 10−2 | 2.61 (0.03) × 102 | 1.69 (0.02) × 10−3 | 1.43 (0.01) × 101 | 3.57 (0.03) × 10−4 | 3.30 (0.03) |
| 0.5 | 4.46 (0.08) × 10−2 | 3.05 (0.04) × 102 | 2.46 (0.02) × 10−3 | 1.84 (0.01) × 101 | 4.59 (0.04) × 10−4 | 3.75 (0.03) |
| 0.6 | 5.69 (0.15) × 10−2 | 3.36 (0.06) × 102 | 3.18 (0.03) × 10−3 | 2.10 (0.02) × 101 | 5.45 (0.03) × 10−4 | 3.95 (0.02) |
| 0.7 | 6.72 (0.14) × 10−2 | 3.56 (0.06) × 102 | 4.01 (0.05) × 10−3 | 2.41 (0.03) × 101 | 6.09 (0.03) × 10−4 | 4.01 (0.02) |
| 0.8 | 7.60 (0.10) × 10−2 | 3.63 (0.03) × 102 | 4.68 (0.03) × 10−3 | 2.54 (0.02) × 101 | 6.47 (0.04) × 10−4 | 3.85 (0.02) |
| 0.9 | 8.00 (0.14) × 10−2 | 3.55 (0.05) × 102 | 4.86 (0.08) × 10−3 | 2.41 (0.04) × 101 | 6.51 (0.04) × 10−4 | 3.54 (0.02) |
| 1.0 | 7.70 (0.11) × 10−2 | 3.27 (0.04) × 102 | 4.63 (0.14) × 10−3 | 2.14 (0.07) × 101 | 6.12 (0.07) × 10−4 | 3.09 (0.03) |
| x*NMP | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| x | c (mg/mL) | x | c (mg/mL) | x | c (mg/mL) | |
| 0.1 | 3.86 (0.05) × 10−3 | 5.66 (0.07) × 101 | 4.43 (0.05) × 10−4 | 5.75 (0.06) | 1.12 (0.01) × 10−4 | 1.58 (0.02) |
| 0.2 | 1.05 (0.01) × 10−2 | 1.11 (0.01) × 102 | 1.23 (0.01) × 10−3 | 1.21 (0.01) × 101 | 3.05 (0.03) × 10−4 | 3.29 (0.03) |
| 0.3 | 1.98 (0.04) × 10−2 | 1.61 (0.03) × 102 | 2.32 (0.03) × 10−3 | 1.82 (0.02) × 101 | 5.74 (0.10) × 10−4 | 4.94 (0.08) |
| 0.4 | 3.09 (0.04) × 10−2 | 2.05 (0.02) × 102 | 3.53 (0.04) × 10−3 | 2.33 (0.02) × 101 | 8.92 (0.10) × 10−4 | 6.49 (0.07) |
| 0.5 | 5.43 (0.07) × 10−2 | 2.48 (3.03) × 102 | 5.24 (0.06) × 10−3 | 2.95 (0.03) × 101 | 1.24 (0.01) × 10−3 | 7.72 (0.07) |
| 0.6 | 5.77 (0.07) × 10−2 | 2.72 (0.03) × 102 | 6.71 (0.08) × 10−3 | 3.30 (0.04) × 101 | 1.60 (0.02) × 10−3 | 8.71 (0.11) |
| 0.7 | 6.87 (0.11) × 10−2 | 2.85 (0.03) × 102 | 7.95 (0.08) × 10−3 | 3.45 (0.04) × 101 | 1.99 (0.02) × 10−3 | 9.56 (0.10) |
| 0.8 | 7.76 (0.08) × 10−2 | 2.94 (0.03) × 102 | 8.96 (0.09) × 10−3 | 3.59 (0.04) × 101 | 2.29 (0.02) × 10−3 | 1.02 (0.01) × 101 |
| 0.9 | 7.96 (0.10) × 10−2 | 2.82 (0.03) × 102 | 9.53 (0.11) × 10−3 | 3.49 (0.04) × 101 | 2.40 (0.03) × 10−3 | 9.71 (0.12) |
| 1.0 | 8.06 (0.25) × 10−2 | 2.65 (0.07) × 102 | 9.80 (0.13) × 10−3 | 3.27 (0.04) × 101 | 2.44 (0.06) × 10−3 | 8.99 (0.25) |
| Solvent | HTPI | EI |
|---|---|---|
| DMSO | 0.1300 | 0.2600 |
| 4FM | 0.2535 | 0.5080 |
| ethanol | 0.2680 | 0.5540 |
| γ-nonanoic lactone | 0.2860 | 0.6440 |
| triglyme | 0.2910 | 0.5830 |
| acetone | 0.3260 | 0.6620 |
| diglyme | 0.3495 | 0.6990 |
| methanol | 0.3675 | 0.7420 |
| tetraglyme | 0.3355 | 0.7640 |
| ε-caprolactone | 0.4405 | 0.8830 |
| NMP | 0.4830 | 0.9730 |
| dioxane | 0.4500 | 0.9840 |
| γ-heptalactone | 0.5500 | 1.1100 |
| 24DMP | 0.5900 | 1.3100 |
| acetonitryl | 0.7650 | 1.9000 |
| 1-propanol | 1.0100 | 2.0900 |
| DMF | 0.6750 | 2.1600 |
| 1-butanol | 2.3900 | 4.8600 |
| x*DMSO | HTPI | % HTPI x1.0 DMSO | EI | x*NMP | HTPI | % HTPI x1.0 NMP | EI |
|---|---|---|---|---|---|---|---|
| 0.1 | 0.0490 | 37.7 | 0.0981 | 0.1 | 0.1890 | 39.1 | 0.3820 |
| 0.2 | 0.0725 | 55.8 | 0.1450 | 0.2 | 0.2840 | 58.8 | 0.5720 |
| 0.3 | 0.0881 | 67.8 | 0.1760 | 0.3 | 0.3420 | 70.8 | 0.6890 |
| 0.4 | 0.0993 | 76.4 | 0.1990 | 0.4 | 0.3820 | 79.1 | 0.7690 |
| 0.5 | 0.1080 | 83.1 | 0.2150 | 0.5 | 0.4100 | 84.9 | 0.8260 |
| 0.6 | 0.1140 | 87.7 | 0.2280 | 0.6 | 0.4320 | 89.4 | 0.8700 |
| 0.7 | 0.1190 | 91.5 | 0.2390 | 0.7 | 0.4490 | 93.0 | 0.9040 |
| 0.8 | 0.1240 | 95.4 | 0.2470 | 0.8 | 0.4630 | 95.9 | 0.9310 |
| 0.9 | 0.1270 | 97.7 | 0.2540 | 0.9 | 0.4740 | 98.1 | 0.9540 |
| 1.0 | 0.1300 | 100.0 | 0.2600 | 1.0 | 0.4830 | 100.0 | 0.9730 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czeleń, P.; Jeliński, T.; Skotnicka, A.; Szefler, B.; Szupryczyński, K. ADMET and Solubility Analysis of New 5-Nitroisatine-Based Inhibitors of CDK2 Enzymes. Biomedicines 2023, 11, 3019. https://doi.org/10.3390/biomedicines11113019
Czeleń P, Jeliński T, Skotnicka A, Szefler B, Szupryczyński K. ADMET and Solubility Analysis of New 5-Nitroisatine-Based Inhibitors of CDK2 Enzymes. Biomedicines. 2023; 11(11):3019. https://doi.org/10.3390/biomedicines11113019
Chicago/Turabian StyleCzeleń, Przemysław, Tomasz Jeliński, Agnieszka Skotnicka, Beata Szefler, and Kamil Szupryczyński. 2023. "ADMET and Solubility Analysis of New 5-Nitroisatine-Based Inhibitors of CDK2 Enzymes" Biomedicines 11, no. 11: 3019. https://doi.org/10.3390/biomedicines11113019
APA StyleCzeleń, P., Jeliński, T., Skotnicka, A., Szefler, B., & Szupryczyński, K. (2023). ADMET and Solubility Analysis of New 5-Nitroisatine-Based Inhibitors of CDK2 Enzymes. Biomedicines, 11(11), 3019. https://doi.org/10.3390/biomedicines11113019