
Three Diseases Mediated by Different Immunopathologic Mechanisms—ANCA-Associated Vasculitis, Anti-Glomerular Basement Membrane Disease, and Immune Complex-Mediated Glomerulonephritis—A Common Clinical and Histopathologic Picture: Rapidly Progressive Crescentic Glomerulonephritis

 ,
, 
Abstract
:1. Introduction
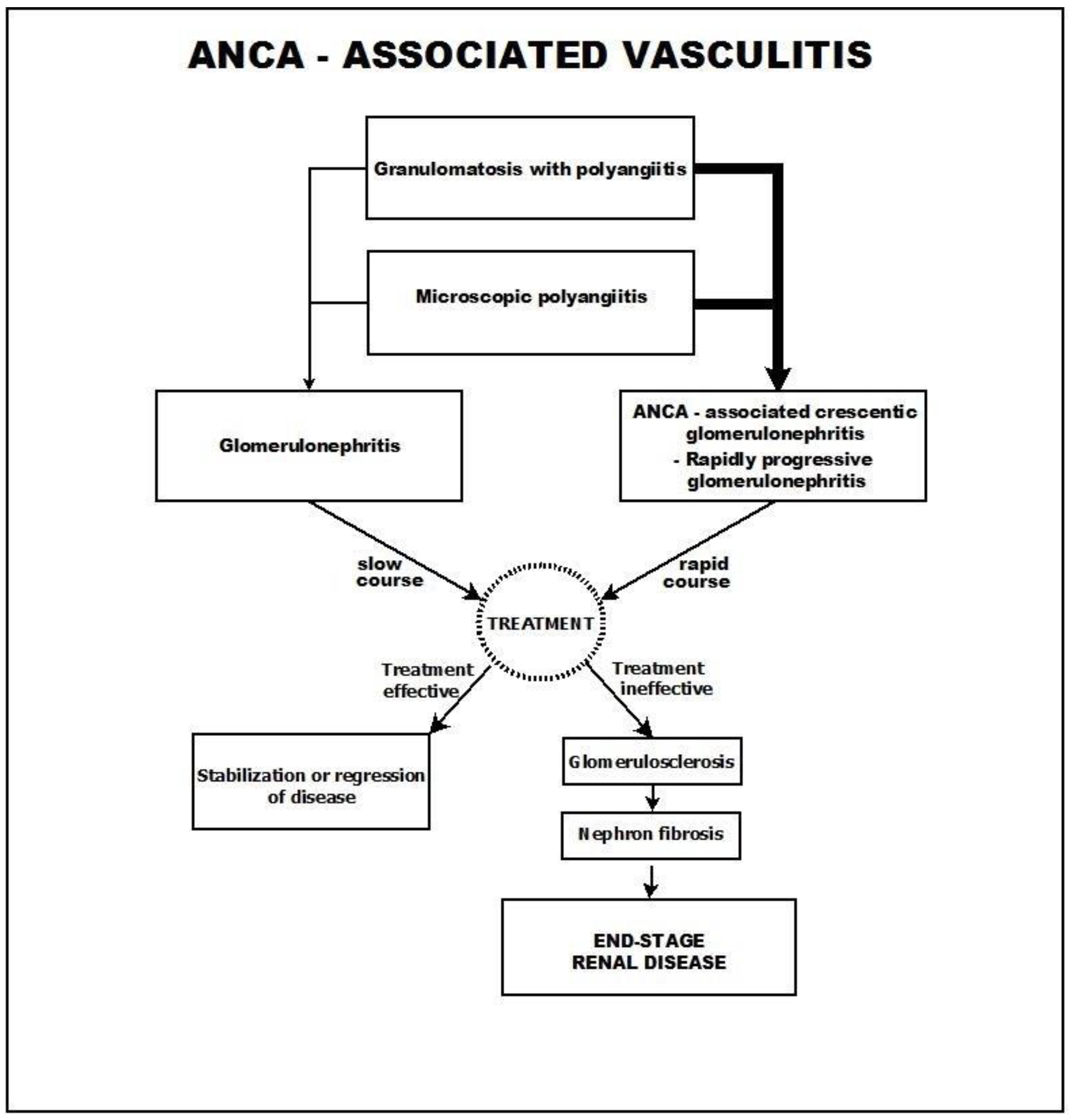
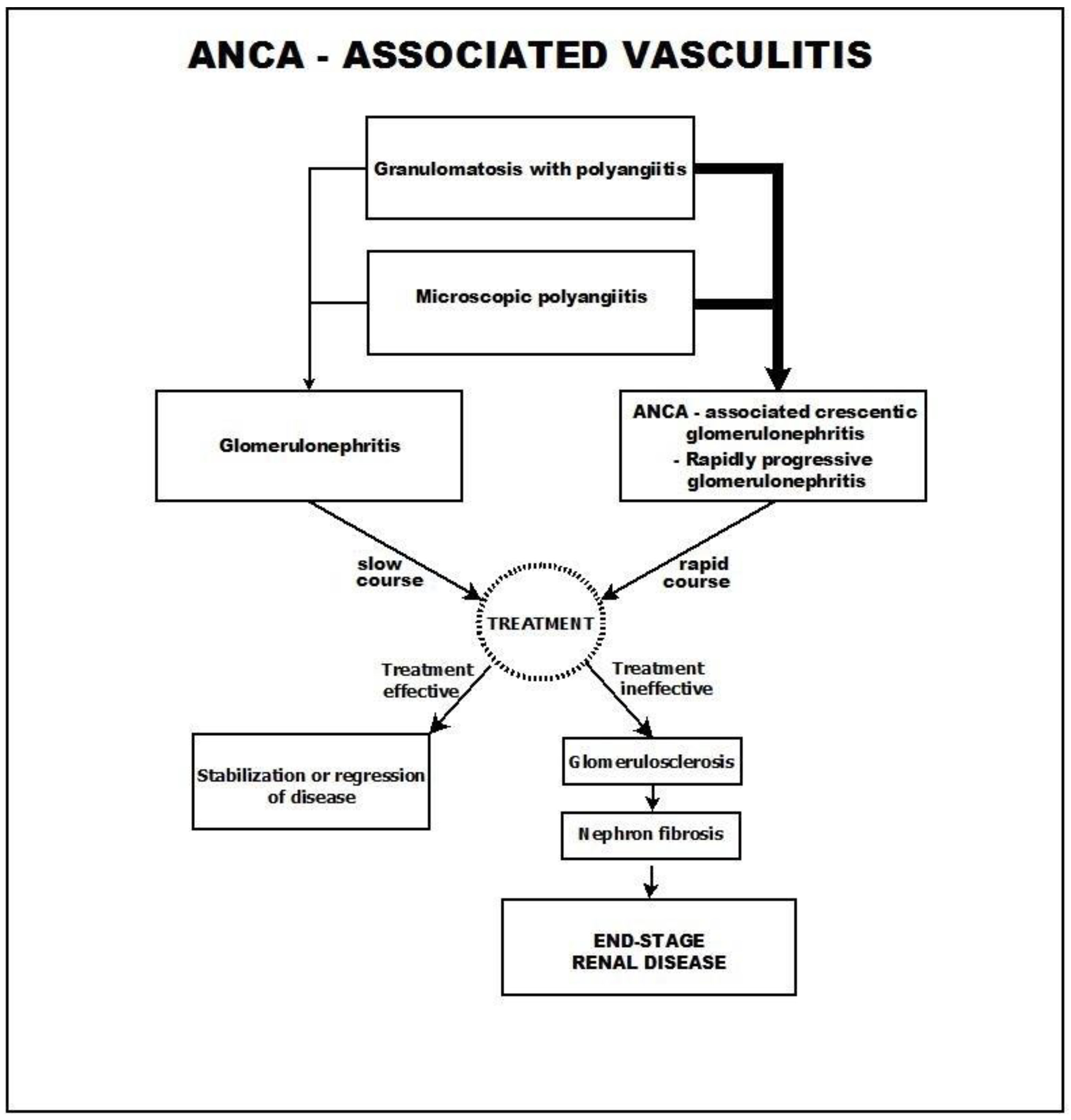
2. ANCA-Associated Vasculitis
3. Anti-Glomerular Basement Membrane (GBM) Disease
4. Immune Complex-Mediated Crescentic Glomerulonephritis
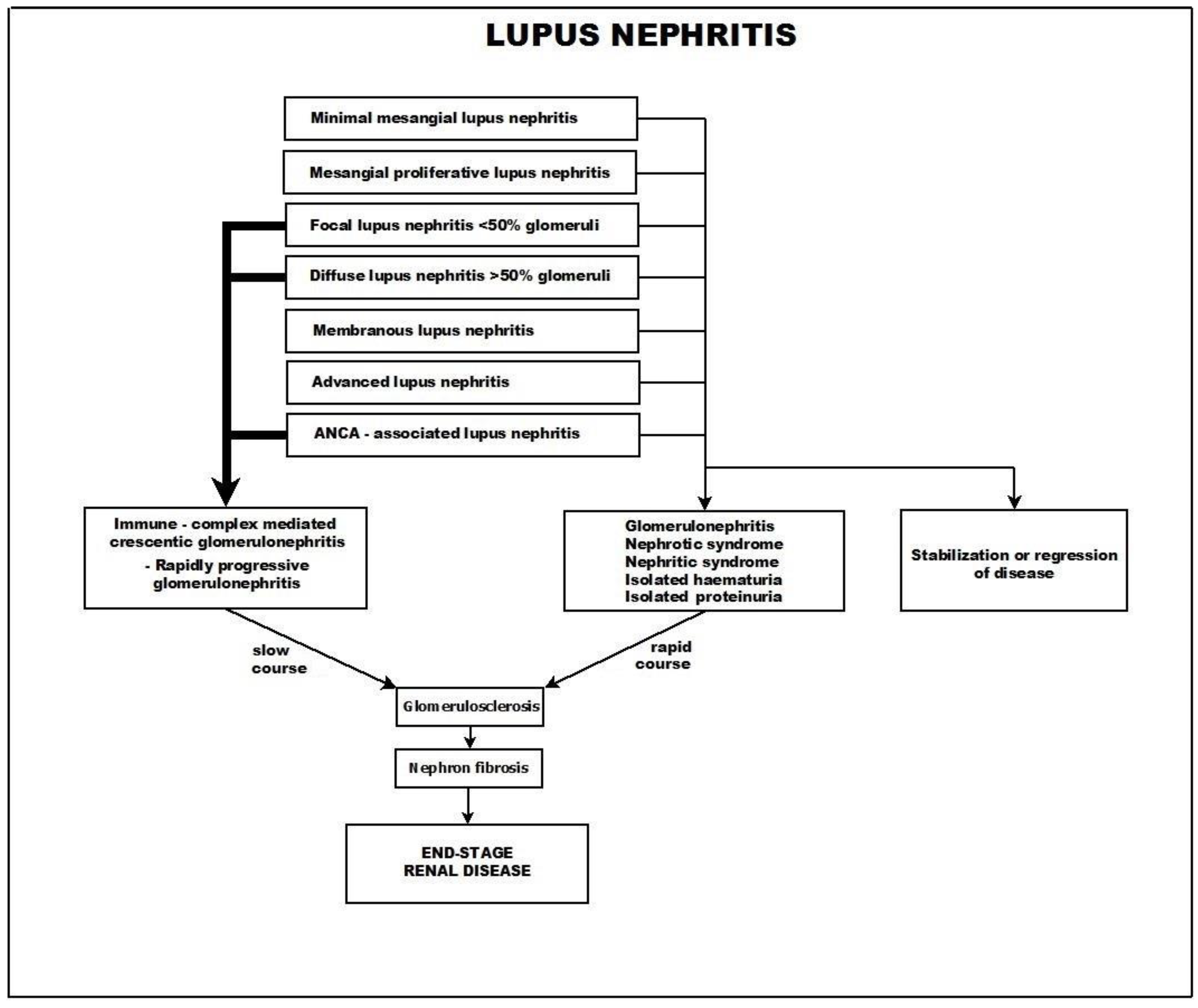
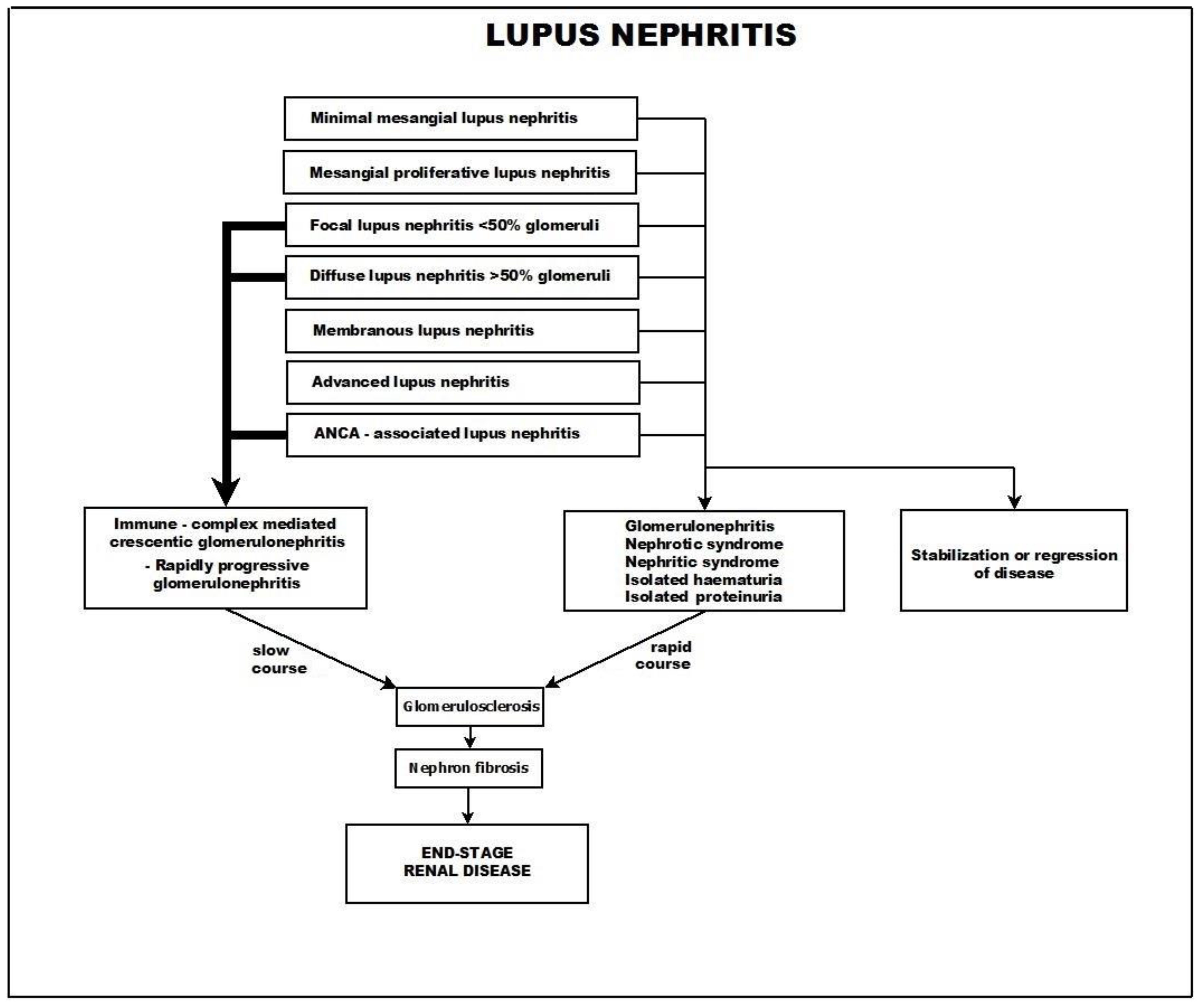
4.1. Crescentic Rapidly Progressive Glomerulonephritis in Lupus Nephritis
- Class I—minimal mesangial lupus nephritis,
- Class II—mesangial proliferative lupus nephritis,
- Class III—focal lupus nephritis (<50% of glomeruli),
- Class IV—diffuse lupus nephritis (>50% of glomeruli),
- Class V—membranous lupus nephritis, and
- Class VI—advanced sclerosing lupus nephritis.
- Reactive oxygen species are also produced within the framework of the oxidative stress that accompanies the inflammatory processes triggered by immune complexes and the complement system.
- Chemokines are released from endothelial cells. Thus, CCL5/RANTES has an important role in recruitment of inflammatory cells to areas of injury [91]. Other chemokines produced by the endothelium may also play a role in lupus nephritis, including fractalkine [92]; interleukin 8, which is an inflammatory cytokine with additional chemokine properties [93]; CXCL10, which is considered a biomarker in lupus patients [94]; and chemoattractant protein-1 [95]. Activation of mesangial cells by nucleosome-containing immune complexes produces chemokines (e.g., CCL2, CCL7, CXCL1, CXCL2, and CXCL7) that have chemotactic roles and produces neutrophil, macrophage, and T and B cell infiltration [96]. The chemokine CXCL13, which is produced during lupus nephritis, has been analyzed as a new SLE and lupus nephritis biomarker [97].
- T cells can cause disruption of the glomerular basement membrane by means of the granzymes they produce [98].
- B cells have multiple functions in lupus. One of them is the production of anti-nuclear and polyreactive autoantibodies [88].
4.2. Other Forms of Immune Complex-Mediated Crescentic Glomerulonephritis
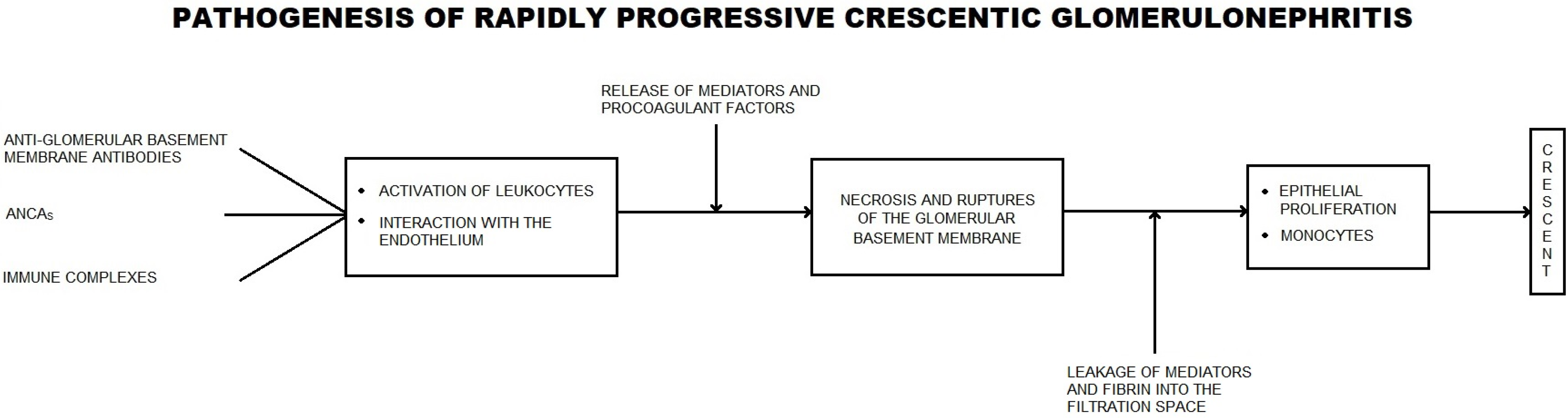
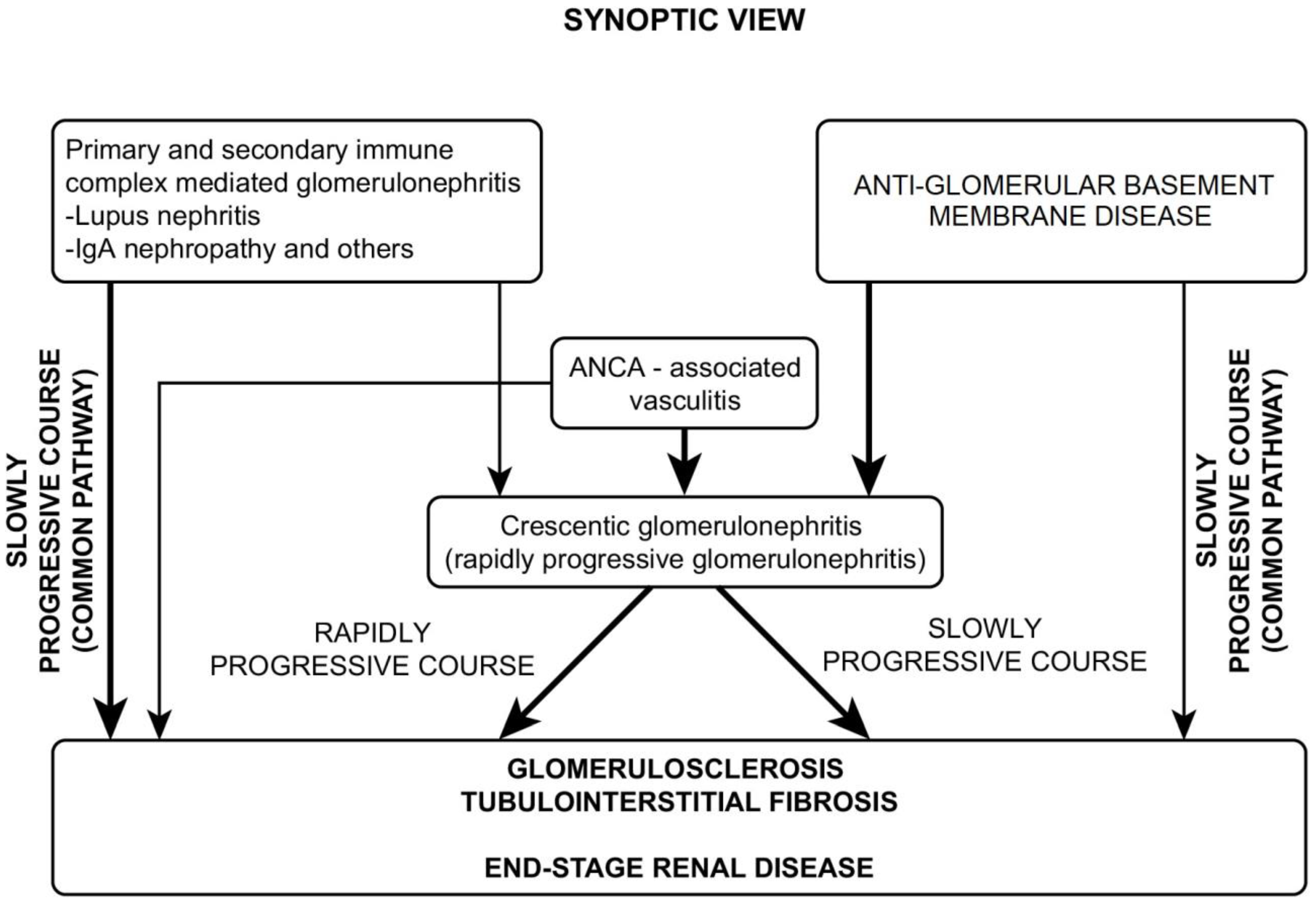
5. Crescentic Glomerulonepthritis—A Synthesis
5.1. Bowman’s Capsule Lesions
5.2. Parietal Epithelial Cells (PECs)
5.3. Renal Progenitor Cells
5.4. Podocytes
5.5. Macrophages
5.6. Dendritic Cells
5.7. T Cells
5.8. Neutrophils
5.9. Fibroblasts
6. Course and Outcome
7. Conclusions
- A common course, in which the glomerulosclerosis process advances slowly and progressively and is usually accompanied by tubulo-interstitial fibrosis.
- A different course, in which the presenting picture rapidly progresses toward crescentic glomerulonephritis.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gluhovschi, G.; Trandafirescu, V.; Solovan, C.; Lazăr, E.; Gluhovschi, C.; Petrica, L.; Bob, F.; Bozdog, G.; Gadalean, F.; Cornianu, M.; et al. Has the diversity of clinical and biological manifestations of systemic lupus erythematosus a correspondent in the diversity of immune mechanisms? Observations based on a Rowell’s syndrome case associated with arthritis and nephritis. Rom. J. Intern. Med. 2012, 50, 249–255. [Google Scholar] [PubMed]
- Jennette, J. Rapidly progressive and crescentic glomerulonephritis. Kidney Int. 2003, 63, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C. Overview of the 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Clin. Exp. Nephrol. 2013, 17, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.A.; Lane, S.E.; Bentham, G.; Scott, D.G. Epidemiology of systemic vasculitis: A ten-year study in the United Kingdom. Arthritis Rheum. 2000, 43, 414–419. [Google Scholar] [CrossRef]
- McKinley, E.F.; Willcocks, L.C.; Broecker, V.; Smith, K.G. The immunopathology of ANCA-associated vasculitis. Semin. Immunopathol. 2014, 36, 461–478. [Google Scholar] [CrossRef]
- Berden, A.E.; Ferrario, F.; Hagen, E.C.; Jayne, D.R.; Jennette, J.C.; Joh, K.; Neumann, I.; Noël, L.H.; Pusey, C.D.; Waldherr, R.; et al. Histopathologic Classification of ANCA-Associated Glomerulonephritis. J. Am. Soc. Nephrol. 2010, 21, 1628–1636. [Google Scholar] [CrossRef]
- Schreiber, A.; Kettritz, R. The neutrophil in antineutrophil cytoplasmic autoantibody-associated vasculitis. J. Leukoc. Biol. 2013, 94, 623–631. [Google Scholar] [CrossRef]
- Ryu, M.; Migliorini, A.; Miosge, N.; Gross, O.; Shankland, S.; Brinkkoetter, P.T.; Hagmann, H.; Romagnani, P.; Liapis, H.; Anders, H.J. Plasma leakage through glomerular basement membrane ruptures triggers the proliferation of parietal epithelial cells and crescent formation in non-inflammatory glomerular injury. J. Pathol. 2012, 228, 482–494. [Google Scholar] [CrossRef]
- Lan, H.Y.; Nikolic-Paterson, D.J.; Atkins, R.C. Involvement of activated periglomerular leucocytes in the rupture of Bowman’s capsule and glomerular crescent progression in experimental glomerulonephritis. Lab. Investig. 1997, 67, 743–751. [Google Scholar]
- Dringenberg, C.; Apenberg, S.; Andrassy, K. p-ANCA with myeloperoxidase antibodies and c-ANCA with proteinase 3 antibodies define a different vasculitis entity in patients with renal involvement. Adv. Exp. Med. Biol. 1993, 336, 445–447. [Google Scholar]
- Jennette, J.C.; Falk, R.J. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat. Rev. Rheumatol. 2014, 10, 463–473. [Google Scholar] [CrossRef]
- Gaudin, P.B.; Askin, B.; Falk, R.J.; Jennette, J.C. The pathologic spectrum of pulmonary lesions in patients with anti-neutrophil cytoplasmic autoantibodies specific for anti-proteinase 3 and anti-myeloperoxidase. Am. J. Clin. Pathol. 1995, 104, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Kallenberg, C.G.; Heeringa, P. Complement system activation in ANCA vasculitis: A translational success story? Mol. Immunol. 2015, 68, 535–536. [Google Scholar] [CrossRef]
- Van Daalen, E.; Ferrario, F.; Noël, L.H.; Waldherr, R.; Hagen, E.C.; Bruijn, J.A.; Bajema, I.M. Twenty-five years of RENHIS: A history of histopathological studies within EUVAS. Nephrol. Dial. Transplant. 2015, 30 (Suppl. 1), i31–i36. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Zhao, M.H.; Segelmark, M.; Hellmark, T. Natural autoantibodies to myeloperoxidase, proteinase 3, and the glomerular basement membrane are present in normal individuals. Kidney Int. 2010, 78, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J. ANCAs are also antimonocyte cytoplasmic autoantibodies. Clin. J. Am. Soc. Nephrol. 2015, 10, 4–6. [Google Scholar] [CrossRef]
- Kelsey, R. Vasculitis: Epitope specificity responsible for MPO-ANCA pathogenicity. Nat. Rev. Nephrol. 2013, 9, 310. [Google Scholar] [CrossRef]
- Kallenberg, C.G. Pathogenesis of ANCA-associated vasulitis, un update. Clin. Rev. Allergy Immunol. 2011, 41, 224–231. [Google Scholar] [CrossRef]
- Kain, R.; Tadema, H.; McKinney, E.F.; Benharkou, A.; Brandes, R.; Peschel, A.; Hubert, V.; Feenstra, T.; Sengölge, G.; Stegeman, C.; et al. High prevalence of autoantibodies to hLAMP-2 in anti-neutrophil cytoplasmic antibody-associated vasculitis. J. Am. Soc. Nephrol. 2012, 23, 556–566. [Google Scholar] [CrossRef]
- Furuta, S.; Jayne, D.R. Antineutrophyl cytoplasm antibody-associated vasculitis: Recent developments. Kidney Int. 2013, 84, 244–249. [Google Scholar] [CrossRef]
- Flint, S.M.; Savage, C.O.S. Anti–LAMP-2 Autoantibodies in ANCA-Associated Pauci-Immune Glomerulonephritis. J. Am. Soc. Nephrol. 2012, 22, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Kain, R.; Exner, M.; Brandes Ziebermayr, R.; Cunningham, D.; Alderson, C.A.; Davidovits, A.; Raab, I.; Jahn, R.; Ashour, O.; Spitzauer, S.; et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat. Med. 2008, 14, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Kain, R.; Firmin, D.; Rees, A.J. Pathogenesis of small vessel vasculitis associated with autoantibodies to neutrophil cytoplasmic antigens: New insight from animal models. Curr. Opin. Rheumatol. 2010, 22, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Van Timmeren, M.M.; Heeringa, P.; Kallenberg, C.G. Infectious triggers for vasculitis. Curr. Opin. Rheumatol. 2014, 26, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Stegeman, C.A.; Tervaert, J.W.; deJong, P.E.; Kallenberg, C.G. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. N. Engl. J. Med. 1996, 335, 16–20. [Google Scholar] [CrossRef]
- Pendergraft, W.F., 3rd; Preston, G.A.; Shah, R.R.; Tropsha, A.; Carter, C.W., Jr.; Jennette, J.C.; Falk, R.J. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat. Med. 2004, 10, 172–179. [Google Scholar] [CrossRef]
- Guilpain, P.; Mouthon, L. Antiendotelial cells autoantibodies in vasculitis-associated systemic diseases. Clin. Rev. Allergy Immunol. 2008, 35, 59–65. [Google Scholar] [CrossRef]
- Kallenberg, C.G.; Stegeman, C.A.; Abdulahad, W.H.; Heeringa, P. Pathogenesis of ANCA-associated vasculitis: New possibilities for intervention. Am. J. Kidney Dis. 2013, 62, 1167–1187. [Google Scholar] [CrossRef]
- Morgan, M.D.; Day, C.J.; Piper, K.P.; Khan, N.; Harper, L.; Moss, P.A.; Savage, C.O. Patients with Wegener’s granulomatosis demonstrate a relative deficiency and functional impairment of T regulatory cells. Immunology 2010, 130, 64–73. [Google Scholar] [CrossRef]
- Kallenberg, C.G.; Hauser, T. B-cell therapy in antineutrophil cytoplasmic antibody-associated vasculitis. Nephrol. Dial. Transplant. 2015, 30 (Suppl. 1), i119–i122. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, D.; Kurz, T.; Motamedi, A.T.; Hellmark, T.; Eriksson, P.; Segelmark, M. Increased levels of neutrophil extracellular trap in the circulation of patients with small vasculitis, but an inverse correlation to anti-neutrophil cytoplasmic antibodies during remission. Rheumatology 2015, 54, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, K.M.; Lo, C.Y.; Summers, S.A.; Elgass, K.D.; McMillan, P.J.; Longano, A.; Ford, S.L.; Gan, P.Y.; Kerr, P.G.; Kitching, A.R.; et al. Renal participation of myeloperoxidase in antineutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Kidney Int. 2015, 85, 1030–1046. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, S.M.; Ohlsson, S.; Söderberg, D.; Gunnarsson, L.; Pettersson, Å.; Segelmark, M.; Hellmark, T. Neutrophils from vasculitis patients exhibit an increased propensity for activation by anti-neutrophil cytoplasmic antibodies. Clin. Exp. Immunol. 2014, 176, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Yamada, M.; Sudo, Y.; Kojima, T.; Tomiyasu, T.; Yoshikawa, N.; Oda, T.; Yamada, M. Myeloperoxidase anti-neutrophil cytoplasmic antibody affinity is associated with the formation of neutrophil extracellular traps in the kidney and vasculitis activity in myeloperoxidase anti-neutrophil cytoplasmic antibody-associated microscopic polyangiitis. Nephrology 2016, 21, 624–629. [Google Scholar] [CrossRef]
- Tang, S.; Zang, Y.; Yin, S.W.; Gao, X.J.; Shi, W.W.; Wang, Y.; Huang, X.; Wang LZhao, J.H.; Huang, Y.J.; Shan, L.Y.; et al. Neutrophil trap formation is associated with autophagy-related signaling in ANCA-associated vasculitis. Clin. Exp. Immunol. 2015, 180, 408–418. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, X.; Cai, J.; Xie, L.; Wang, W.; Tang, S.; Yin, S.; Gao, X.; Zhang, J.; Zhao, J.; et al. Clinicopathologic Characteristics and Outcomes of Lupus Nephritis With Antineutrophil Cytoplasmic Antibody: A Retrospective Study. Medicine 2016, 95, e2580. [Google Scholar] [CrossRef]
- Alberici, F.; Martorana, D.; Vaglio, A. Genetic aspects of anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrol. Dial. Transplant. 2015, 30 (Suppl. 1), i37–i45. [Google Scholar] [CrossRef]
- Monach, P.A.; Merkel, P.A. Genetics of vasculitis. Curr. Opin. Rheumatol. 2010, 22, 157–163. [Google Scholar] [CrossRef]
- Ciavatta, D.J.; Yang, J.; Preston, G.A.; Badhwar, A.K.; Xiao, H.; Hewins, P.; Nester, C.M.; Pendergraft WF 3rd Magnuson, T.R.; Jennette, J.C.; Falk, R.J. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J. Clin. Investig. 2010, 120, 3209–3219. [Google Scholar] [CrossRef]
- Pendergraft, W.F., 3rd; Nachman, P.H. Recent pathogenetic advances in ANCA-associated vasculitis. Presse Med. 2015, 44 Pt. 2, e223–e229. [Google Scholar] [CrossRef]
- Silvarino, R.; Noboa, O.; Cervera, R. Anti-glomerular basement membrane antibodies. Isr. Med. Assoc. J. 2014, 16, 723–732. [Google Scholar]
- Berg, L. Goodpasture syndrome. Crit. Care Nurse 2005, 25, 50–58. [Google Scholar] [CrossRef]
- Hellmark, T.; Segelmark, M. Diagnosis and classification of Goodpasture’s disease (anti-GBM). J. Autoimmun. 2014, 48–49, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. Advances in the pathogenesis of Goodpasture’s disease: From epitopes to autoantibodies to effector T cells. J. Autoimmun. 2008, 31, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ooi, J.D.; Chang, J.; O’Sullivan, K.M.; Pedchenko, V.; Hudson, B.G.; Vandenbark, A.A.; Fugger, L.; Holdsworth, S.R.; Kitching, A.R. The HLA-DRB1*15:01-restricted Goodpasture’s T cell epitope induces GN. J. Am. Soc. Nephrol. 2013, 24, 419–431. [Google Scholar] [CrossRef]
- Borza, D.B.; Bondar, O.; Todd, P.; Sundaramoorthy, M.; Sado, Y.; Ninomiya, Y.; Hudson, B.G. Quaternary organization of the goodpasture autoantigen, the alpha 3(IV) collagen chain. Sequestration of two cryptic autoepitopes by intrapromoter interactions with the alpha4 and alpha5 NC1 domains. Biol. Chem. 2002, 277, 40075–40083. [Google Scholar] [CrossRef]
- Borza, D.B.; Hudson, B.G. Molecular characterization of the target antigens of anti glomerular basement membrane antibody disease. Springer Semin. Immunopathol. 2003, 24, 345–361. [Google Scholar]
- Kambham, N. Crescentic Glomerulonephritis:an update on Pauci-immune and Anti-GBM diseases. Adv. Anat. Pathol. 2012, 19, 111–124. [Google Scholar] [CrossRef]
- Lahmer, T.; Heemann, U. Anti-glomerular basement membrane antibody disease: A rare autoimmune disorder affecting the kidney and the lung. Autoimmun. Rev. 2012, 12, 169–173. [Google Scholar] [CrossRef]
- Pedchenko, V.; Bondar, O.; Fogo, A.B.; Vanacore, R.; Voziyan, P.; Kitching, A.R.; Wieslander, J.; Kashtan, C.; Borza, D.B.; Neilson, E.G.; et al. Molecular architecture of the Goodpasture autoantigen in anti-GBM nephritis. N. Engl. J. Med. 2010, 363, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.Y.; Cui, Z.; Yang, R.; Hu, S.Y.; Zhao, M.H. Antibodies against linear epitopes on the Goodpasture autoantigen and kidney injury. Clin. J. Am. Soc. Nephrol. 2012, 7, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.; Haxby, J.; Juggapah, J.K.; Evans, D.J.; Pusey, C.D. Identification of a nephritogenic immunodominant B and T cell epitope in experimental autoimmune glomerulonephritis. Clin. Exp. Immunol. 2009, 155, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Abbate, M.; Kalluri, R.; Corna, D.; Yamaguchi, N.; McCluskey, R.T.; Hudson, B.G.; Andres, G.; Zoja, C.; Remuzzi, G. Experimental Goodpasture’s syndrome in Wistar-Kyoto rats immunized with alpha3 chain of type IV collagen. Kidney Int. 1998, 54, 1550–1561. [Google Scholar] [CrossRef]
- Kalluri, R.; Melendez, E.; Rumpf, K.W.; Sattler, K.; Müller, G.A.; Strutz, F.; Neilson, E.G. Specificity of circulating and tissue-bound autoantibodies in Goodpasture syndrome. Proc. Assoc. Am. Physicians 1996, 108, 134–139. [Google Scholar]
- Bygren, P.; Wieslander, J.; Heinegard, D. Glomerulonephritis induced in sheep by immunisation with human glomerular basement membrane. Kidney Int. 1987, 31, 25–31. [Google Scholar] [CrossRef]
- Couser, W.G. Pathogenesis and treatment of glomerulonephritis-an update. J. Bras. Nefrol. 2016, 38, 107–122. [Google Scholar] [CrossRef]
- Hünemörder, S.; Treder, J.; Ahrens, S.; Schumacher, V.; Paust, H.J.; Menter, T.; Matthys, P.; Kamradt, T.; Meyer-Schwesinger, C.; Panzer, U.; et al. TH1 and TH17 cells promote crescent formation in experimental autoimmune glomerulonephritis. J. Pathol. 2015, 237, 62–71. [Google Scholar] [CrossRef]
- Hopfer, H.; Hünemörder, S.; Treder, J.; Turner, J.E.; Paust, H.J.; Meyer-Schwesinger, C.; Hopfer, U.; Sachs, M.; Peters, A.; Bucher-Kocaoglu, B.; et al. Glomerulopathy induced by immunization with a peptide derived from the goodpasture antigen α3IV-NC1. J. Immunol. 2015, 15, 3646–3655. [Google Scholar] [CrossRef]
- Kalluri, R.; Cantley, L.G.; Kerjaschki, D.; Neilson, E.G. Reactive oxygen species expose cryptic epitopes associated with autoimmune goodpasture syndrome. J. Biol. Chem. 2000, 275, 20027–20032. [Google Scholar] [CrossRef]
- Wu, J.; Hicks, J.; Borillo, J.; Glass, W.F., 2nd; Lou, Y.H. CD4(+) T cells specific to a glomerular basement membrane antigen mediate glomerulonephritis. J. Clin. Investig. 2002, 109, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Peto, P.; Salama, A.D. Update on antiglomerular basement membrane disease. Curr. Opin. Rheumatol. 2011, 23, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Hellmark, T.; Niles, J.L.; Collins, A.B.; McCluskey, R.T.; Brunmark, C. Comparison of anti-GBM antibodies in sera with or without ANCA. J. AmSoc Nephrol. 1997, 8, 376–385. [Google Scholar] [CrossRef]
- Levy, J.B.; Hammad, T.; Couthart, A.; Dougan, T.; Pusey, C.D. Clinical features and outcome of patients with both ANCA and anti—GBM antibodies. Kidney Int. 2004, 66, 1356–1540. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-N.; Cui, Z.; Wang, J.; Hu, S.-Y.; Jia, X.-Y.; Guan, Z.; Chen, M.; Xie, C.; Zhao, M.-H. Autoantibodies against linear epitopes of myeloperoxidase in anti–glomerular basement membrane disease. Clin. J. Am. Soc. Nephrol. 2016, 6, 775–784. [Google Scholar] [CrossRef]
- Serratrice, J.; Chiche, L.; Dussol, B.; Granel, B.; Daniel, L.; Jego-Desplat, S.; Disdier, P.; Swiader, L.; Berland, Y.; Weiller, P.J. Sequential development of perinuclear ANCA-associated vasculitis and anti-glomerular basement membrane glomerulonephritis. Am. J. Kidney Dis. 2004, 43, e26–e30. [Google Scholar] [CrossRef]
- Rutgers, A.; Slot, M.; vanPassen, P.; van Breda Vriesman, P.; Heeringa, P.; CohenTervaert, J.W. Coexistence of anti-glomerular basement membrane antibodies and myeloperoxidase-ANCAs in crescentic glomerulonephritis. Am. Kidney Dis. 2005, 48, 253–262. [Google Scholar] [CrossRef]
- Srivastava, A.; Rao, G.K.; Segal, P.E.; Shah, M.; Geetha, D. Characteristics and outcome of crescentic glomerulonephritis in patients with both antineutrophil cytoplasmic antibody and anti-glomerular basement membrane antibody. Clin. Rheumatol. 2013, 32, 1317–1322. [Google Scholar] [CrossRef]
- Liu, Y.; Anders, H.J. Lupus nephritis: From pathogenesis to targets for biologic treatment. Nephron Clin. Pract. 2014, 128, 224–231. [Google Scholar] [CrossRef]
- Sumethkul, V.; Chalermanyakom, P.; Changsirikulchai, S.; Ranadinahmed, P. Lupus nephritis: A challenging cause of rapidly progressive crescentic glomerulonephritis. Lupus 2009, 9, 424–428. [Google Scholar] [CrossRef]
- Schärer, K.; Krmar, R.; Querfeld, U.; Ruder, H.; Waldherr, R.; Schaefer, F. Clinical outcome of Schönlein-Henoch purpura nephritis in children. Pediatr. Nephrol. 1999, 13, 816–823. [Google Scholar] [CrossRef]
- Rodriguez, E.F.; Nasr, S.H.; Larsen, C.P.; Sethi, S.; Filder, M.E.; Cornell, L.D. Membranous nephropathy with crescents: A series of 19 cases. Am. J. Kidney Dis. 2014, 64, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Puri, K.; Hari, P.; Dinda, A.K.; Bagga, A. Etiology and outcome of crescentic glomerulonephritis. Indian. Pediatr. 2013, 50, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Wong, C.F.; Shawki, H.; Kapoor, N.; Pandya, B.K. Rapidly deteriorating renal function with membranoproliferative glomerulonephritis Type 1 associated with hepatitis C treated successfully with steroids and antiviral therapy: A case report and review of literature. Clin. Nephrol. 2008, 69, 298–301. [Google Scholar] [CrossRef]
- El-Husseini, A.A.; Sheashaa, H.A.; Sabry, A.A.; Moustafa, F.E.; Sobh, M.A. Acute postinfectious crescentic glomerulonephritis: Clinicopathologic presentation and risk factors. Int. Urol. Nephrol. 2005, 37, 603–609. [Google Scholar] [PubMed]
- Balafa, O.; Kalaitzidis, R.; Liapis, G.; Xiromeriti, S.; Zarzoulas, F.; Baltatzis, G.; Elisaf, M. Crescentic glomerulonephritis and membranous nephropathy: A rare coexistence. Int. Urol. Nephrol. 2015, 47, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Yalçinkaya, F.; Ince, E.; Tümer, N.; Ekim, M. The correlation between the clinical, laboratory and histopathological features of childhood membranoproliferative glomerulonephritis and response to treatment. Turk. J. Pediatr. 1992, 34, 135–144. [Google Scholar]
- Nasr, S.H.; Valeri, A.M.; Cornell, L.D.; Fidler, M.E.; Sethi, S.; Leunh, N.; Farvenza, F.C. Fibrillary Glomerulonephritis: A Report of 66 Cases from a Single Institution. Clin. J. Am. Soc. Nephrol. 2011, 6, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Köhler, H.; Fries, J.; Thoenes, W.; Meyer zum Büschenfelde, K.H. Rapidly progressive glomerulonephritis in IgA/IgG cryoglobulinemia. Nephron 1985, 41, 258–261. [Google Scholar] [CrossRef]
- Connolly, C.E.; BGallagher, B. Acute crescentic glomerulonephritis as a complication of a Staphylococcus aureus abscess of hip joint prosthesis. J. Clin. Pathol. 1987, 40, 1486. [Google Scholar] [CrossRef]
- Dooley, M.A. Clinical manifestations of lupus nephritis. In Lupus Nephritis, 2nd ed.; Lewis, E.J., Schwartz, M.M., Korbet, S.M., Chan, D.T.M., Eds.; Oxford University Press: Oxford, UK, 2011; pp. 1–34. [Google Scholar]
- Nasr, S.H.; D’Agati, V.D.; Park, H.R.; Sterman, P.; Goyzueta, J.D.; Dressler, R.M.; Haziett Pursell, R.N.; Caputo Markowitz, G.S. Necrotizing and crescentic lupus nephritis with antineutrophil cytoplasmic antibody seropositivity. Clin. J. Am. Soc. Nephrol. 2008, 3, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Korbet, S.M.; Schwartz, M.M. Pathology, pathogenesis and clinical features of severe lupus nephritis. In Lupus Nephritis, 2nd ed.; Lewis, E.J., Schwartz, M.M., Korbet, S.M., Chan, D.T.M., Eds.; Oxford University Press: Oxford, UK, 2011; pp. 129–167. [Google Scholar]
- Weening, J.J.; D’Agati, V.D.; Swartz, M.M.; Seshan, S.V.; Alpers, C.E.; Appel, G.B.; Balow, J.E.; Bruijn, J.A.; Cook, T.; Ferrario, F.; et al. The classification of glomerulonephritis in systemic lupus erythematosus revised. Kidney Int. 2004, 65, 521–530. [Google Scholar] [CrossRef]
- Leffler, J.; Ciacma, K.; Guilstrand, B.; Bengsston, A.A.; Martin, M.; Blom, A.M. A subset of patients with systemic lupus erythematosus fail to degrade DNA from multiple clinically relevant sources. Arthritis Res. Ther. 2015, 17, 205. [Google Scholar] [CrossRef] [PubMed]
- Nowling, T.K.; Gilkeson, G.S. Mechanism of tissue injury in lupus nephritis. Arthritis Res. Ther. 2015, 13, 250. [Google Scholar] [CrossRef] [PubMed]
- Vlahakos, D.V.; Fetser, M.H.; Adams, S.; Katz, M.; Ucci, A.A.; Barrett, H.J.; Datta, S.K.; Madaio, M.P. Anti-DNA antibodies from immune deposits at distinct glomerular and vascular sites. Kidney Int. 1992, 41, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.H. T cells and B cells in Lupus Nephritis. Semin. Nephrol. 2007, 27, 47–58. [Google Scholar] [CrossRef]
- Lech, M.; Anders, H.-J. The pathogenesis of lupus nephritis. J. Am. Soc. Nephrol. 2013, 24, 1357–1366. [Google Scholar] [CrossRef]
- Niederer, H.A.; Clatworthy, M.R.; Willcocks, L.C.; Smith, K.G. FcgammaRIIB, FcgamaRIIIB, and systemic lupus erythematosus. Ann. NY Acad. Sci. 2010, 1183, 69–88. [Google Scholar] [CrossRef]
- Lennard Richard, M.L.; Sato, S.; Suzuki, E.; Williams, S.; Nowling, T.K.; Zhang, X.K. The Fl transcription factor regulates the expression of CCL5/RANTES. J. Immunol. 2014, 193, 2661–2668. [Google Scholar] [CrossRef]
- Inoue, A.; Hasegawa, H.; Kohno, M.; Ito, M.R.; Terada, M.; Imai, T.; Yoshie, O.; Nose, M.; Fujita, S. Antagonist of fractalkine (CX3CL1) delays the initiation and ameliorates the progression of lupus nephritis in MRL/lpr mice. Arthritis Rheum. 2005, 52, 1522–1533. [Google Scholar] [CrossRef]
- Niemir, Z.I.; Stein, H.; Ciechanowicz, A.; Olejniczak, P.; Dworacki, G.; Ritz, E.; Waldherr, R.; Czekalski, S. The in situ expression of interleukin-8 in the normal human kidney and in different morphological forms of glomerulonephritis. Am. J. Kidney Dis. 2004, 43, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Marie, M.A.; Abu Khalil, R.E.; Habib, H.M. Urinary CXCL10: A marker of nephritis in lupus patients. Reumatismo. 2014, 65, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Karam, E.; Williams, S.; Watson, D.K.; Gilkeson, G.; Zhang, X.K. Fli-1 transcription factor affects glomerulonephritis development by regulating expression of monocyte chemoattractant protein-1 in endothelial cells in the kidney. Clin. Immunol. 2012, 145, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Kanapathippillai, P.; Hedberg, A.; Fenton, C.G.; Fenton, K.A. Nucleosomes contribute to increase mesangial cell chemokine expression during development of lupus nephritis. Cytokine 2013, 62, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, L.; Worthmann, K.; Haller, H.; Schiffer, M. CXCL 13 as a new biomarker of systemic lupus erythematosus and lupus nephritis-from bench to bedside ? Clin. Exp. Immunol. 2015, 179, 85–89. [Google Scholar] [CrossRef]
- Bailey, N.C.; Kelly, C.J. Nephritogenic T cells use granzyme C as a cytotoxic mediator. Eur. J. Immunol. 1997, 27, 2302–2309. [Google Scholar] [CrossRef]
- Chen, S.; Tang Zhang, Y.; Liu, Z.; Zhang, H.; Hu, W.; Liu, Z. Significance of histological crescent formation in patients with diffuse proliferative lupus nephritis. Am. J. Nephrol. 2013, 38, 445–452. [Google Scholar] [CrossRef]
- Knight, J.S.; Kaplan, M.J. Lupus neutrophils:’NET’ gain in understanding lupus pathogenesis. Curr. Opin. Rheumatol. 2012, 24, 441–450. [Google Scholar] [CrossRef]
- Yu, Y.; Su, K. Neutrophil extracellular traps and systemic lupus erythematosus. J. Clin. Cell immunol. 2013, 4, 139–146. [Google Scholar] [CrossRef]
- Bosch, X. Systemic lupus erythematosus and neutrophil. N. Engl. J. Med. 2011, 365, 758–760. [Google Scholar] [CrossRef]
- Grayson, P.; Kaplan, M.Y. At the bench neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune disease. J. Leukoc. Biol. 2015, 99, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, A.A.; Waris, S.; Lakhani, A.; Kadikoy, H.; Haque, W.; Truong, L.D. True vasculitis in lupus nephritis. Clin. Nephrol. 2010, 74, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, R.; Shah, N.; Abel, M.; Oliver, J.D., 3rd; Lewin, M. Pauci-Immune Necrotizing and Crescentic Glomerulonephritis with Membranous Lupus Nephritis, Fifteen Years after Initial Diagnosis of Secondary Membranous Nephropathy. Case Rep. Nephrol. 2015, 2015, 120762. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, V.D.; Badakere, S.S.; Bichile, L.S.; Almeida, A.F. Anti-neutrophil cytoplasmic antibodies (ANCA) in systemic lupus erythematosus: Prevalence, clinical associations and correlation with other autoantibodies. J. Assoc. Physicians India. 2004, 52, 533–537. [Google Scholar]
- Marshall, S.; Dressler, R.; D’Agati, V. Membranous lupus nephritis with antineutrophil cytoplasmic antibody-associated segmental necrotizing and crescentic glomerulonephritis. Am. J. Kidney Dis. 1997, 29, 119–124. [Google Scholar] [CrossRef]
- Tesar, V. Rare transformation in repeat renal biopsies suggests a different pathogenesis of different pathogenesis of segmental and global lesions in proliferative lupus nephritis. Nephrol. Dial. Transpl. 2013, 28, 2929–2932. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Baradaran, A.; Nasri, H.; Rafieian-Kopaei, M.; Baradaran, A.; Nasri, H. Significance of extracapillary proliferation in IgA-nephropathy patients with regard to clinical and histopathological variables. Hippokratia. 2013, 17, 258–261. [Google Scholar]
- Davin, J.C. Henoch-Schonlein purpura nephritis: Pathophysiology, treatment, and future strategy. Clin. J. Am. Soc. Nephrol. 2011, 6, 3. [Google Scholar] [CrossRef]
- Raff, A.; Hebert, T.; Pullman, J.; Coco, M. Crescentic post-streptococcal glomerulonephritis with nephrotic syndrome in the adult: Is aggressive therapy warranted? Clin. Nephrol. 2005, 63, 375–380. [Google Scholar] [CrossRef]
- Alsaad, K.; Oudah, N.; Al Ameer, A.; Fakeeh, K.; Al Jomaih, A.; Al Sayyari, A. Glomerulonephritis with crescents in children: Etiology and predictors of renal outcome. ISRN Pediatr. 2011, 2011, 507298. [Google Scholar] [CrossRef]
- Tarzi, R.M.; Cook, H.T.; Pusey, C.D. Crescentic glomerulonephritis: New aspects of pathogenesis. Semin. Nephrol. 2011, 31, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Van Roeyen, C.R.C.; Eitner, F.; Boor, P.; Boor, P.; Moeller, M.J.; Raffetseder, U.; Hanssen, L.; Bücher, E.; Villa, L.; Banas, M.C.; et al. Induction of progressive glomerulonephritis by podocyte-specific overexpression of platelet-derived growth factor-D. Kidney Int. 2011, 80, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- Tipping, P.G.; Erlich, J.H.; Apostolopoulos, J.; Mackman, N.; Loskutoff, D.; Holdsworth, S.R. Glomerular tissue factor expression in crescentic glomerulonephritis. Correlations between antigen, activity, and mRNA. Am. J. Pathol. 1995, 147, 736–748. [Google Scholar]
- Erlich, J.H.; Apostolopoulos, J.; Wun, T.C.; Kretzmer, K.K.; Holdsworth, S.R.; Tipping, P.G. Renal expression of tissue factor pathway inhibitor and evidence for a role in crescentic glomerulonephritis in rabbits. J. Clin. Investig. 1996, 98, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.M.; Moran Simpson, I.J.; Peters, D.K. Defibrination with ancrod in nephrotoxic nephritis in rabbits. Kidney Int. 1976, 5, 343–347. [Google Scholar] [CrossRef]
- Kitching, A.R.; Holdsworth, S.R.; Ploplis, V.A.; Plow, E.F.; Collen, D.; Carmeliet, P.; Tipping, P.G. Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J. Exp. Med. 1997, 185, 963–968. [Google Scholar] [CrossRef]
- Kitching, A.R.; Kong, Y.Z.; Huang, X.R.; Davenport, P.; Edgtton, K.L.; Camellet, P.; Holdworth, S.P.; Tipping, P.G. Plasminogen activator inhibitor-1 is a significant determinant of renal injury in experimental crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2003, 14, 1487–1495. [Google Scholar] [CrossRef]
- Atkins, R.C.; Nikolic-Pedersen, D.J.; Song, Q.; Lan, H.Y. Modulators of crescentic glomerulonephritis. J. Am. Soc. Nephrol. 1996, 7, 2271–2278. [Google Scholar] [CrossRef]
- Boucher, A.; Droz, D.; Adafer, E.; Noel, L.H. Relationship between the integrity of Bowman’s capsule and the composition of cellular crescents in human crescentic glomerulonephritis. Lab. Investig. 1987, 56, 526–533. [Google Scholar]
- Ward, H.H.; Romero, E.; Welford, A.; Pickett, G.; Bacalao, R.; Cattone MH2nd Ness, S.A.; Wanddinger-Ness ARotbak, T. Adult human CD133/1(+) kidney cells isolated from papilla integrate into developing kidney tubules. Biochem. Biophys. Acta. 2011, 1812, 1344–1357. [Google Scholar] [CrossRef]
- Shankland, S.J.; Anders, H.J.; Romagnani, P. Glomerular parietal epithelial cells in kidney physiology, pathology, and repair. Curr. Opin. Nephrol. Hypertens. 2013, 22, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Appel, D.; Kershaw, D.B.; Smeets, B.; Yuan, C.; Fuss, A.; Feye, B.; Elger, M.; Kriz, W.; Floege, J.; Moeller, M.J. Recruitment of podocytes from glomerular parietal epithelial cells. J. Am. Soc. Nephrol. 2009, 20, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Sagrinati, C.; Netti, G.S.; Mazzinghi, B.; Larari, E.; Liottar, F.; Frosali, F.; Ronconi, F.; Meini, C.; Gacci, M.; Squecco, R.; et al. Isolated and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J. Am. Soc. Nephrol. 2006, 17, 2443–2456. [Google Scholar] [CrossRef] [PubMed]
- Ronconi, F.; Sagrinati, C.; Angelott, M.L.; Lazzeri, E.; Mazzinghi, B.; Ballerini, L.; Parente, E.; Becherucci, F.; Gacci, M.; Carini, M.; et al. Regeneration of glomerular podocytes by human renal progenitors. J. Am. Soc. Nephrol. 2009, 20, 322–332. [Google Scholar] [CrossRef]
- Bussolati, B.; Camussi, G. New insights into renal progenitor cells and kidney disease and kidney disease by studying CD133. Adv. Exp. Med. Biol. 2013, 777, 113–123. [Google Scholar]
- Smeets, B.; Angelotti, M.; Rizo, P. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 2593–2603. [Google Scholar] [CrossRef]
- Wiese, C.; Rolletschek, A.; Kania Blyszeczuk, P.; Tarasov, K.; Tarasova, Y.; Werste, P.; Boheler, K.; Wobus, A. Nestin expression a property of multi-lineage progenitor cells? Cell Mol. Life Sci. 2004, 61, 2510–2522. [Google Scholar] [CrossRef]
- Le Hir, M.; Keller, C.; Eschman, V.; Hahnel, B.; Hosser, H.; Kriz, W. Podocyte bridges between the tuft and Bowman’s capsule an early event in experimental crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2001, 12, 2060–2071. [Google Scholar] [CrossRef]
- Moeller, M.; Soofi, A.; Hartman, I.; Le-Hir, M.; Wiggins, R.; Kriz, W.; Holzman, L. Podocytes populate cellular crescents in a murine model of inflammatory glomerulonephritis. J. Am. Soc. Nephrol. 2004, 15, 61–67. [Google Scholar] [CrossRef]
- Bariety, J.; Bruneval, P.; Meyrier, A.; Mandet, C.; Hill, G.; Jacquet, L. Podocyte involvement in human immune crescentic glomerulonephritis. Kidney Int. 2005, 68, 1109–1119. [Google Scholar] [CrossRef]
- Han, Y.; Ma, F.Y.; Tesch, G.H.; Manthey, C.L.; Nikolic-Paterson, G.J. Role of macrophages in fibrotic phase of rat crescentic glomerulonephritis. Am. J. Physiol. Renal Physiol. 2013, 304, F1043–F1053. [Google Scholar] [CrossRef]
- Evers, B.D.; Engel, D.R.; Böhner, A.M.; Tittel, A.P.; Krause, T.A.; Heuser, C.; Garbi, N.; Kastenmüller, W.; Mack, M.; Tiegs, G.; et al. CD103+ Kidney Dendritic Cells Protect against Crescentic GN by Maintaining IL-10-Producing Regulatory T Cells. J. Am. Soc. Nephrol. 2016, 27, 3368. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.A.; Campbell, E.J. The cell biology of leucocyte-mediated proteolysis. J. Leukoc. 1999, 65, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Kitching, A.R.; Holdworth, S.R.; Tipping, P.G. Crescentic glomerulonephritis-a manifestation of nephritogenic Th 1 response. Histol. Histopathol. 2000, 15, 993–1003. [Google Scholar] [PubMed]
- Odobasic, D.; Gan, P.Y.; Summers, S.A.; Semple, T.J.; Muljadi, R.S.; Iwakura, Y.; Kitching, A.R.; Holdsworth, S.R. Interleukin 17A promotes early but attenuates established disease in crescentic glomerulonephritis in mice. Am. J. Pathol. 2011, 179, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Paust, H.J.; Turner, J.E.; Riedel, J.A.; Disteldorf, E.; Peters, A.; Schmidt, T.; Krebs, C.; Velden, J.; Mittrücker, H.-W.; Steinmetz, O.M.; et al. Chemokines play a critical role in cross-regulation of Th 1 and Th17 immune responses in murine crescentic glomerulonephritis. Kidney Int. 2001, 82, 74–83. [Google Scholar]
- Ramani, K.; Pawaria, S.; Maers, K.; Gaffen, S.L.; Biswas, P.S. An essential role of interleukin-17 receptor signaling in the development of autoimmune glomerulonephritis. J. Leukoc. Biol. 2014, 96, 463–472. [Google Scholar] [CrossRef]
- Ostmann, A.; Paust, H.J.; Panzer, U.; Wegscheid, C.; Kapfer, S.; Huger, S.; Flawell, R.A.; Erhardt, A.; Tiiegs, G. Regulatory T cell-derive IL 10 ameliorated crescentic GN. J. Am. Soc. Nephrol. 2013, 24, 930–942. [Google Scholar] [CrossRef]
- Tipping, P.G.; Holdsworth, S.R. T cells in crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2006, 17, 1253–1263. [Google Scholar] [CrossRef]
- Chen, Y.X.; Chen, N. Pathogenesis of rapidly progressive glomerulonephritis. What do we learn? Contrib. Nephrol. 2013, 181, 207–215. [Google Scholar]
- Mihara, K.; Ramachandran, R.; Renaux, B.; Saifeddine, M.; Hollenberg, M.D. Neutrophil elastase and proteinase-3 trigger G protein-biased signaling through proteinase-activated receptor-1 (PAR1). J. Biol. Chem. 2013, 288, 32979–32990. [Google Scholar] [CrossRef] [PubMed]
- Wiedow, O.; Meyer-Hoffert, U. Neutrophil serine proteases: Potential key regulators of cell signalling during inflammation. J. Intern. Med. 2005, 257, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Hoffert, U. Neutrophil-derived serine proteases modulate innate immune responses. Front. Biosci. 2009, 14, 3409–3418. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fc-receptors as regulators of immunity. Adv. Immunol. 2007, 96, 179–204. [Google Scholar] [PubMed]
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Müller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef]
- Nakazawa, D.; Tomaru, U.; Ishizu, A. Possible implication of disordered neutrophil extracellular traps in the pathogenesis of MPO-ANCA-associated vasculitis. Clin. Exp. Nephrol. 2013, 17, 631–633. [Google Scholar] [CrossRef]
- Goumenos, D. Myofibroblasts and the progression of crescentic glomerulonephritis. Nephrol. Dial. Transpl. 1998, 304, 1652–1661. [Google Scholar] [CrossRef]
- Ng, Y.Y.; Fan, J.M.; Mu, W.; Nikolic-Paterson, D.J.; Yang, W.C.; Huang, T.P.; Atkins, R.C.; Lan, H.Y. Glomerular epithelial-myofibroblast transdifferentiation in the evolution of glomerular crescentic formation. Nephrol. Dial. Transpl. 1999, 14, 2860–2872. [Google Scholar] [CrossRef]
- Kriz, W.; Gretz, N.; Lemley, K.V. Progression of glomerular diseases: Is the podocyte the culprit? Kidney Int. 1998, 54, 654–697. [Google Scholar] [CrossRef]
- McAdoo, S.P.; Tanna, A.; Randone, O.; Tam, F.W.; Tarzi, R.M.; Levy, J.B.; Griffith, M.; Lightstone, L.; Cook, H.T.; Cairns, T.; et al. Necrotizing and crescentic glomerulonephritis presenting with preserved renal function in patients with underlying multisystem autoimmune disease: A retrospective cases series. Rheumatology 2015, 54, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Greenhall, G.H.; Salama, A.D. What is new in the management of rapidly progressive glomerulonephritis? Clin. Kidney J. 2015, 8, 143–150. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IMMUNE COMPLEX-MEDIATED CRESCENTIC GLOMERULONEPHRITIS | |
|---|---|
| 1. | Lupus nephritis |
| 2. | IgA nephropathy |
| 3. | IgA vasculitis |
| 4. | Hepatitis B and C secondary glomerulonephritis |
| 5. | Post-infectious glomerulonephritis |
| 6. | Membranous nephropathy |
| 7. | Membranoproliferative glomerulonephritis |
| 8. | Fibrillary nephropathy |
| 9. | Cryoglobulinemia glomerulonephritis |
| 10. | Abscesses |
| 11. | Idiopathic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gluhovschi, C.; Gadalean, F.; Velciov, S.; Nistor, M.; Petrica, L. Three Diseases Mediated by Different Immunopathologic Mechanisms—ANCA-Associated Vasculitis, Anti-Glomerular Basement Membrane Disease, and Immune Complex-Mediated Glomerulonephritis—A Common Clinical and Histopathologic Picture: Rapidly Progressive Crescentic Glomerulonephritis. Biomedicines 2023, 11, 2978. https://doi.org/10.3390/biomedicines11112978
Gluhovschi C, Gadalean F, Velciov S, Nistor M, Petrica L. Three Diseases Mediated by Different Immunopathologic Mechanisms—ANCA-Associated Vasculitis, Anti-Glomerular Basement Membrane Disease, and Immune Complex-Mediated Glomerulonephritis—A Common Clinical and Histopathologic Picture: Rapidly Progressive Crescentic Glomerulonephritis. Biomedicines. 2023; 11(11):2978. https://doi.org/10.3390/biomedicines11112978
Chicago/Turabian StyleGluhovschi, Cristina, Florica Gadalean, Silvia Velciov, Mirabela Nistor, and Ligia Petrica. 2023. "Three Diseases Mediated by Different Immunopathologic Mechanisms—ANCA-Associated Vasculitis, Anti-Glomerular Basement Membrane Disease, and Immune Complex-Mediated Glomerulonephritis—A Common Clinical and Histopathologic Picture: Rapidly Progressive Crescentic Glomerulonephritis" Biomedicines 11, no. 11: 2978. https://doi.org/10.3390/biomedicines11112978
APA StyleGluhovschi, C., Gadalean, F., Velciov, S., Nistor, M., & Petrica, L. (2023). Three Diseases Mediated by Different Immunopathologic Mechanisms—ANCA-Associated Vasculitis, Anti-Glomerular Basement Membrane Disease, and Immune Complex-Mediated Glomerulonephritis—A Common Clinical and Histopathologic Picture: Rapidly Progressive Crescentic Glomerulonephritis. Biomedicines, 11(11), 2978. https://doi.org/10.3390/biomedicines11112978