
Identification of Neurotransmission and Synaptic Biological Processes Disrupted in Autism Spectrum Disorder Using Interaction Networks and Community Detection Analysis

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of a List of Neurotransmission and Synaptic ASD Candidate Genes
2.2. Datasets Analysed in This Study
2.3. Identification of Ultra-Rare Loss-of-Function SNVs Targeting Neurotransmission and Synaptic Genes in ASD Exomic Datasets
2.4. Construction of the Protein–Protein Interaction Network Spanned by Neurotransmission and Synaptic Genes Affected in ASD Probands
2.5. Protein–Protein Interaction Network Community Detection
2.6. Protein–Protein Interaction Network Gene Validation in Independent Datasets
2.7. Brain Regional Specificity of Gene Expression of Network Communities
3. Results
3.1. Generating a List of Neurotransmission and Synaptic ASD Candidate Genes
3.2. Identification of Ultra-Rare Loss-of-Function SNVs Targeting Neurotransmission and Synaptic Genes in ASD Exomic Datasets
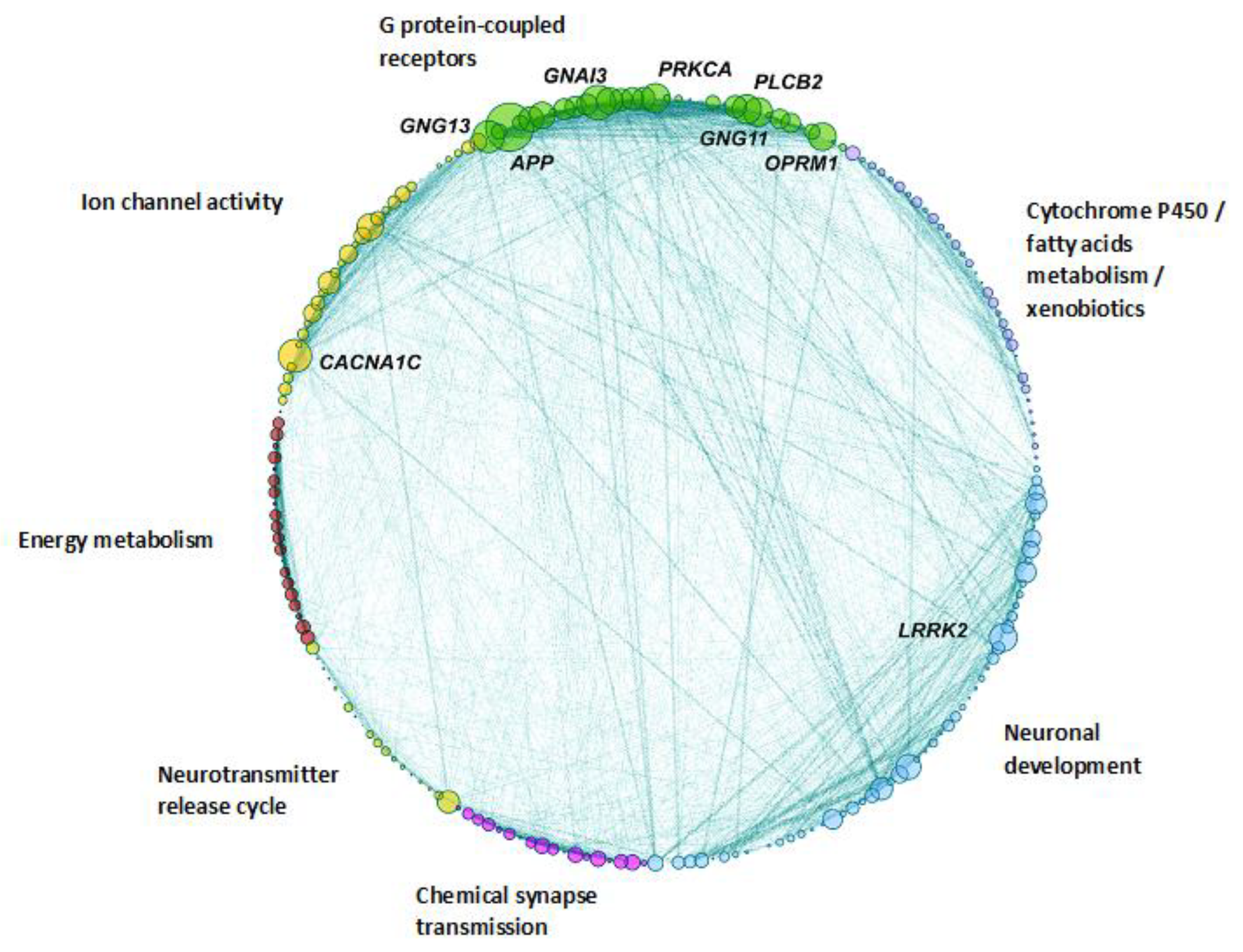
3.3. Construction of the Protein–Protein Interaction Network Spanned by NS Genes Affected in ASD Probands and Network Community Detection
3.4. Protein–Protein Interaction Network Gene Validation in Independent Datasets
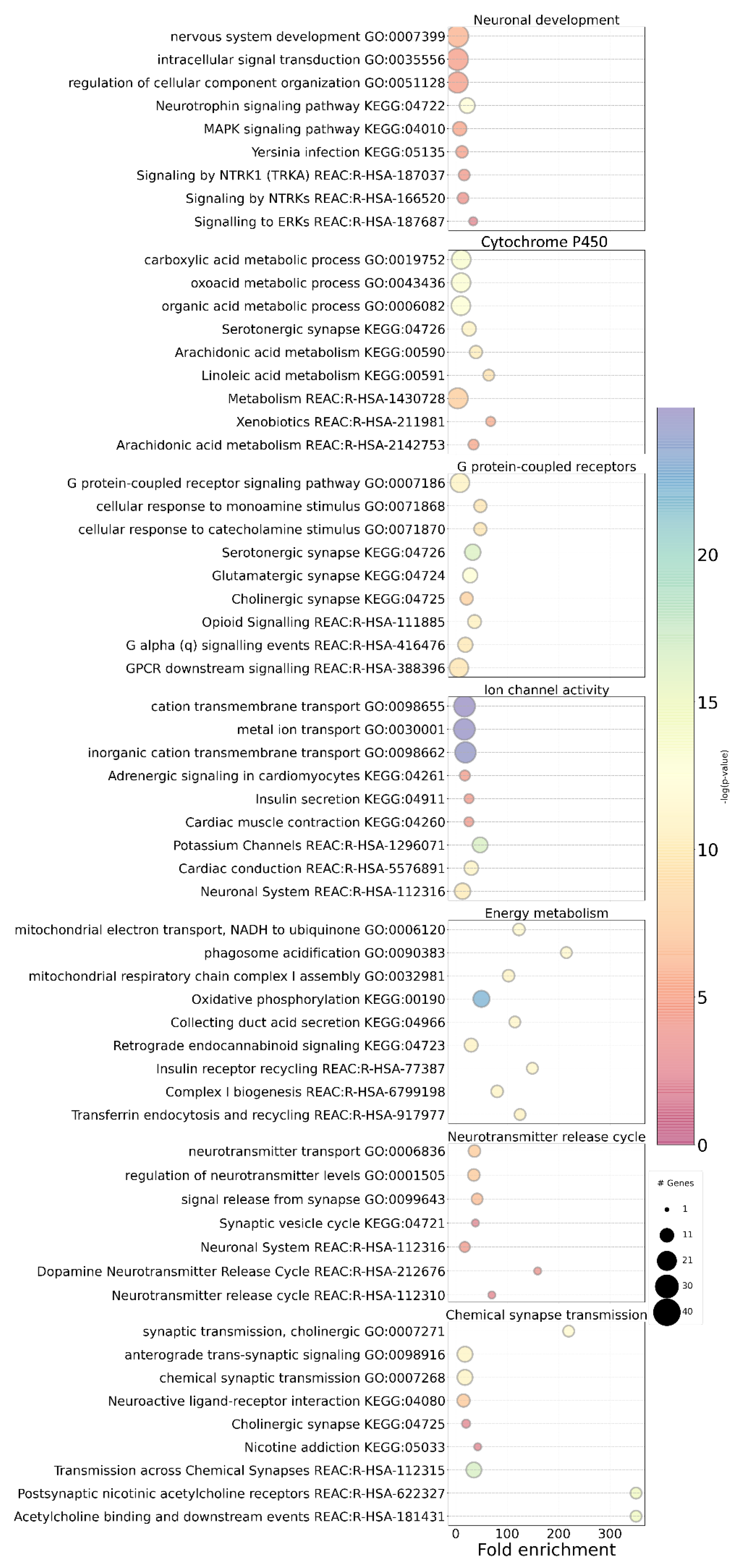
3.5. Brain Regional Specificity of Gene Expression of Network Communities
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013.
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The Familial Risk of Autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of Autism Spectrum Disorder in a UK Population-Based Twin Sample. JAMA Psychiatry 2015, 72, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of Autism Spectrum Disorders: A Meta-Analysis of Twin Studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef]
- Wigdor, E.M.; Weiner, D.J.; Grove, J.; Fu, J.M.; Thompson, W.K.; Carey, C.E.; Baya, N.; van der Merwe, C.; Walters, R.K.; Satterstrom, F.K.; et al. The female protective effect against autism spectrum disorder. Cell Genom. 2022, 2, 100134. [Google Scholar] [CrossRef]
- Genovese, A.; Butler, M.G. Clinical Assessment, Genetics, and Treatment Approaches in Autism Spectrum Disorder (ASD). Int. J. Mol. Sci. 2020, 21, E4726. [Google Scholar] [CrossRef] [PubMed]
- Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-Analysis of GWAS of over 16,000 Individuals with Autism Spectrum Disorder Highlights a Novel Locus at 10q24.32 and a Significant Overlap with Schizophrenia. Mol. Autism. 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Zhou, X.; Feliciano, P.; Shu, C.; Wang, T.; Astrovskaya, I.; Hall, J.B.; Obiajulu, J.U.; Wright, J.R.; Murali, S.C.; Xu, S.X.; et al. Integrating de Novo and Inherited Variants in 42,607 Autism Cases Identifies Mutations in New Moderate-Risk Genes. Nat. Genet. 2022, 54, 1305–1319. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.; Martiniano, H.F.M.C.M.; Vicente, A.M.; Couto, F.M. Identifying Disease Genes Using Machine Learning and Gene Functional Similarities, Assessed through Gene Ontology. PLoS ONE 2018, 13, e0208626. [Google Scholar] [CrossRef]
- Vilela, J.; Asif, M.; Marques, A.R.; Santos, J.X.; Rasga, C.; Vicente, A.; Martiniano, H. Biomedical Knowledge Graph Embeddings for Personalized Medicine: Predicting Disease-Gene Associations. Expert Syst. 2023, 40, e13181. [Google Scholar] [CrossRef]
- Asif, M.; Martiniano, H.F.M.C.; Lamurias, A.; Kausar, S.; Couto, F.M. DGH-GO: Dissecting the Genetic Heterogeneity of Complex Diseases Using Gene Ontology. BMC Bioinform. 2023, 24, 171. [Google Scholar] [CrossRef]
- Beopoulos, A.; Géa, M.; Fasano, A.; Iris, F. Autism spectrum disorders pathogenesis: Toward a comprehensive model based on neuroanatomic and neurodevelopment considerations. Front Neurosci. 2022, 16, 988735. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, Transcriptional and Chromatin Genes Disrupted in Autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, B.S.; Geschwind, D.H. Advances in Autism Genetics: On the Threshold of a New Neurobiology. Nat. Rev. Genet. 2008, 9, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Lionel, A.C.; Vaags, A.K.; Sato, D.; Gazzellone, M.J.; Mitchell, E.B.; Chen, H.Y.; Costain, G.; Walker, S.; Egger, G.; Thiruvahindrapuram, B.; et al. Rare Exonic Deletions Implicate the Synaptic Organizer Gephyrin (GPHN) in Risk for Autism, Schizophrenia and Seizures. Hum. Mol. Genet. 2013, 22, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-Analysis of SHANK Mutations in Autism Spectrum Disorders: A Gradient of Severity in Cognitive Impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [PubMed]
- Quaak, I.; Brouns, M.R.; de Bor, M.V. The Dynamics of Autism Spectrum Disorders: How Neurotoxic Compounds and Neurotransmitters Interact. Int. J. Environ. Res. Public. Health 2013, 10, 3384–3408. [Google Scholar] [CrossRef]
- Marotta, R.; Risoleo, M.C.; Messina, G.; Parisi, L.; Carotenuto, M.; Vetri, L.; Roccella, M. The Neurochemistry of Autism. Brain Sci. 2020, 10, 163. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-Linked Genes Encoding Neuroligins NLGN3 and NLGN4 Are Associated with Autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef]
- Zoghbi, H.Y. Postnatal Neurodevelopmental Disorders: Meeting at the Synapse? Science 2003, 302, 826–830. [Google Scholar] [CrossRef]
- Kaizuka, T.; Takumi, T. Postsynaptic Density Proteins and Their Involvement in Neurodevelopmental Disorders. J. Biochem. 2018, 163, 447–455. [Google Scholar] [CrossRef]
- Van Spronsen, M.; Hoogenraad, C.C. Synapse Pathology in Psychiatric and Neurologic Disease. Curr. Neurol. Neurosci. Rep. 2010, 10, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sundaram, S.K.; Sivaswamy, L.; Behen, M.E.; Makki, M.I.; Ager, J.; Janisse, J.; Chugani, H.T.; Chugani, D.C. Alterations in Frontal Lobe Tracts and Corpus Callosum in Young Children with Autism Spectrum Disorder. Cereb. Cortex 2010, 20, 2103–2113. [Google Scholar] [CrossRef] [PubMed]
- Piton, A.; Gauthier, J.; Hamdan, F.F.; Lafrenière, R.G.; Yang, Y.; Henrion, E.; Laurent, S.; Noreau, A.; Thibodeau, P.; Karemera, L.; et al. Systematic Resequencing of X-Chromosome Synaptic Genes in Autism Spectrum Disorder and Schizophrenia. Mol. Psychiatry 2011, 16, 867–880. [Google Scholar] [CrossRef]
- Uzunova, G.; Pallanti, S.; Hollander, E. Excitatory/Inhibitory Imbalance in Autism Spectrum Disorders: Implications for Interventions and Therapeutics. World J. Biol. Psychiatry 2016, 17, 174–186. [Google Scholar] [CrossRef]
- Gao, R.; Penzes, P. Common Mechanisms of Excitatory and Inhibitory Imbalance in Schizophrenia and Autism Spectrum Disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef] [PubMed]
- Toro, R.; Konyukh, M.; Delorme, R.; Leblond, C.; Chaste, P.; Fauchereau, F.; Coleman, M.; Leboyer, M.; Gillberg, C.; Bourgeron, T. Key Role for Gene Dosage and Synaptic Homeostasis in Autism Spectrum Disorders. Trends Genet. 2010, 26, 363–372. [Google Scholar] [CrossRef]
- Penzes, P.; Buonanno, A.; Passafaro, M.; Sala, C.; Sweet, R.A. Developmental Vulnerability of Synapses and Circuits Associated with Neuropsychiatric Disorders. J. Neurochem. 2013, 126, 165–182. [Google Scholar] [CrossRef]
- Hutsler, J.J.; Zhang, H. Increased Dendritic Spine Densities on Cortical Projection Neurons in Autism Spectrum Disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef]
- Chattopadhyaya, B.; Cristo, G.D. GABAergic Circuit Dysfunctions in Neurodevelopmental Disorders. Front. Psychiatry 2012, 3, 51. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic Autism Exomes Reveal a Highly Interconnected Protein Network of de Novo Mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Stessman, H.A.; Boyle, E.A.; Witherspoon, K.T.; Martin, B.; Lee, C.; Vives, L.; Baker, C.; Hiatt, J.B.; Nickerson, D.A.; et al. Recurrent de Novo Mutations Implicate Novel Genes Underlying Simplex Autism Risk. Nat. Commun. 2014, 5, 5595. [Google Scholar] [CrossRef]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most Genetic Risk for Autism Resides with Common Variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of Common Genetic Risk Variants for Autism Spectrum Disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef]
- Wilfert, A.B.; Turner, T.N.; Murali, S.C.; Hsieh, P.; Sulovari, A.; Wang, T.; Coe, B.P.; Guo, H.; Hoekzema, K.; Bakken, T.E.; et al. Recent Ultra-Rare Inherited Variants Implicate Novel Autism Candidate Risk Genes. Nat. Genet. 2021, 53, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Levy, D.; Allen, J.; Ye, K.; Ronemus, M.; Lee, Y.; Yamrom, B.; Wigler, M. Low Load for Disruptive Mutations in Autism Genes and Their Biased Transmission. Proc. Natl. Acad. Sci. USA 2015, 112, E5600–E5607. [Google Scholar] [CrossRef]
- Krumm, N.; Turner, T.N.; Baker, C.; Vives, L.; Mohajeri, K.; Witherspoon, K.; Raja, A.; Coe, B.P.; Stessman, H.A.; He, Z.-X.; et al. Excess of Rare, Inherited Truncating Mutations in Autism. Nat. Genet. 2015, 47, 582–588. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO: Online Access to Ontology and Annotation Data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward Understanding the Origin and Evolution of Cellular Organisms. Protein Sci. Publ. Protein Soc. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Pirooznia, M.; Wang, T.; Avramopoulos, D.; Valle, D.; Thomas, G.; Huganir, R.L.; Goes, F.S.; Potash, J.B.; Zandi, P.P. SynaptomeDB: An Ontology-Based Knowledgebase for Synaptic Genes. Bioinformatics 2012, 28, 897–899. [Google Scholar] [CrossRef] [PubMed]
- Von Eichborn, J.; Dunkel, M.; Gohlke, B.O.; Preissner, S.C.; Hoffmann, M.F.; Bauer, J.M.J.; Armstrong, J.D.; Schaefer, M.H.; Andrade-Navarro, M.A.; Le Novere, N.; et al. SynSysNet: Integration of Experimental Data on Synaptic Protein-Protein Interactions with Drug-Target Relations. Nucleic Acids Res. 2013, 41, D834–D840. [Google Scholar] [CrossRef]
- Banerjee-Basu, S.; Packer, A. SFARI Gene: An Evolving Database for the Autism Research Community. Dis. Model. Mech. 2010, 3, 133–135. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Daly, M.J.; Devlin, B.; Lehner, T.; Roeder, K.; State, M.W. The Autism Sequencing Consortium: Large Scale, High Throughput Sequencing in Autism Spectrum Disorders. Neuron 2012, 76, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.; Tammimies, K.; Pellecchia, G.; Alipanahi, B.; Hu, P.; Wang, Z.; Pinto, D.; Lau, L.; Nalpathamkalam, T.; Marshall, C.R.; et al. Brain-Expressed Exons under Purifying Selection Are Enriched for de Novo Mutations in Autism Spectrum Disorder. Nat. Genet. 2014, 46, 742–747. [Google Scholar] [CrossRef]
- Alonso-Gonzalez, A.; Rodriguez-Fontenla, C.; Carracedo, A. De Novo Mutations (DNMs) in Autism Spectrum Disorder (ASD): Pathway and Network Analysis. Front. Genet. 2018, 9, 406. [Google Scholar] [CrossRef]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional Impact of Global Rare Copy Number Variation in Autism Spectrum Disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of Genes and Cellular Pathways Dysregulated in Autism Spectrum Disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef]
- Autism Genome Project Consortium; Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.-Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; et al. Mapping Autism Risk Loci Using Genetic Linkage and Chromosomal Rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar] [CrossRef]
- Anney, R.; Klei, L.; Pinto, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; Sykes, N.; Pagnamenta, A.T.; et al. A Genome-Wide Scan for Common Alleles Affecting Risk for Autism. Hum. Mol. Genet. 2010, 19, 4072–4082. [Google Scholar] [CrossRef]
- Anney, R.; Klei, L.; Pinto, D.; Almeida, J.; Bacchelli, E.; Baird, G.; Bolshakova, N.; Bölte, S.; Bolton, P.F.; Bourgeron, T.; et al. Individual Common Variants Exert Weak Effects on the Risk for Autism Spectrum Disorders. Hum. Mol. Genet. 2012, 21, 4781–4792. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing Well-Connected Communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef]
- Rossetti, G.; Milli, L.; Cazabet, R. CDLIB: A Python Library to Extract, Compare and Evaluate Communities from Complex Networks. Appl. Netw. Sci. 2019, 4, 52. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A Web Server for Functional Enrichment Analysis and Conversions of Gene Lists (2019 Update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC Browser: Displaying Reference Data Information from over 60,000 Exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef]
- Bastian, F.B.; Roux, J.; Niknejad, A.; Comte, A.; Fonseca Costa, S.S.; de Farias, T.M.; Moretti, S.; Parmentier, G.; de Laval, V.R.; Rosikiewicz, M.; et al. The Bgee Suite: Integrated Curated Expression Atlas and Comparative Transcriptomics in Animals. Nucleic Acids Res. 2021, 49, D831–D847. [Google Scholar] [CrossRef]
- Haendel, M.A.; Balhoff, J.P.; Bastian, F.B.; Blackburn, D.C.; Blake, J.A.; Bradford, Y.; Comte, A.; Dahdul, W.M.; Dececchi, T.A.; Druzinsky, R.E.; et al. Unification of Multi-Species Vertebrate Anatomy Ontologies for Comparative Biology in Uberon. J. Biomed. Semant. 2014, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Waskom, M. Seaborn: Statistical Data Visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Vilela, J.; Martiniano, H.; Marques, A.R.; Santos, J.X.; Rasga, C.; Oliveira, G.; Vicente, A.M. Disease Similarity Network Analysis of Autism Spectrum Disorder and Comorbid Brain Disorders. Front. Mol. Neurosci. 2022, 15, 932305. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Merico, D.; Thiruvahindrapuram, B.; Wei, J.; Lionel, A.C.; Sato, D.; Rickaby, J.; Lu, C.; Szatmari, P.; Roberts, W.; et al. A Discovery Resource of Rare Copy Number Variations in Individuals with Autism Spectrum Disorder. G3 Genes Genomes Genet. 2012, 2, 1665–1685. [Google Scholar] [CrossRef]
- Yuan, H.; Dougherty, J.D. Investigation of Maternal Genotype Effects in Autism by Genome-Wide Association. Autism Res. Off. J. Int. Soc. Autism Res. 2014, 7, 245–253. [Google Scholar] [CrossRef]
- Levy, I.; Horvath, A.; Azevedo, M.; de Alexandre, R.B.; Stratakis, C.A. Phosphodiesterase Function and Endocrine Cells: Links to Human Disease and Roles in Tumor Development and Treatment. Curr. Opin. Pharmacol. 2011, 11, 689–697. [Google Scholar] [CrossRef]
- Qin, W.; Zhou, A.; Zuo, X.; Jia, L.; Li, F.; Wang, Q.; Li, Y.; Wei, Y.; Jin, H.; Cruchaga, C.; et al. Exome Sequencing Revealed PDE11A as a Novel Candidate Gene for Early-Onset Alzheimer’s Disease. Hum. Mol. Genet. 2021, 30, 811–822. [Google Scholar] [CrossRef]
- Hegde, S.; Ji, H.; Oliver, D.; Patel, N.S.; Poupore, N.; Shtutman, M.; Kelly, M.P. PDE11A Regulates Social Behaviors and Is a Key Mechanism by Which Social Experience Sculpts the Brain. Neuroscience 2016, 335, 151–169. [Google Scholar] [CrossRef]
- Luo, H.-R.; Wu, G.-S.; Dong, C.; Arcos-Burgos, M.; Ribeiro, L.; Licinio, J.; Wong, M.-L. Association of PDE11A Global Haplotype with Major Depression and Antidepressant Drug Response. Neuropsychiatr. Dis. Treat. 2009, 5, 163–170. [Google Scholar] [CrossRef]
- Mertens, J.; Wang, Q.-W.; Kim, Y.; Yu, D.X.; Pham, S.; Yang, B.; Zheng, Y.; Diffenderfer, K.E.; Zhang, J.; Soltani, S.; et al. Differential Responses to Lithium in Hyperexcitable Neurons from Patients with Bipolar Disorder. Nature 2015, 527, 95–99. [Google Scholar] [CrossRef]
- Fukuda, M.; Mikoshiba, K. Synaptotagmin-like Protein 1-3: A Novel Family of C-Terminal-Type Tandem C2 Proteins. Biochem. Biophys. Res. Commun. 2001, 281, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Yang, L.; Liu, K.; Ji, X.; Tang, C.; Li, W.; Ma, L.; Mei, Y.; Peng, T.; Feng, B.; et al. Transcriptional Networks Identify Synaptotagmin-like 3 as a Regulator of Cortical Neuronal Migration during Early Neurodevelopment. Cell Rep. 2021, 34, 108802. [Google Scholar] [CrossRef]
- Prokopenko, D.; Morgan, S.L.; Mullin, K.; Hofmann, O.; Chapman, B.; Kirchner, R.; Alzheimer’s Disease Neuroimaging Initiative (ADNI); Amberkar, S.; Wohlers, I.; Lange, C.; et al. Whole-Genome Sequencing Reveals New Alzheimer’s Disease-Associated Rare Variants in Loci Related to Synaptic Function and Neuronal Development. Alzheimers Dement. J. Alzheimers Assoc. 2021, 17, 1509–1527. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De Novo Mutations Revealed by Whole-Exome Sequencing Are Strongly Associated with Autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- Veeramah, K.R.; O’Brien, J.E.; Meisler, M.H.; Cheng, X.; Dib-Hajj, S.D.; Waxman, S.G.; Talwar, D.; Girirajan, S.; Eichler, E.E.; Restifo, L.L.; et al. De Novo Pathogenic SCN8A Mutation Identified by Whole-Genome Sequencing of a Family Quartet Affected by Infantile Epileptic Encephalopathy and SUDEP. Am. J. Hum. Genet. 2012, 90, 502–510. [Google Scholar] [CrossRef]
- Schmunk, G.; Gargus, J.J. Channelopathy Pathogenesis in Autism Spectrum Disorders. Front. Genet. 2013, 4, 222. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Frei, J.A.; Kilander, M.B.C.; Shen, W.; Blatt, G.J. A Subset of Autism-Associated Genes Regulate the Structural Stability of Neurons. Front. Cell. Neurosci. 2016, 10, 263. [Google Scholar] [CrossRef]
- Lee, E.-J.; Choi, S.Y.; Kim, E. NMDA Receptor Dysfunction in Autism Spectrum Disorders. Curr. Opin. Pharmacol. 2015, 20, 8–13. [Google Scholar] [CrossRef]
- Ferguson, C.S.; Tyndale, R.F. Cytochromes P450 in the Brain: Emerging Evidence for Biological Significance. Trends Pharmacol. Sci. 2011, 32, 708–714. [Google Scholar] [CrossRef]
- Kuban, W.; Daniel, W.A. Cytochrome P450 Expression and Regulation in the Brain. Drug Metab. Rev. 2021, 53, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Charry, L.; Nardi, L.; Methner, A.; Schmeisser, M.J. Abnormalities of Synaptic Mitochondria in Autism Spectrum Disorder and Related Neurodevelopmental Disorders. J. Mol. Med. 2021, 99, 161–178. [Google Scholar] [CrossRef]
- Smith, H.L.; Bourne, J.N.; Cao, G.; Chirillo, M.A.; Ostroff, L.E.; Watson, D.J.; Harris, K.M. Mitochondrial Support of Persistent Presynaptic Vesicle Mobilization with Age-Dependent Synaptic Growth after LTP. eLife 2016, 5, e15275. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Schwarcz, A.D.; Dénes, Á. Mitochondrial Ultrastructure Is Coupled to Synaptic Performance at Axonal Release Sites. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Chauhan, A.; Gu, F.; Essa, M.M.; Wegiel, J.; Kaur, K.; Brown, W.T.; Chauhan, V. Brain Region-Specific Deficit in Mitochondrial Electron Transport Chain Complexes in Children with Autism. J. Neurochem. 2011, 117, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Anitha, A.; Nakamura, K.; Thanseem, I.; Matsuzaki, H.; Miyachi, T.; Tsujii, M.; Iwata, Y.; Suzuki, K.; Sugiyama, T.; Mori, N. Downregulation of the Expression of Mitochondrial Electron Transport Complex Genes in Autism Brains. Brain Pathol. 2012, 23, 294–302. [Google Scholar] [CrossRef]
- Oliveira, G.; Diogo, L.; Grazina, M.; Garcia, P.; Ataíde, A.; Marques, C.; Miguel, T.; Borges, L.; Vicente, A.M.; Oliveira, C.R. Mitochondrial Dysfunction in Autism Spectrum Disorders: A Population-Based Study. Dev. Med. Child Neurol. 2005, 47, 185–189. [Google Scholar] [CrossRef]
- Correia, C.; Coutinho, A.M.; Diogo, L.; Grazina, M.; Marques, C.; Miguel, T.; Ataíde, A.; Almeida, J.; Borges, L.; Oliveira, C.; et al. Brief Report: High Frequency of Biochemical Markers for Mitochondrial Dysfunction in Autism: No Association with the Mitochondrial Aspartate/Glutamate Carrier SLC25A12 Gene. J. Autism Dev. Disord. 2006, 36, 1137–1140. [Google Scholar] [CrossRef]
- Weissman, J.R.; Kelley, R.I.; Bauman, M.L.; Cohen, B.H.; Murray, K.F.; Mitchell, R.L.; Kern, R.L.; Natowicz, M.R. Mitochondrial Disease in Autism Spectrum Disorder Patients: A Cohort Analysis. PLoS ONE 2008, 3, e3815. [Google Scholar] [CrossRef]
- Haas, R.H. Autism and Mitochondrial Disease. Dev. Disabil. Res. Rev. 2010, 16, 144–153. [Google Scholar] [CrossRef]
- Frye, R.E.; Rossignol, D.A. Mitochondrial Dysfunction Can Connect the Diverse Medical Symptoms Associated with Autism Spectrum Disorders. Pediatr. Res. 2011, 69 Pt 2, 41R–47R. [Google Scholar] [CrossRef]
- Goldenthal, M.J.; Damle, S.; Sheth, S.; Shah, N.; Melvin, J.; Jethva, R.; Hardison, H.; Marks, H.; Legido, A. Mitochondrial Enzyme Dysfunction in Autism Spectrum Disorders; a Novel Biomarker Revealed from Buccal Swab Analysis. Biomark. Med. 2015, 9, 957–965. [Google Scholar] [CrossRef]
- Basal Ganglia and Autism—A Translational Perspective—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/28730641/ (accessed on 29 August 2023).
- Estes, A.; Shaw, D.W.W.; Sparks, B.F.; Friedman, S.; Giedd, J.N.; Dawson, G.; Bryan, M.; Dager, S.R. Basal Ganglia Morphometry and Repetitive Behavior in Young Children with Autism Spectrum Disorder. Autism Res. Off. J. Int. Soc. Autism Res. 2011, 4, 212–220. [Google Scholar] [CrossRef]
- Becker, E.B.E.; Stoodley, C.J. Autism Spectrum Disorder and the Cerebellum. Int. Rev. Neurobiol. 2013, 113, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, E.; Dowling, S.; Phelps, M.; Findling, R.L. Pharmacotherapy of emotional and behavioral symptoms associated with autism spectrum disorder in children and adolescents. Dialogues Clin. Neurosci. 2017, 19, 395–402. [Google Scholar] [CrossRef]
- Mano-Sousa, B.J.; Pedrosa, A.M.; Alves, B.C.; Galduróz, J.C.F.; Belo, V.S.; Chaves, V.E.; Duarte-Almeida, J.M. Effects of Risperidone in Autistic Children and Young Adults: A Systematic Review and Meta-Analysis. Curr. Neuropharmacol. 2021, 19, 538–552. [Google Scholar] [CrossRef]
- Hollander, E.; Soorya, L.; Chaplin, W.; Anagnostou, E.; Taylor, B.P.; Ferretti, C.J.; Wasserman, S.; Swanson, E.; Settipani, C. A double-blind placebo-controlled trial of fluoxetine for repetitive behaviors and global severity in adult autism spectrum disorders. Am. J. Psychiatry 2012, 169, 292–299. [Google Scholar] [CrossRef]
- Mandic-Maravic, V.; Grujicic, R.; Milutinovic, L.; Munjiza-Jovanovic, A.; Pejovic-Milovancevic, M. Dopamine in Autism Spectrum Disorders-Focus on D2/D3 Partial Agonists and Their Possible Use in Treatment. Front. Psychiatry 2022, 12, 787097. [Google Scholar] [CrossRef]
- More, R.P.; Warrier, V.; Brunel, H.; Buckingham, C.; Smith, P.; Allison, C.; Holt, R.; Bradshaw, C.R.; Baron-Cohen, S. Identifying rare genetic variants in 21 highly multiplex autism families: The role of diagnosis and autistic traits. Mol. Psychiatry 2023, 28, 2148–2157. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilela, J.; Martiniano, H.; Marques, A.R.; Santos, J.X.; Asif, M.; Rasga, C.; Oliveira, G.; Vicente, A.M. Identification of Neurotransmission and Synaptic Biological Processes Disrupted in Autism Spectrum Disorder Using Interaction Networks and Community Detection Analysis. Biomedicines 2023, 11, 2971. https://doi.org/10.3390/biomedicines11112971
Vilela J, Martiniano H, Marques AR, Santos JX, Asif M, Rasga C, Oliveira G, Vicente AM. Identification of Neurotransmission and Synaptic Biological Processes Disrupted in Autism Spectrum Disorder Using Interaction Networks and Community Detection Analysis. Biomedicines. 2023; 11(11):2971. https://doi.org/10.3390/biomedicines11112971
Chicago/Turabian StyleVilela, Joana, Hugo Martiniano, Ana Rita Marques, João Xavier Santos, Muhammad Asif, Célia Rasga, Guiomar Oliveira, and Astrid Moura Vicente. 2023. "Identification of Neurotransmission and Synaptic Biological Processes Disrupted in Autism Spectrum Disorder Using Interaction Networks and Community Detection Analysis" Biomedicines 11, no. 11: 2971. https://doi.org/10.3390/biomedicines11112971
APA StyleVilela, J., Martiniano, H., Marques, A. R., Santos, J. X., Asif, M., Rasga, C., Oliveira, G., & Vicente, A. M. (2023). Identification of Neurotransmission and Synaptic Biological Processes Disrupted in Autism Spectrum Disorder Using Interaction Networks and Community Detection Analysis. Biomedicines, 11(11), 2971. https://doi.org/10.3390/biomedicines11112971