


An Overview of Recent Advances in the Neuroprotective Potentials of Fisetin against Diverse Insults in Neurological Diseases and the Underlying Signaling Pathways


Abstract
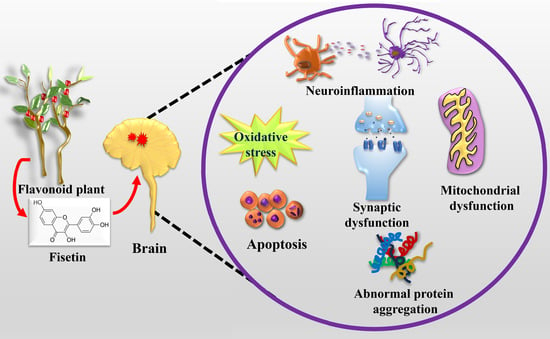
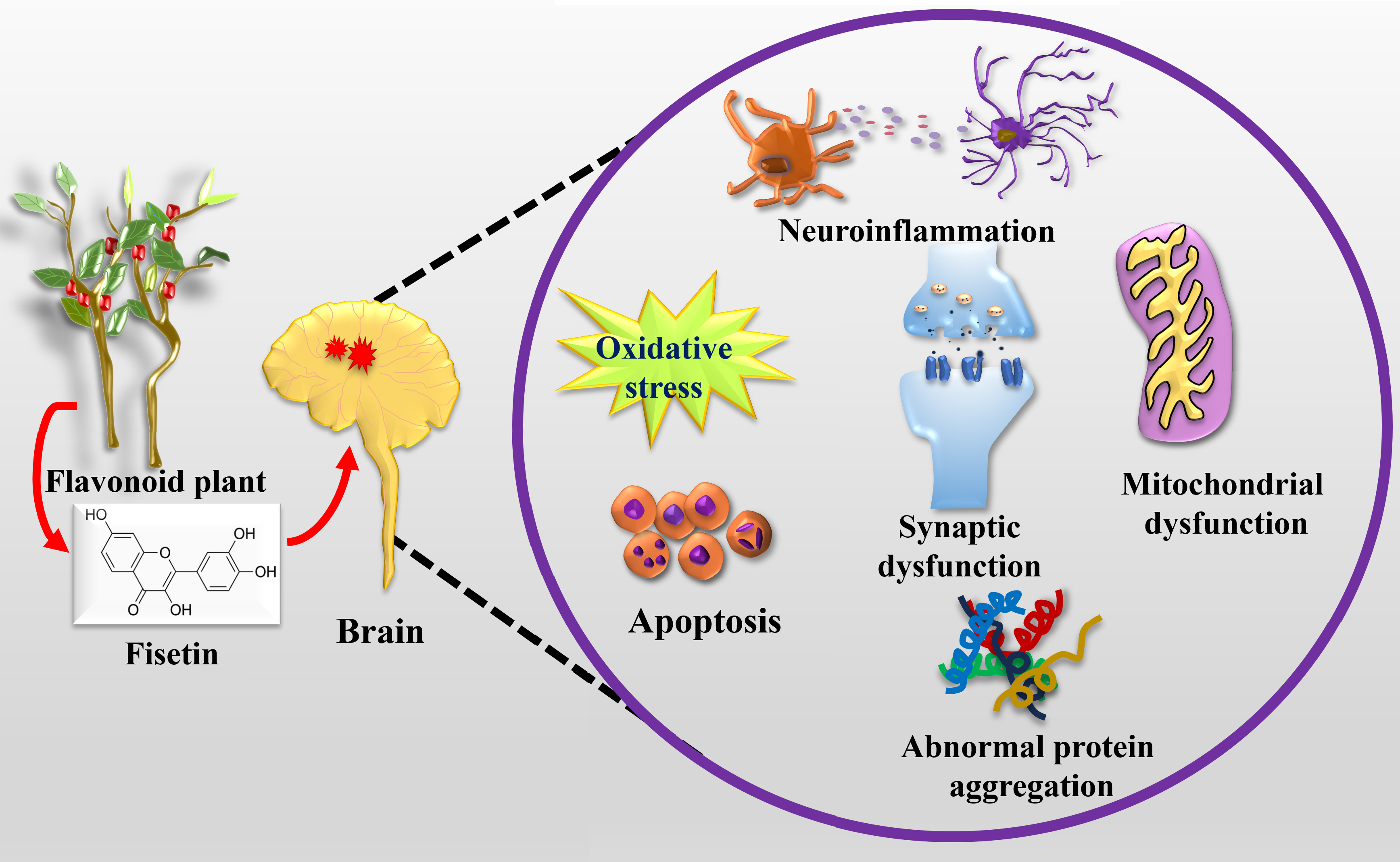
1. Introduction
2. Fisetin Structure, Physicochemical Attributes, Pharmacokinetics and Toxicity
3. Neuroprotective Potentials of Fisetin
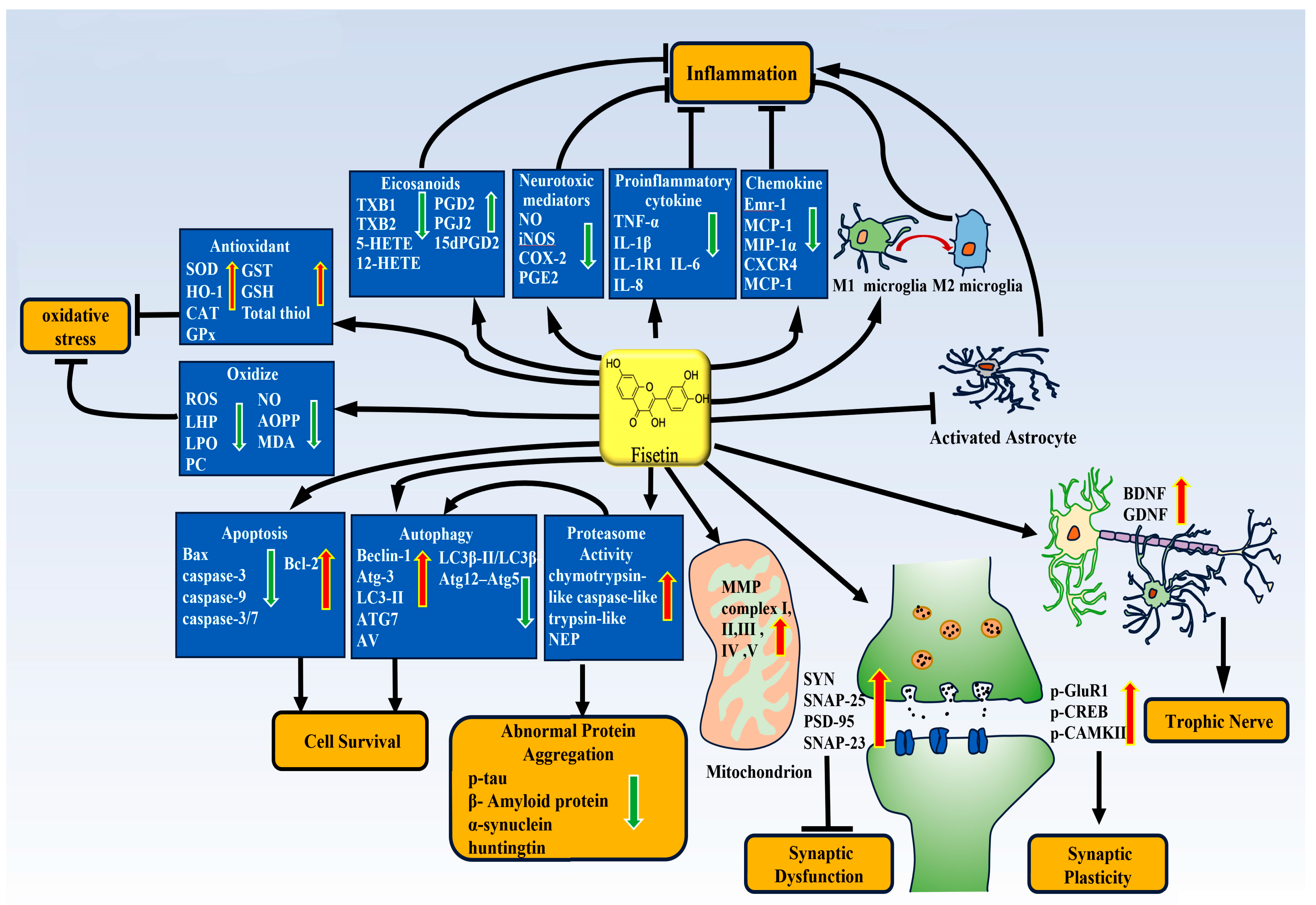
3.1. Fisetin and Neuroinflammation
3.1.1. Microglia-Dependent Neuroinflammation in Nervous System Diseases
3.1.2. The Anti-Inflammatory Effect of Fisetin and Molecular Mechanism
3.2. Fisetin and Antioxidative Stress
3.3. Fisetin and Autophagy
3.4. Fisetin and Cyclin-Dependent Kinase 5 (Cdk5)
3.5. Fisetin and Apoptosis
3.6. Fisetin Improves Synaptic Function
3.7. Fisetin and Proteasome Activity and Abnormal Protein Aggregation
3.8. Fisetin and Mitochondrial Function
3.9. Fisetin and Neurotrophic Effects
3.10. Fisetin and Target of Sirtuins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effects of Fisetin | Suggested Mechanism | References |
|---|---|---|
| Anti-inflammation | Suppress inflammatory factors and chemokine such as TNF-α, IL-1β, IL-6, IL-8, iNOS, COX-2, PGE2, IL-18, Emr-1, MCP-1m, MIP-1α, CXCR4, MCP-1 | [49,51,54,57,76] |
| Reduce the expression of TXB1, TXB2, 5-HETE, 12-HETE and increase the expression of PGD2, PGJ2, 15dPGD2 | [58] | |
| Reduce the polarization of M1 phenotype and production of inflammatory mediators in microglia | [7,47,52,53,72] | |
| Inhibit the activation of astrocytes and reduce the expression of GFAP | [53,57] | |
| Reduce the level of p25 | [58] | |
| NLRP-3, ASC and cleaved-caspase-1 ↓ | [51] | |
| NF-κB activation ↓ | [51,52,57] | |
| TLR4/MyD88/NF-κB ↓ | [81] | |
| Phosphorylation of JNK ↓ | [52] | |
| Phosphorylation of ERK ↓ | [72] | |
| IL-1R/TLR Axis ↓ | [54] | |
| Antioxidative stress | Reduce oxide production such as ROS, LHP, LPO, PC, NO, AOPP, MDA | [60,65,69,73,74,75,76] |
| Increase the production of antioxidants such as SOD, HO-1, CAT, GPx, GSH, GST, Total thiol | [60,69,70,72,73,74,76] | |
| Nrf2-ARE ↑ | [51,74] | |
| HIF-1α/HRE ↑; phosphorylation of MEK1/2 and p38 MAPK; ↑ PI3K/Akt ↑ | [75] | |
| Hyperphosphorylation of ERK and phosphorylation of c-Myc ↓; Nrf2 ↑ | [70] | |
| Phosphorylation of ERK, JNK (c-JUN NH2-terminal protein kinase), and p38 MAPK ↑; SIRT1 ↑; Nrf2 ↑ | [69] | |
| Regulation of autophagy | Upregulation of autophagy genes (Atg-3 and Beclin-1) | [76] |
| Increase these autophagy-related proteins LC3-II and Beclin-1 | [76] | |
| Increase the number of autophagic vesicles and ATG proteins including Beclin-1 and ATG7 | [84] | |
| Stimulate autophagic degradation of phosphorylated tau via the activation of TFEB and Nrf2 transcription factors ↑ | [84] | |
| Reduce the ratio of LC3β-II/LC3β-I and the formation of Atg12-Atg5 conjugation | [69] | |
| mTORC1 ↓ | [84] | |
| Antiapoptosis | Reduce the expression of proapoptotic proteins such as BAX, caspase-3, caspase-9 and caspase-3/7 | [74,77,81,94,95] |
| Increased the expression of antiapoptotic proteins such as Bcl-2 | [16,81,109,112,115,116,117,119] | |
| PI3K-Akt ↑ | [77] | |
| Increase in the activity of proteasomes and reduction in the aggregation of abnormal proteins | Increase the chymotrypsin-like activity of the proteasome | [16,109] |
| Increase the expression of NEP in the brain and reduce the deposition of phosphorylated-tau | [81] | |
| Decrease α-synuclein expression | [115] | |
| Reduce the percentage of cells containing α-synuclein inclusions as well as their size and subcellular localization | [116] | |
| Increase the inhibitory ratio toα-synuclein fibrillation | [117] | |
| Inhibit the expression of mutant Huntington protein | [119] | |
| Activation of Ras-ERK ↑ | [119] | |
| Improvement of mitochondrial function | Reduce loss of mitochondrial membrane potential | [76] |
| Increase mitochondrial enzyme activity | [120] | |
| Restore the level of brain NADH-dehydrogenase, brain mitochondrial SDH level, brain mitochondrial MTT (Complex-III), and brain mitochondrial cytochrome oxidase (Complex-IV) | [99] | |
| Reverse MeHg-induced mitochondrial swelling and decrease in mitochondrial ETC (complex I, II, IV & V) activity | [121] | |
| Improvement of synaptic function | Increase the levels of both presynaptic (SYN and SNAP-25) and postsynaptic proteins (PSD-95, SNAP-23, p-GluR1, p-CREB and p-CAMKII) | [81,97] |
| Restore the levels of PSD-95 phosphorylation and PSD-95-related protein drebrin | [58] | |
| Reverse the increase in brain AChE activity | [76,99] | |
| Reverse the repressed synaptophysin and Gria1 genes and increase the phosphorylation and surface expression of AMPAR GluA1 subunit | [102,103] | |
| Regulate the expression of genes and proteins involved in synaptic transmission and plasticity and prevent the downregulation of neurogranin, dendritic protein, synaptic fusion protein 1A, Lin-7 homolog A, Complexin-2 and Exolyst complex component 8 | [104] | |
| Prevent the decrease in three proteins linked with synaptic functions such as activity-regulated cytoskeleton-associated protein, Homer, and synapse-associated protein 102 | [105] | |
| Improvement of memory and cognition | Upregulate expression of neurotrophic factors such as BDNF and GDNF | [102,104] |
| Increased expression of synaptic proteins and improved synaptic function | [97,102,103,104,105] | |
| Reduce the level of AChE | [53,76,99] | |
| Activation of Ras-ERK ↑ | [119] | |
| Phosphorylation of ERK/CREB ↑ | [15,103] | |
| Activation of PI3K/Akt/CREB ↑ | [102] |
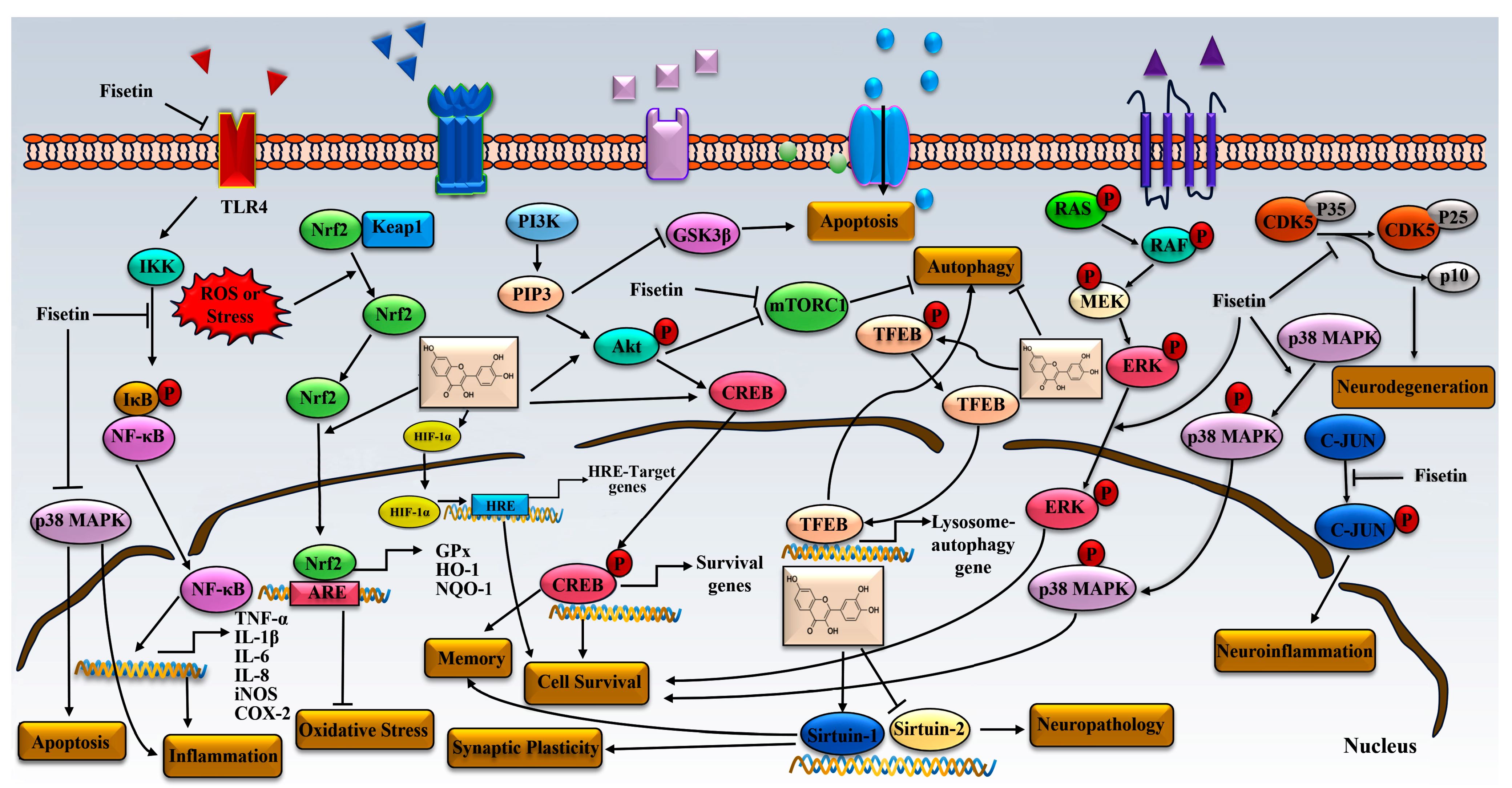
4. Signal Pathway
4.1. Fisetin and NF-κB Pathway
4.2. Fisetin and Keap1/Nrf2/ARE Pathway
4.3. Fisetin and PI3K-Akt
4.4. Fisetin and MAPK Pathway
4.5. Fisetin and TFEB
5. Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| HD | Huntington’s disease |
| ALS | amyotrophic lateral sclerosis |
| TBI | traumatic brain injury |
| CNS | central nervous system |
| BBB | blood–brain barrier |
| Aβ | amyloid beta-peptide |
| NO | nitric oxide |
| iNOS | inducible nitric oxide synthase |
| TLR 4 | Toll-like receptor 4 |
| COX | cyclooxygenase |
| TNF-α | tumor necrosis factor α |
| GFAP | glial fibrillary acidic protein |
| PGD2 | prostaglandin D2 |
| PGE2 | prostaglandin E2 |
| HO-1 | heme oxygenase-1 |
| IL-1 β | interleukin 1 β |
| IL-6 | Interleukin 6 |
| MeHg | methyl mercury |
| LPS | lipopolysaccharide |
| ROS | radical oxygen species |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| Nox | NADPH oxidase |
| BDNF | brain-derived neurotrophic factor |
| GDNF | Glial-cell-derived neurotrophic factor |
| MDA | malondialdehyde |
| GSH | glutathione |
| GCL | glutamate-cysteine ligase |
| GCLC | glutamate-cysteine ligase catalytic |
| GCLM | glutamate-cysteine ligase modifier subunit |
| SOD | superoxide dismutase |
| Nrf2 | NF-E2 related factor 2 |
| mTOR | mammalian or mechanistic target of rapamycin |
| MDA | malondialdehyde |
| Nrf2 | NF-E2-related factor 2 |
| Cdk5 | Cyclin-dependent kinase 5 |
| SNAP-25 | synaptosomal-associated protein 25 |
| PSD-95 | postsynaptic density protein 95 |
| CaMKII | Calcium–calmodulin (CaM)-dependent protein kinase II |
| NEP | neprilysin |
| CREB | cAMP response element-binding protein |
| AChE | acetylcholinesterase |
| NF-κB | nuclear factor kappa B |
| MAPK | mitogen-activated protein kinase |
| IKK | IκB kinases |
| ARE | antioxidant-response element |
| Keap1 | Kelch-like ECH-associated protein 1 |
| PI3K | Phosphoinositide 3-kinase |
| TFEB | Transcription factor EB |
References
- Ravula, A.R.; Teegala, S.B.; Kalakotla, S.; Pasangulapati, J.P.; Perumal, V.; Boyina, H.K. Fisetin, potential flavonoid with multifarious targets for treating neurological disorders: An updated review. Eur. J. Pharmacol. 2021, 910, 174492. [Google Scholar] [CrossRef]
- Maher, P. Preventing and Treating Neurological Disorders with the Flavonol Fisetin. Brain Plast. 2021, 6, 155–166. [Google Scholar] [CrossRef]
- Jang, H.S.; Kook, S.H.; Son, Y.O.; Kim, J.G.; Jeon, Y.M.; Jang, Y.S.; Choi, K.C.; Kim, J.; Han, S.K.; Lee, K.Y.; et al. Flavonoids purified from Rhus verniciflua Stokes actively inhibit cell growth and induce apoptosis in human osteosarcoma cells. Biochim. Biophys. Acta 2005, 1726, 309–316. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Demchuk, O.M. New Perspectives for Fisetin. Front. Chem. 2019, 7, 697. [Google Scholar] [CrossRef]
- Pal, H.C.; Pearlman, R.L.; Afaq, F. Fisetin and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 213–244. [Google Scholar] [CrossRef]
- Ding, H.; Li, Y.; Chen, S.; Wen, Y.; Zhang, S.; Luo, E.; Li, X.; Zhong, W.; Zeng, H. Fisetin ameliorates cognitive impairment by activating mitophagy and suppressing neuroinflammation in rats with sepsis-associated encephalopathy. CNS Neurosci. Ther. 2022, 28, 247–258. [Google Scholar] [CrossRef]
- Tsurudome, N.; Minami, Y.; Kajiya, K. Fisetin, a major component derived from mulberry (Morus australis Poir.) leaves, prevents vascular abnormal contraction. Biofactors 2022, 48, 56–66. [Google Scholar] [CrossRef]
- Jash, S.K.; Mondal, S. Bioactive flavonoid fisetin—A molecule of pharmacological interest. Cardiovasc. Dis. 2014, 5, 010314. [Google Scholar]
- Kimira, M.; Arai, Y.; Shimoi, K.; Watanabe, S. Japanese intake of flavonoids and isoflavonoids from foods. J. Epidemiol. 1998, 8, 168–175. [Google Scholar] [CrossRef]
- Prabhu, K.; Bhute, A.S. Plant based natural dyes and mordants: A Review. J. Nat. Prod. Plant Resour. 2012, 2, 649–664. [Google Scholar]
- Kashyap, D.; Sharma, A.; Sak, K.; Tuli, H.S.; Buttar, H.S.; Bishayee, A. Fisetin: A bioactive phytochemical with potential for cancer prevention and pharmacotherapy. Life Sci. 2018, 194, 75–87. [Google Scholar] [CrossRef]
- Hassan, S.S.U.; Samanta, S.; Dash, R.; Karpiński, T.M.; Habibi, E.; Sadiq, A.; Ahmadi, A.; Bunagu, S. The neuroprotective effects of fisetin, a natural flavonoid in neurodegenerative diseases: Focus on the role of oxidative stress. Front. Pharmacol. 2022, 13, 1015835. [Google Scholar] [CrossRef]
- Tordera, M.; Ferrándiz, M.L.; Alcaraz, M.J. Influence of anti-inflammatory flavonoids on degranulation and arachidonic acid release in rat neutrophils. Z. Naturforsch C J. Biosci. 1994, 49, 235–240. [Google Scholar] [CrossRef]
- Maher, P.; Akaishi, T.; Abe, K. Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc. Natl. Acad. Sci. USA 2006, 103, 16568–16573. [Google Scholar] [CrossRef]
- Maher, P. Modulation of multiple pathways involved in the maintenance of neuronal function during aging by fisetin. Genes. Nutr. 2009, 4, 297–307. [Google Scholar] [CrossRef]
- Krasieva, T.B.; Ehren, J.; O’Sullivan, T.; Tromberg, B.J.; Maher, P. Cell and brain tissue imaging of the flavonoid fisetin using label-free two-photon microscopy. Neurochem. Int. 2015, 89, 243–248. [Google Scholar] [CrossRef]
- He, W.B.; Abe, K.; Akaishi, T. Oral administration of fisetin promotes the induction of hippocampal long-term potentiation in vivo. J. Pharmacol. Sci. 2018, 136, 42–45. [Google Scholar] [CrossRef]
- Jo, J.H.; Jo, J.J.; Lee, J.M.; Lee, S. Identification of absolute conversion to geraldol from fisetin and pharmacokinetics in mouse. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2016, 1038, 95–100. [Google Scholar] [CrossRef]
- Maher, P. Fisetin acts on multiple pathways to reduce the impact of age and disease on CNS function. Front. Biosci. (Sch. Ed.) 2015, 7, 58. [Google Scholar]
- Shia, C.S.; Tsai, S.Y.; Kuo, S.C.; Hou, Y.C.; Chao, P.D. Metabolism and pharmacokinetics of 3,3′,4′,7-tetrahydroxyflavone (fisetin), 5-hydroxyflavone, and 7-hydroxyflavone and antihemolysis effects of fisetin and its serum metabolites. J. Agric. Food Chem. 2009, 57, 83–89. [Google Scholar] [CrossRef]
- Sari, E.N.; Soysal, Y. Molecular and Therapeutic Effects of Fisetin Flavonoid in Diseases. J. Basic. Clin. Health Sci. 2020, 4, 190–196. [Google Scholar] [CrossRef]
- Guzzo, M.R.; Uemi, M.; Donate, P.M.; Nikolaou, S.; Machado, A.E.; Okano, L.T. Study of the complexation of fisetin with cyclodextrins. J. Phys. Chem. A 2006, 110, 10545–10551. [Google Scholar] [CrossRef]
- Mignet, N.; Seguin, J.; Ramos Romano, M.; Brullé, L.; Touil, Y.S.; Scherman, D.; Bessodes, M.; Chabot, G.G. Development of a liposomal formulation of the natural flavonoid fisetin. Int. J. Pharm. 2012, 423, 69–76. [Google Scholar] [CrossRef]
- Sowa, M.; Ślepokura, K.; Matczak-Jon, E. Cocrystals of fisetin, luteolin and genistein with pyridinecarboxamide coformers: Crystal structures, analysis of intermolecular interactions, spectral and thermal characterization. CrystEngComm 2013, 15, 7696–7708. [Google Scholar] [CrossRef]
- Sowa, M.; Ślepokura, K.; Matczak-Jon, E. Improving solubility of fisetin by cocrystallization. CrystEngComm 2014, 16, 10592–10601. [Google Scholar] [CrossRef]
- Ragelle, H.; Crauste-Manciet, S.; Seguin, J.; Brossard, D.; Scherman, D.; Arnaud, P.; Chabot, G.G. Nanoemulsion formulation of fisetin improves bioavailability and antitumour activity in mice. Int. J. Pharm. 2012, 427, 452–459. [Google Scholar] [CrossRef]
- Liu, W.Y.; Lin, C.C.; Hsieh, Y.S.; Wu, Y.T. Nanoformulation Development to Improve the Biopharmaceutical Properties of Fisetin Using Design of Experiment Approach. Molecules 2021, 26, 3031. [Google Scholar] [CrossRef]
- de Andrade, E.W.V.; Dupont, S.; Beney, L.; Hoskin, R.T.; da Silva Pedrini, M.R. Osmoporation is a versatile technique to encapsulate fisetin using the probiotic bacteria Lactobacillus acidophilus. Appl. Microbiol. Biotechnol. 2022, 106, 1031–1044. [Google Scholar] [CrossRef]
- Elsallabi, O.; Patruno, A.; Pesce, M.; Cataldi, A.; Carradori, S.; Gallorini, M. Fisetin as a Senotherapeutic Agent: Biopharmaceutical Properties and Crosstalk between Cell Senescence and Neuroprotection. Molecules 2022, 27, 738. [Google Scholar] [CrossRef]
- Carregosa, D.; Carecho, R.; Figueira, I.; Santos, C.N. Low-Molecular Weight Metabolites from Polyphenols as Effectors for Attenuating Neuroinflammation. J. Agric. Food Chem. 2020, 68, 1790–1807. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Islam, M.S.; Quispe, C.; Hossain, R.; Islam, M.T.; Al-Harrasi, A.; Al-Rawahi, A.; Martorell, M.; Mamurova, A.; Seilkhan, A.; Altybaeva, N.; et al. Neuropharmacological Effects of Quercetin: A Literature-Based Review. Front. Pharmacol. 2021, 12, 665031. [Google Scholar] [CrossRef]
- An, J.; Chen, B.; Kang, X.; Zhang, R.; Guo, Y.; Zhao, J.; Yang, H. Neuroprotective effects of natural compounds on LPS-induced inflammatory responses in microglia. Am. J. Transl. Res. 2020, 12, 2353–2378. [Google Scholar]
- Molteni, M.; Rossetti, C. Neurodegenerative diseases: The immunological perspective. J. Neuroimmunol. 2017, 313, 109–115. [Google Scholar] [CrossRef]
- Dheen, S.T.; Kaur, C.; Ling, E.A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Zhang, B.; Wei, Y.Z.; Wang, G.Q.; Li, D.D.; Shi, J.S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell. Neurosci. 2018, 12, 531. [Google Scholar] [CrossRef]
- Gupta, N.; Shyamasundar, S.; Patnala, R.; Karthikeyan, A.; Arumugam, T.V.; Ling, E.-A.; Dheen, S.T. Recent progress in therapeutic strategies for microglia-mediated neuroinflammation in neuropathologies. Expert. Opin. Ther. Targets 2018, 22, 765–781. [Google Scholar] [CrossRef]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases—An update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef]
- Zheng, L.T.; Ock, J.; Kwon, B.M.; Suk, K. Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. Int. Immunopharmacol. 2008, 8, 484–494. [Google Scholar] [CrossRef]
- Chuang, J.Y.; Chang, P.C.; Shen, Y.C.; Lin, C.; Tsai, C.F.; Chen, J.H.; Yeh, W.L.; Wu, L.H.; Lin, H.Y.; Liu, Y.S.; et al. Regulatory effects of fisetin on microglial activation. Molecules 2014, 19, 8820–8839. [Google Scholar] [CrossRef]
- Zhang, P.; Cui, J. Neuroprotective Effect of Fisetin Against the Cerebral Ischemia-Reperfusion Damage via Suppression of Oxidative Stress and Inflammatory Parameters. Inflammation 2021, 44, 1490–1506. [Google Scholar] [CrossRef]
- Chen, C.; Yao, L.; Cui, J.; Liu, B. Fisetin Protects against Intracerebral Hemorrhage-Induced Neuroinflammation in Aged Mice. Cerebrovasc. Dis. 2018, 45, 154–161. [Google Scholar] [CrossRef]
- Cordaro, M.; D’Amico, R.; Fusco, R.; Peritore, A.F.; Genovese, T.; Interdonato, L.; Franco, G.; Arangia, A.; Gugliandolo, E.; Crupi, R.; et al. Discovering the Effects of Fisetin on NF-κB/NLRP-3/NRF-2 Molecular Pathways in a Mouse Model of Vascular Dementia Induced by Repeated Bilateral Carotid Occlusion. Biomedicines 2022, 10, 1448. [Google Scholar] [CrossRef]
- Gelderblom, M.; Leypoldt, F.; Lewerenz, J.; Birkenmayer, G.; Orozco, D.; Ludewig, P.; Thundyil, J.; Arumugam, T.V.; Gerloff, C.; Tolosa, E.; et al. The flavonoid fisetin attenuates postischemic immune cell infiltration, activation and infarct size after transient cerebral middle artery occlusion in mice. J. Cereb. Blood Flow. Metab. 2012, 32, 835–843. [Google Scholar] [CrossRef]
- Prakash, D.; Gopinath, K.; Sudhandiran, G. Fisetin enhances behavioral performances and attenuates reactive gliosis and inflammation during aluminum chloride-induced neurotoxicity. Neuromol. Med. 2013, 15, 192–208. [Google Scholar] [CrossRef]
- Khatoon, S.; Agarwal, N.B.; Samim, M.; Alam, O. Neuroprotective Effect of Fisetin through Suppression of IL-1R/TLR Axis and Apoptosis in Pentylenetetrazole-Induced Kindling in Mice. Front. Neurol. 2021, 12, 689069. [Google Scholar] [CrossRef]
- Wang, N.; Yao, F.; Li, K.; Zhang, L.; Yin, G.; Du, M.; Wu, B. Fisetin regulates astrocyte migration and proliferation in vitro. Int. J. Mol. Med. 2017, 39, 783–790. [Google Scholar] [CrossRef]
- Li, D.; Liu, X.; Liu, T.; Liu, H.; Tong, L.; Jia, S.; Wang, Y.F. Neurochemical regulation of the expression and function of glial fibrillary acidic protein in astrocytes. Glia 2020, 68, 878–897. [Google Scholar] [CrossRef]
- Xu, M.-X.; Ge, C.-X.; Li, Q.; Lou, D.-S.; Hu, L.-F.; Sun, Y.; Xiong, M.-X.; Lai, L.-L.; Zhong, S.-Y.; Yi, C.; et al. Fisetin nanoparticles protect against PM2.5 exposure-induced neuroinflammation by down-regulation of astrocytes activation related NF-κB signaling pathway. J. Funct. Foods 2020, 65, 103716. [Google Scholar] [CrossRef]
- Currais, A.; Prior, M.; Dargusch, R.; Armando, A.; Ehren, J.; Schubert, D.; Quehenberger, O.; Maher, P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 2014, 13, 379–390. [Google Scholar] [CrossRef]
- Sun, Y. Free radicals, antioxidant enzymes, and carcinogenesis. Free Radic. Biol. Med. 1990, 8, 583–599. [Google Scholar] [CrossRef]
- Naeimi, A.F.; Alizadeh, M. Antioxidant properties of the flavonoid fisetin: An updated review of in vivo and in vitro studies. Trends Food Sci. Technol. 2017, 70, 34–44. [Google Scholar] [CrossRef]
- Mani, S. Production of reactive oxygen species and its implication in human diseases. In Free Radicals in Human Health and Disease; Springer: New Delhi, India, 2015; pp. 3–15. [Google Scholar]
- Wang, H.; Patterson, C. Atherosclerosis: Risks, Mechanisms, and Therapies; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325. [Google Scholar] [CrossRef]
- Tsatsakis, A.; Docea, A.O.; Calina, D.; Tsarouhas, K.; Zamfira, L.M.; Mitrut, R.; Sharifi-Rad, J.; Kovatsi, L.; Siokas, V.; Dardiotis, E.; et al. A Mechanistic and Pathophysiological Approach for Stroke Associated with Drugs of Abuse. J. Clin. Med. 2019, 8, 1295. [Google Scholar] [CrossRef]
- Das, J.; Singh, R.; Sharma, D. Antiepileptic effect of fisetin in iron-induced experimental model of traumatic epilepsy in rats in the light of electrophysiological, biochemical, and behavioral observations. Nutr. Neurosci. 2017, 20, 255–264. [Google Scholar] [CrossRef]
- Ehren, J.L.; Maher, P. Concurrent regulation of the transcription factors Nrf2 and ATF4 mediates the enhancement of glutathione levels by the flavonoid fisetin. Biochem. Pharmacol. 2013, 85, 1816–1826. [Google Scholar] [CrossRef]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Yen, J.H.; Wu, P.S.; Chen, S.F.; Wu, M.J. Fisetin Protects PC12 Cells from Tunicamycin-Mediated Cell Death via Reactive Oxygen Species Scavenging and Modulation of Nrf2-Driven Gene Expression, SIRT1 and MAPK Signaling in PC12 Cells. Int. J. Mol. Sci. 2017, 18, 852. [Google Scholar] [CrossRef]
- Burdo, J.; Schubert, D.; Maher, P. Glutathione production is regulated via distinct pathways in stressed and non-stressed cortical neurons. Brain Res. 2008, 1189, 12–22. [Google Scholar] [CrossRef]
- Liu, L.; Liu, R.; Liu, Y.; Li, G.; Chen, Q.; Liu, X.; Ma, S. Cystine-glutamate antiporter xCT as a therapeutic target for cancer. Cell Biochem. Funct. 2021, 39, 174–179. [Google Scholar] [CrossRef]
- Cho, N.; Lee, K.Y.; Huh, J.; Choi, J.H.; Yang, H.; Jeong, E.J.; Kim, H.P.; Sung, S.H. Cognitive-enhancing effects of Rhus verniciflua bark extract and its active flavonoids with neuroprotective and anti-inflammatory activities. Food Chem. Toxicol. 2013, 58, 355–361. [Google Scholar] [CrossRef]
- Jacob, S.; Thangarajan, S. Effect of Gestational Intake of Fisetin (3,3′,4′,7-Tetrahydroxyflavone) on Developmental Methyl Mercury Neurotoxicity in F1 Generation Rats. Biol. Trace Elem. Res. 2017, 177, 297–315. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Zhou, Y.; Zhu, Y.; Fei, M. Fisetin alleviates oxidative stress after traumatic brain injury via the Nrf2-ARE pathway. Neurochem. Int. 2018, 118, 304–313. [Google Scholar] [CrossRef]
- Chen, P.Y.; Ho, Y.R.; Wu, M.J.; Huang, S.P.; Chen, P.K.; Tai, M.H.; Ho, C.T.; Yen, J.H. Cytoprotective effects of fisetin against hypoxia-induced cell death in PC12 cells. Food Funct. 2015, 6, 287–296. [Google Scholar] [CrossRef]
- Singh, S.; Singh, A.K.; Garg, G.; Rizvi, S.I. Fisetin as a caloric restriction mimetic protects rat brain against aging induced oxidative stress, apoptosis and neurodegeneration. Life Sci. 2018, 193, 171–179. [Google Scholar] [CrossRef]
- Watanabe, R.; Kurose, T.; Morishige, Y.; Fujimori, K. Protective Effects of Fisetin Against 6-OHDA-Induced Apoptosis by Activation of PI3K-Akt Signaling in Human Neuroblastoma SH-SY5Y Cells. Neurochem. Res. 2018, 43, 488–499. [Google Scholar] [CrossRef]
- Bar-Yosef, T.; Damri, O.; Agam, G. Dual Role of Autophagy in Diseases of the Central Nervous System. Front. Cell Neurosci. 2019, 13, 196. [Google Scholar] [CrossRef]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef]
- Xilouri, M.; Stefanis, L. Autophagy in the central nervous system: Implications for neurodegenerative disorders. CNS Neurol. Disord. Drug Targets 2010, 9, 701–719. [Google Scholar] [CrossRef]
- Yang, W.; Tian, Z.K.; Yang, H.X.; Feng, Z.J.; Sun, J.M.; Jiang, H.; Cheng, C.; Ming, Q.L.; Liu, C.M. Fisetin improves lead-induced neuroinflammation, apoptosis and synaptic dysfunction in mice associated with the AMPK/SIRT1 and autophagy pathway. Food Chem. Toxicol. 2019, 134, 110824. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef]
- Kim, S.; Choi, K.J.; Cho, S.J.; Yun, S.M.; Jeon, J.P.; Koh, Y.H.; Song, J.; Johnson, G.V.; Jo, C. Fisetin stimulates autophagic degradation of phosphorylated tau via the activation of TFEB and Nrf2 transcription factors. Sci. Rep. 2016, 6, 24933. [Google Scholar] [CrossRef]
- Zhu, J.; Li, W.; Mao, Z. Cdk5: Mediator of neuronal development, death and the response to DNA damage. Mech. Ageing Dev. 2011, 132, 389–394. [Google Scholar] [CrossRef]
- Pao, P.C.; Seo, J.; Lee, A.; Kritskiy, O.; Patnaik, D.; Penney, J.; Raju, R.M.; Geigenmuller, U.; Silva, M.C.; Lucente, D.E.; et al. A Cdk5-derived peptide inhibits Cdk5/p25 activity and improves neurodegenerative phenotypes. Proc. Natl. Acad. Sci. USA 2023, 120, e2217864120. [Google Scholar] [CrossRef]
- Allnutt, A.B.; Waters, A.K.; Kesari, S.; Yenugonda, V.M. Physiological and Pathological Roles of Cdk5: Potential Directions for Therapeutic Targeting in Neurodegenerative Disease. ACS Chem. Neurosci. 2020, 11, 1218–1230. [Google Scholar] [CrossRef]
- Ao, C.; Li, C.; Chen, J.; Tan, J.; Zeng, L. The role of Cdk5 in neurological disorders. Front. Cell Neurosci. 2022, 16, 951202. [Google Scholar] [CrossRef]
- Kesavapany, S.; Zheng, Y.L.; Amin, N.; Pant, H.C. Peptides derived from Cdk5 activator p35, specifically inhibit deregulated activity of Cdk5. Biotechnol. J. 2007, 2, 978–987. [Google Scholar] [CrossRef]
- Muyllaert, D.; Terwel, D.; Kremer, A.; Sennvik, K.; Borghgraef, P.; Devijver, H.; Dewachter, I.; Van Leuven, F. Neurodegeneration and neuroinflammation in cdk5/p25-inducible mice: A model for hippocampal sclerosis and neocortical degeneration. Am. J. Pathol. 2008, 172, 470–485. [Google Scholar] [CrossRef]
- Sundaram, J.R.; Chan, E.S.; Poore, C.P.; Pareek, T.K.; Cheong, W.F.; Shui, G.; Tang, N.; Low, C.M.; Wenk, M.R.; Kesavapany, S. Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J. Neurosci. 2012, 32, 1020–1034. [Google Scholar] [CrossRef]
- Chen, M.; Wu, W.; Liu, D.; Lv, Y.; Deng, H.; Gao, S.; Gu, Y.; Huang, M.; Guo, X.; Liu, B.; et al. Evolution and Structure of API5 and Its Roles in Anti-Apoptosis. Protein Pept. Lett. 2021, 28, 612–622. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Prakash, D.; Sudhandiran, G. Dietary flavonoid fisetin regulates aluminium chloride-induced neuronal apoptosis in cortex and hippocampus of mice brain. J. Nutr. Biochem. 2015, 26, 1527–1539. [Google Scholar] [CrossRef]
- Pak, F.; Oztopcu-Vatan, P. Fisetin effects on cell proliferation and apoptosis in glioma cells. Z. Naturforsch C J. Biosci. 2019, 74, 295–302. [Google Scholar] [CrossRef]
- Hossain, S.; Bhowmick, S.; Jahan, S.; Rozario, L.; Sarkar, M.; Islam, S.; Basunia, M.A.; Rahman, A.; Choudhury, B.K.; Shahjalal, H. Maternal lead exposure decreases the levels of brain development and cognition-related proteins with concomitant upsurges of oxidative stress, inflammatory response and apoptosis in the offspring rats. Neurotoxicology 2016, 56, 150–158. [Google Scholar] [CrossRef]
- Ahmad, A.; Ali, T.; Park, H.Y.; Badshah, H.; Rehman, S.U.; Kim, M.O. Neuroprotective Effect of Fisetin Against Amyloid-Beta-Induced Cognitive/Synaptic Dysfunction, Neuroinflammation, and Neurodegeneration in Adult Mice. Mol. Neurobiol. 2017, 54, 2269–2285. [Google Scholar] [CrossRef]
- Silman, I.; Sussman, J.L. Acetylcholinesterase: ‘classical’ and ‘non-classical’ functions and pharmacology. Curr. Opin. Pharmacol. 2005, 5, 293–302. [Google Scholar] [CrossRef]
- Boyina, H.K.; Jerald, M.K.; Bharatraj, D.K.; Diwan, P.V. Influence of fisetin combined with hesperidin on chronic mild hyperhomocysteinemia induced cognitive dysfunction and oxidative stress in wistar rats. PharmaNutrition 2018, 6, 125–136. [Google Scholar] [CrossRef]
- Kolos, Y.A.; Grigoriyev, I.P.; Korzhevskyi, D.E. A synaptic marker synaptophysin. Morfologiia 2015, 147, 78–82. [Google Scholar]
- Liu, S.J.; Yang, C.; Zhang, Y.; Su, R.Y.; Chen, J.L.; Jiao, M.M.; Chen, H.F.; Zheng, N.; Luo, S.; Chen, Y.B.; et al. Neuroprotective effect of β-asarone against Alzheimer’s disease: Regulation of synaptic plasticity by increased expression of SYP and GluR1. Drug Des. Dev. Ther. 2016, 10, 1461–1469. [Google Scholar] [CrossRef]
- Zhang, S.; Xue, R.; Geng, Y.; Wang, H.; Li, W. Fisetin Prevents HT22 Cells From High Glucose-Induced Neurotoxicity via PI3K/Akt/CREB Signaling Pathway. Front. Neurosci. 2020, 14, 241. [Google Scholar] [CrossRef]
- Zhan, J.Q.; Chen, C.N.; Wu, S.X.; Wu, H.J.; Zou, K.; Xiong, J.W.; Wei, B.; Yang, Y.J. Flavonoid fisetin reverses impaired hippocampal synaptic plasticity and cognitive function by regulating the function of AMPARs in a male rat model of schizophrenia. J. Neurochem. 2021, 158, 413–428. [Google Scholar] [CrossRef]
- Jacob, S.; Sumathi, T. Extenuation of in utero toxic effects of MeHg in the developing neurons by Fisetin via modulating the expression of synaptic transmission and plasticity regulators in hippocampus of the rat offspring. Chem. Biol. Interact. 2019, 305, 3–10. [Google Scholar] [CrossRef]
- Currais, A.; Farrokhi, C.; Dargusch, R.; Armando, A.; Quehenberger, O.; Schubert, D.; Maher, P. Fisetin Reduces the Impact of Aging on Behavior and Physiology in the Rapidly Aging SAMP8 Mouse. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 299–307. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin signalling in neurodegeneration: Mechanisms and therapeutic opportunities. Cell Death Differ. 2021, 28, 570–590. [Google Scholar] [CrossRef]
- Hunter, J.M.; Lesort, M.; Johnson, G.V. Ubiquitin-proteasome system alterations in a striatal cell model of Huntington’s disease. J. Neurosci. Res. 2007, 85, 1774–1788. [Google Scholar] [CrossRef]
- Zheng, J.; Bizzozero, O.A. Decreased activity of the 20S proteasome in the brain white matter and gray matter of patients with multiple sclerosis. J. Neurochem. 2011, 117, 143–153. [Google Scholar] [CrossRef]
- Maher, P. Proteasome inhibitors prevent oxidative stress-induced nerve cell death by a novel mechanism. Biochem. Pharmacol. 2008, 75, 1994–2006. [Google Scholar] [CrossRef][Green Version]
- Osama, A.; Zhang, J.; Yao, J.; Yao, X.; Fang, J. Nrf2: A dark horse in Alzheimer’s disease treatment. Ageing Res. Rev. 2020, 64, 101206. [Google Scholar] [CrossRef]
- Qian, C.; Yang, C.; Lu, M.; Bao, J.; Shen, H.; Deng, B.; Li, S.; Li, W.; Zhang, M.; Cao, C. Activating AhR alleviates cognitive deficits of Alzheimer’s disease model mice by upregulating endogenous Aβ catabolic enzyme Neprilysin. Theranostics 2021, 11, 8797–8812. [Google Scholar] [CrossRef]
- Xiao, S.; Lu, Y.; Wu, Q.; Yang, J.; Chen, J.; Zhong, S.; Eliezer, D.; Tan, Q.; Wu, C. Fisetin inhibits tau aggregation by interacting with the protein and preventing the formation of beta-strands. Int. J. Biol. Macromol. 2021, 178, 381–393. [Google Scholar] [CrossRef]
- Dash, R.; Emran, T.B.; Uddin, M.M.; Islam, A.; Junaid, M. Molecular docking of fisetin with AD associated AChE, ABAD and BACE1 proteins. Bioinformation 2014, 10, 562–568. [Google Scholar] [CrossRef]
- Bezard, E.; Dehay, B. Aggregation and spread of synuclein in Parkinson’s disease. Med. Sci. 2022, 38, 45–51. [Google Scholar] [CrossRef]
- Patel, M.Y.; Panchal, H.V.; Ghribi, O.; Benzeroual, K.E. The neuroprotective effect of fisetin in the MPTP model of Parkinson’s disease. J. Park. Dis. 2012, 2, 287–302. [Google Scholar] [CrossRef]
- Rosado-Ramos, R.; Godinho-Pereira, J.; Marques, D.; Figueira, I.; Fleming Outeiro, T.; Menezes, R.; Nunes Dos Santos, C. Small Molecule Fisetin Modulates Alpha-Synuclein Aggregation. Molecules 2021, 26, 3353. [Google Scholar] [CrossRef]
- Rane, A.R.; Paithankar, H.; Hosur, R.V.; Choudhary, S. Modulation of alpha-synuclein fibrillation by plant metabolites, daidzein, fisetin and scopoletin under physiological conditions. Int. J. Biol. Macromol. 2021, 182, 1278–1291. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Maher, P.; Dargusch, R.; Bodai, L.; Gerard, P.E.; Purcell, J.M.; Marsh, J.L. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum. Mol. Genet. 2011, 20, 261–270. [Google Scholar] [CrossRef]
- Alikatte, K.; Palle, S.; Rajendra Kumar, J.; Pathakala, N. Fisetin Improved Rotenone-Induced Behavioral Deficits, Oxidative Changes, and Mitochondrial Dysfunctions in Rat Model of Parkinson’s Disease. J. Diet. Suppl. 2021, 18, 57–71. [Google Scholar] [CrossRef]
- Jacob, S.; Thangarajan, S. Fisetin impedes developmental methylmercury neurotoxicity via downregulating apoptotic signalling pathway and upregulating Rho GTPase signalling pathway in hippocampus of F1 generation rats. Int. J. Dev. Neurosci. 2018, 69, 88–96. [Google Scholar] [CrossRef]
- Lanni, C.; Stanga, S.; Racchi, M.; Govoni, S. The expanding universe of neurotrophic factors: Therapeutic potential in aging and age-associated disorders. Curr. Pharm. Des. 2010, 16, 698–717. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Villalba, J.M.; Alcaín, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef]
- Fagerli, E.; Escobar, I.; Ferrier, F.J.; Jackson, C.W.; Perez-Lao, E.J.; Perez-Pinzon, M.A. Sirtuins and cognition: Implications for learning and memory in neurological disorders. Front. Physiol. 2022, 13, 908689. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. SIRT1 in neurodevelopment and brain senescence. Neuron 2014, 81, 471–483. [Google Scholar] [CrossRef]
- Chen, X.; Lu, W.; Wu, D. Sirtuin 2 (SIRT2): Confusing Roles in the Pathophysiology of Neurological Disorders. Front. Neurosci. 2021, 15, 614107. [Google Scholar] [CrossRef]
- de Sousa, R.R.; Queiroz, K.C.; Souza, A.C.; Gurgueira, S.A.; Augusto, A.C.; Miranda, M.A.; Peppelenbosch, M.P.; Ferreira, C.V.; Aoyama, H. Phosphoprotein levels, MAPK activities and NFkappaB expression are affected by fisetin. J. Enzyme Inhib. Med. Chem. 2007, 22, 439–444. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Jaeger, B.N.; Parylak, S.L.; Gage, F.H. Mechanisms of dietary flavonoid action in neuronal function and neuroinflammation. Mol. Aspects Med. 2018, 61, 50–62. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Zhou, C.H.; Wang, C.X.; Xie, G.B.; Wu, L.Y.; Wei, Y.X.; Wang, Q.; Zhang, H.S.; Hang, C.H.; Zhou, M.L.; Shi, J.X. Fisetin alleviates early brain injury following experimental subarachnoid hemorrhage in rats possibly by suppressing TLR 4/NF-κB signaling pathway. Brain Res. 2015, 1629, 250–259. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, T.; Leak, R.K.; Chen, J.; Zhang, F. Preventive and Protective Roles of Dietary Nrf2 Activators Against Central Nervous System Diseases. CNS Neurol. Disord. Drug Targets 2017, 16, 326–338. [Google Scholar] [CrossRef]
- de Vries, H.E.; Witte, M.; Hondius, D.; Rozemuller, A.J.; Drukarch, B.; Hoozemans, J.; van Horssen, J. Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic. Biol. Med. 2008, 45, 1375–1383. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, W.; Feng, X.; Yang, F.; Qin, H.; Wu, S.; Hou, D.X.; Chen, J. Nrf2–ARE Signaling Acts as Master Pathway for the Cellular Antioxidant Activity of Fisetin. Molecules 2019, 24, 708. [Google Scholar] [CrossRef]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef]
- Origlia, N.; Arancio, O.; Domenici, L.; Yan, S.S. MAPK, beta-amyloid and synaptic dysfunction: The role of RAGE. Expert. Rev. Neurother. 2009, 9, 1635–1645. [Google Scholar] [CrossRef]
- Wang, T.H.; Wang, S.Y.; Wang, X.D.; Jiang, H.Q.; Yang, Y.Q.; Wang, Y.; Cheng, J.L.; Zhang, C.T.; Liang, W.W.; Feng, H.L. Fisetin Exerts Antioxidant and Neuroprotective Effects in Multiple Mutant hSOD1 Models of Amyotrophic Lateral Sclerosis by Activating ERK. Neuroscience 2018, 379, 152–166. [Google Scholar] [CrossRef]
- Kim, J.; Kwon, J.T.; Kim, H.S.; Han, J.H. CREB and neuronal selection for memory trace. Front. Neural Circuits 2013, 7, 44. [Google Scholar] [CrossRef]
- Falcicchia, C.; Tozzi, F.; Arancio, O.; Watterson, D.M.; Origlia, N. Involvement of p38 MAPK in Synaptic Function and Dysfunction. Int. J. Mol. Sci. 2020, 21, 5624. [Google Scholar] [CrossRef]
- Gu, Z.; Cao, H.; Zuo, C.; Huang, Y.; Miao, J.; Song, Y.; Yang, Y.; Zhu, L.; Wang, F. TFEB in Alzheimer’s disease: From molecular mechanisms to therapeutic implications. Neurobiol. Dis. 2022, 173, 105855. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, X.; Deng, P.; Jiang, Y.; Zhang, L.; He, Y.; Yang, H. An Overview of Recent Advances in the Neuroprotective Potentials of Fisetin against Diverse Insults in Neurological Diseases and the Underlying Signaling Pathways. Biomedicines 2023, 11, 2878. https://doi.org/10.3390/biomedicines11112878
Tang X, Deng P, Jiang Y, Zhang L, He Y, Yang H. An Overview of Recent Advances in the Neuroprotective Potentials of Fisetin against Diverse Insults in Neurological Diseases and the Underlying Signaling Pathways. Biomedicines. 2023; 11(11):2878. https://doi.org/10.3390/biomedicines11112878
Chicago/Turabian StyleTang, Xiangwen, Peng Deng, Yizhen Jiang, Lingling Zhang, Yuqing He, and Hao Yang. 2023. "An Overview of Recent Advances in the Neuroprotective Potentials of Fisetin against Diverse Insults in Neurological Diseases and the Underlying Signaling Pathways" Biomedicines 11, no. 11: 2878. https://doi.org/10.3390/biomedicines11112878
APA StyleTang, X., Deng, P., Jiang, Y., Zhang, L., He, Y., & Yang, H. (2023). An Overview of Recent Advances in the Neuroprotective Potentials of Fisetin against Diverse Insults in Neurological Diseases and the Underlying Signaling Pathways. Biomedicines, 11(11), 2878. https://doi.org/10.3390/biomedicines11112878