
Identifying Potent Nonsense-Mediated mRNA Decay Inhibitors with a Novel Screening System

 , , and
, , and 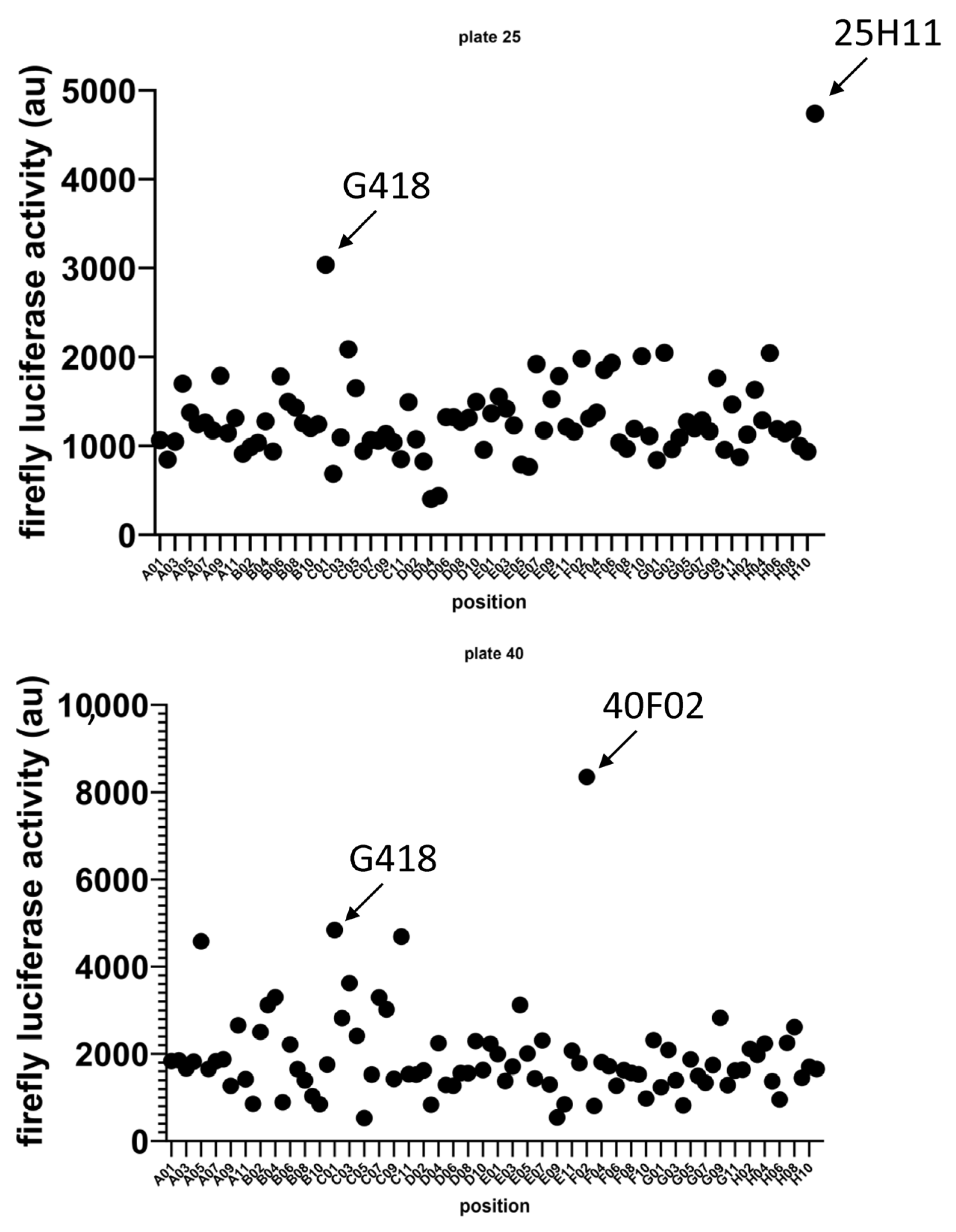{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture Cell
2.2. RNA Extraction and RT-PCR Analysis
2.3. NMD Efficacy Assay
2.4. Toxicity Assay
2.5. Chemicals
3. Results
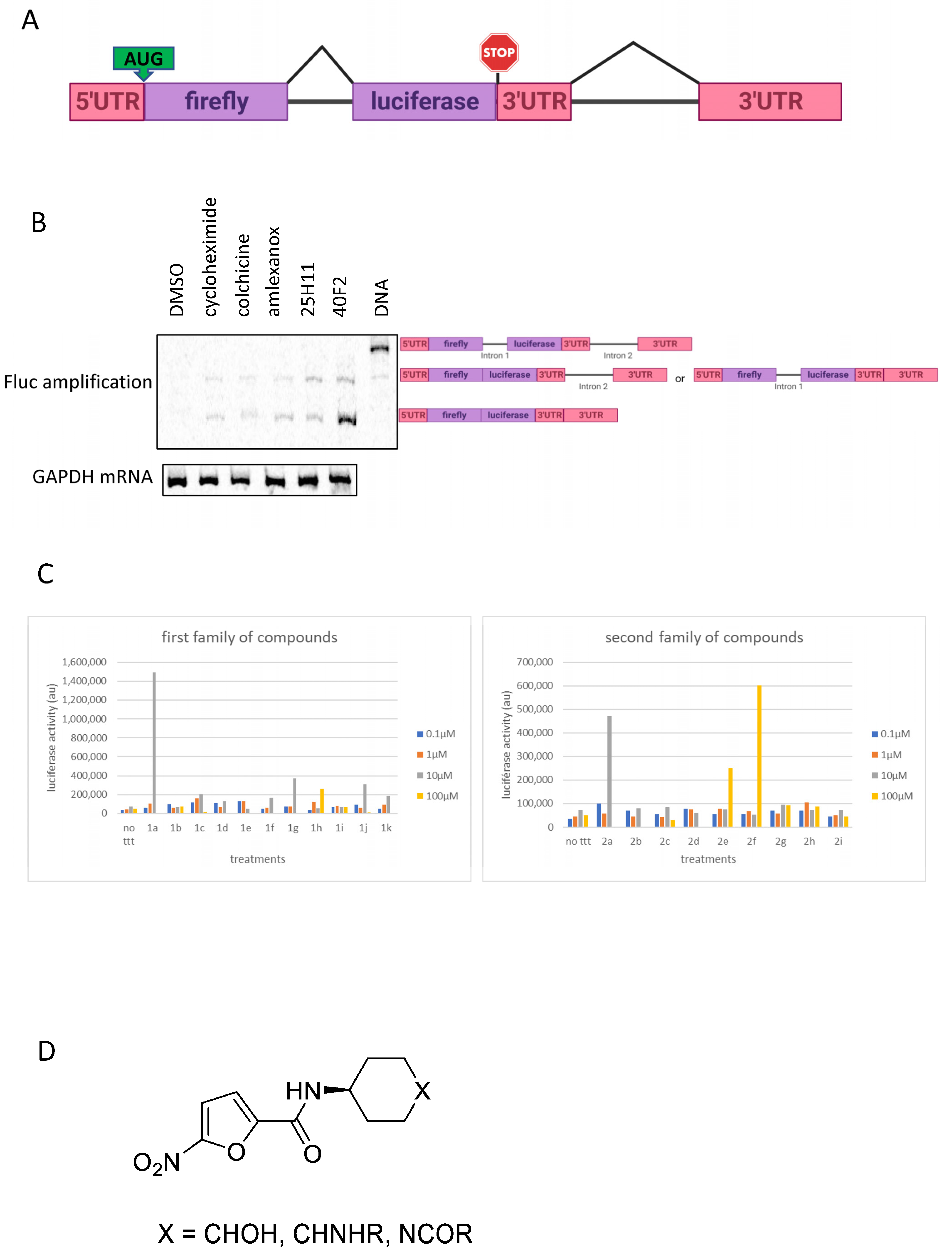
3.1. Identification of Two New Potential NMD Inhibitors
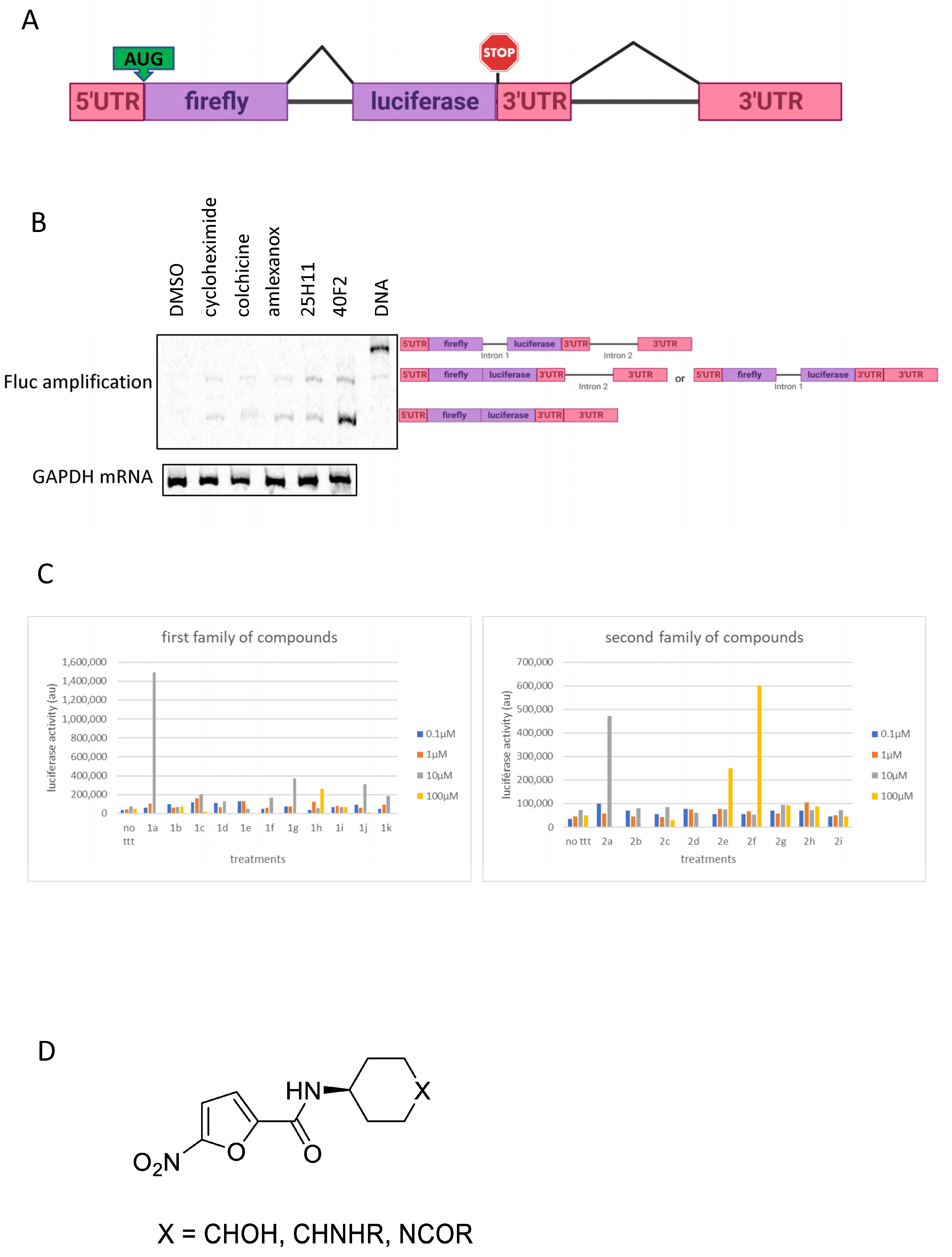
3.2. Validation of 1a and 2a as NMD Inhibitors
3.3. Derivatives of 1a and 2a Retain NMD-Inhibiting Activity
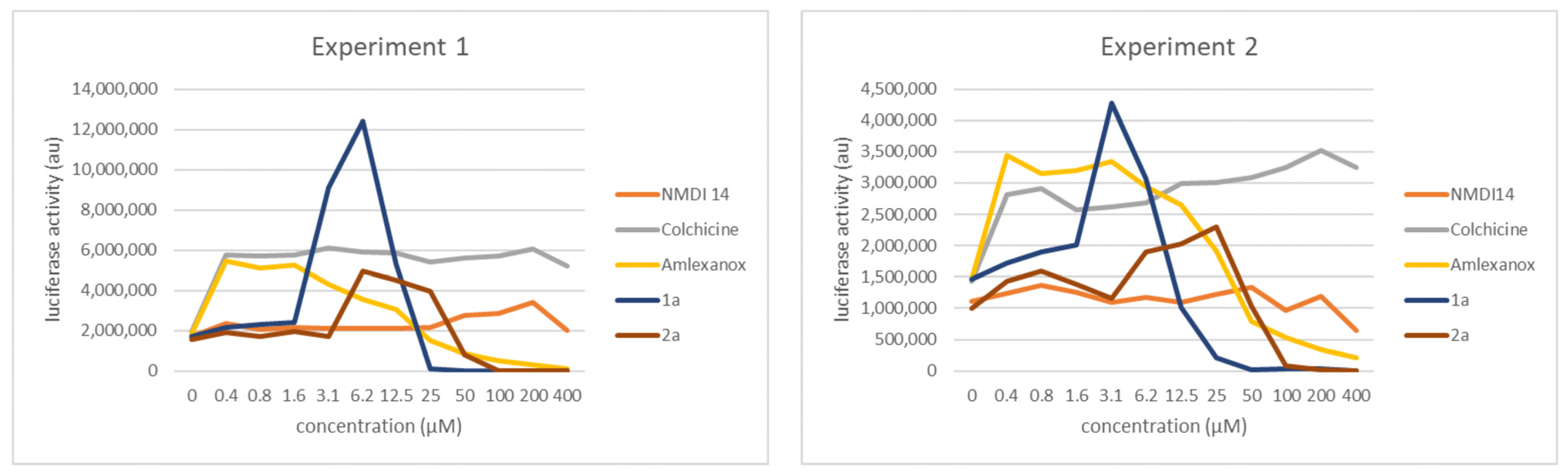
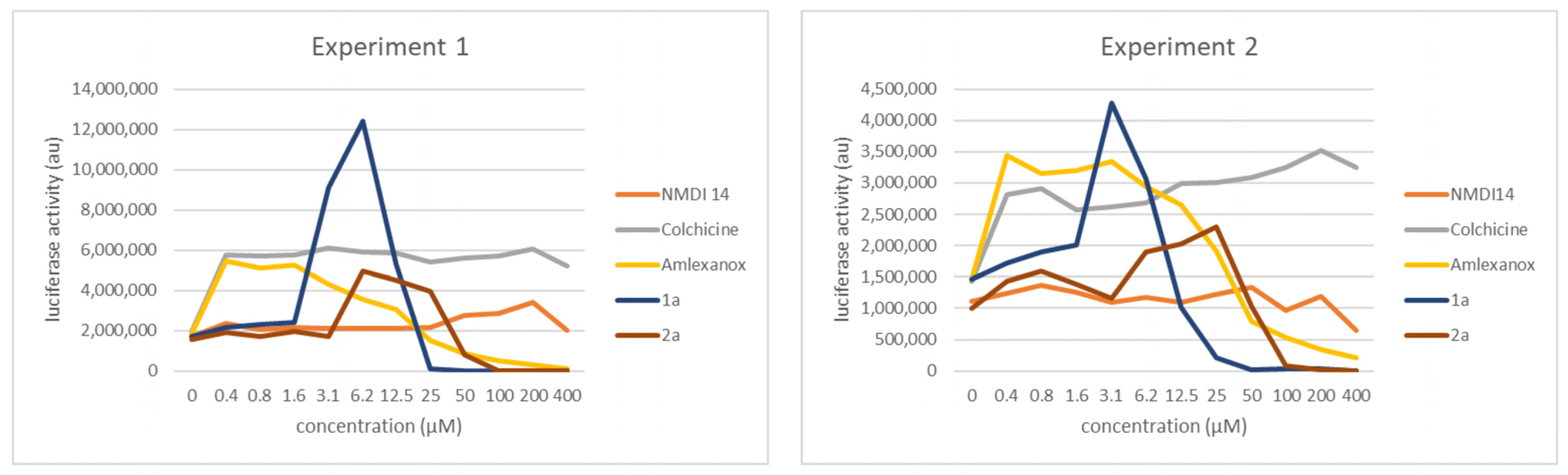
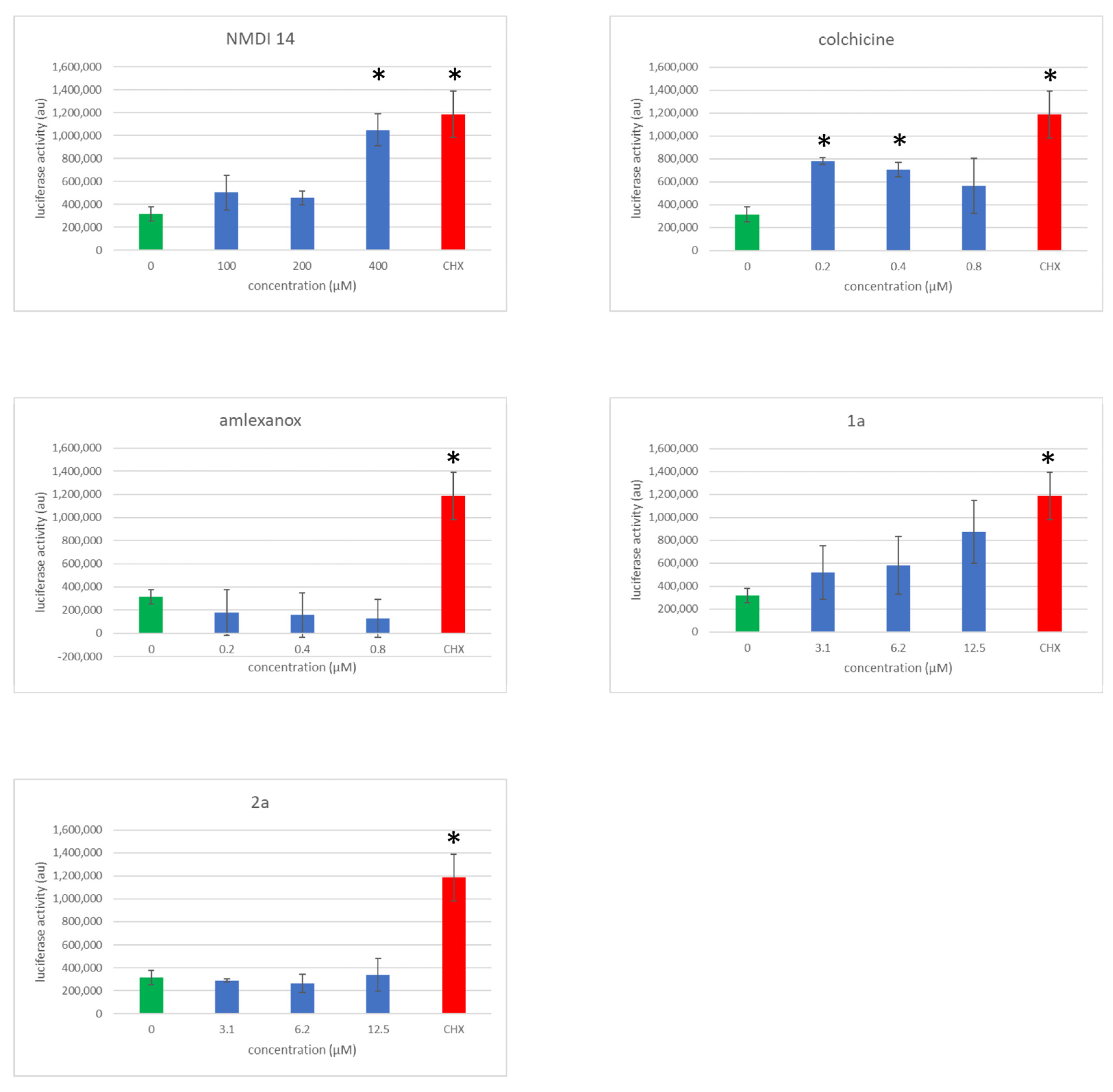
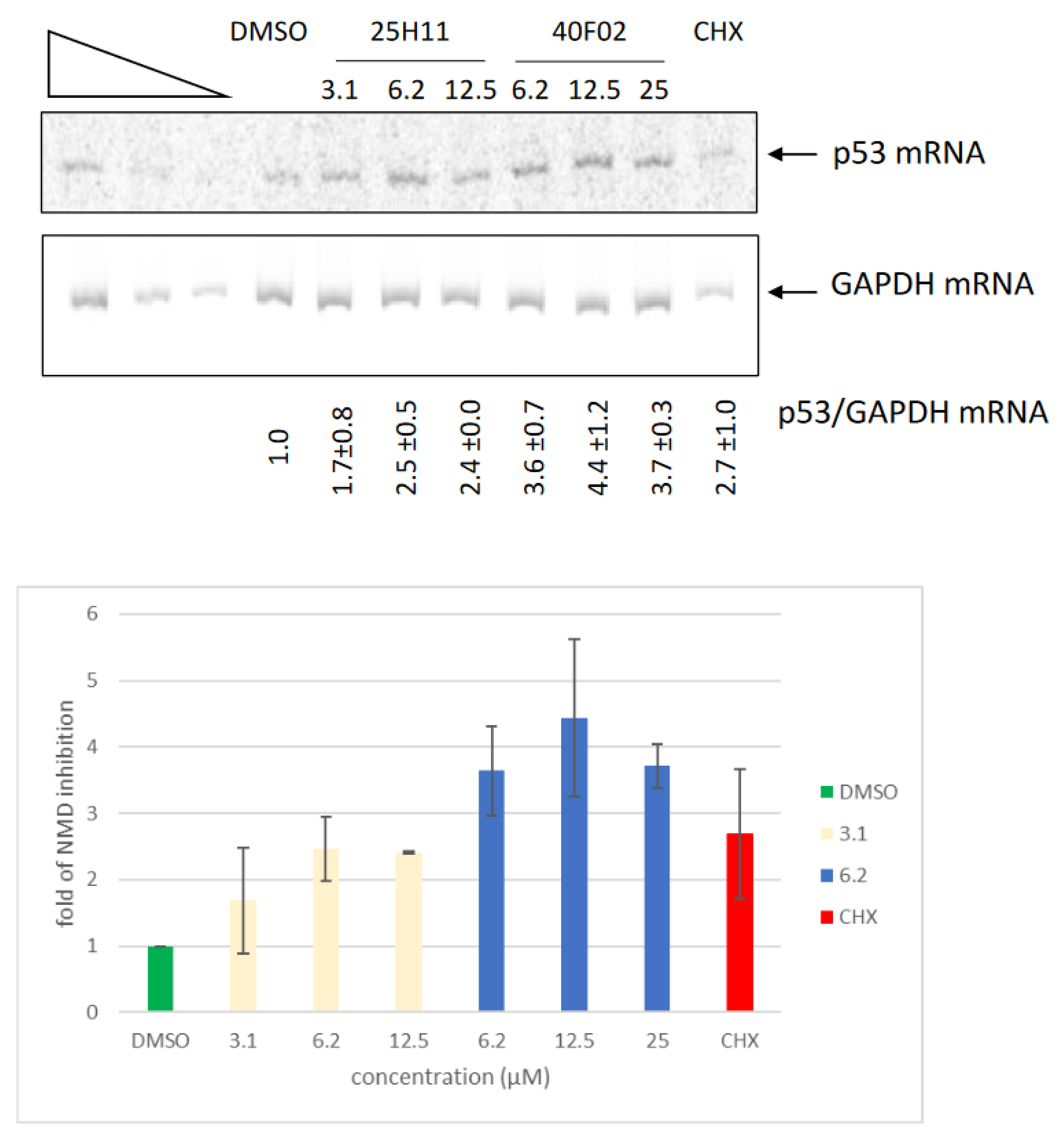
3.4. Comparison of the NMD Inhibition Efficacy of Molecules 1a and 2a versus Other NMD Inhibitors
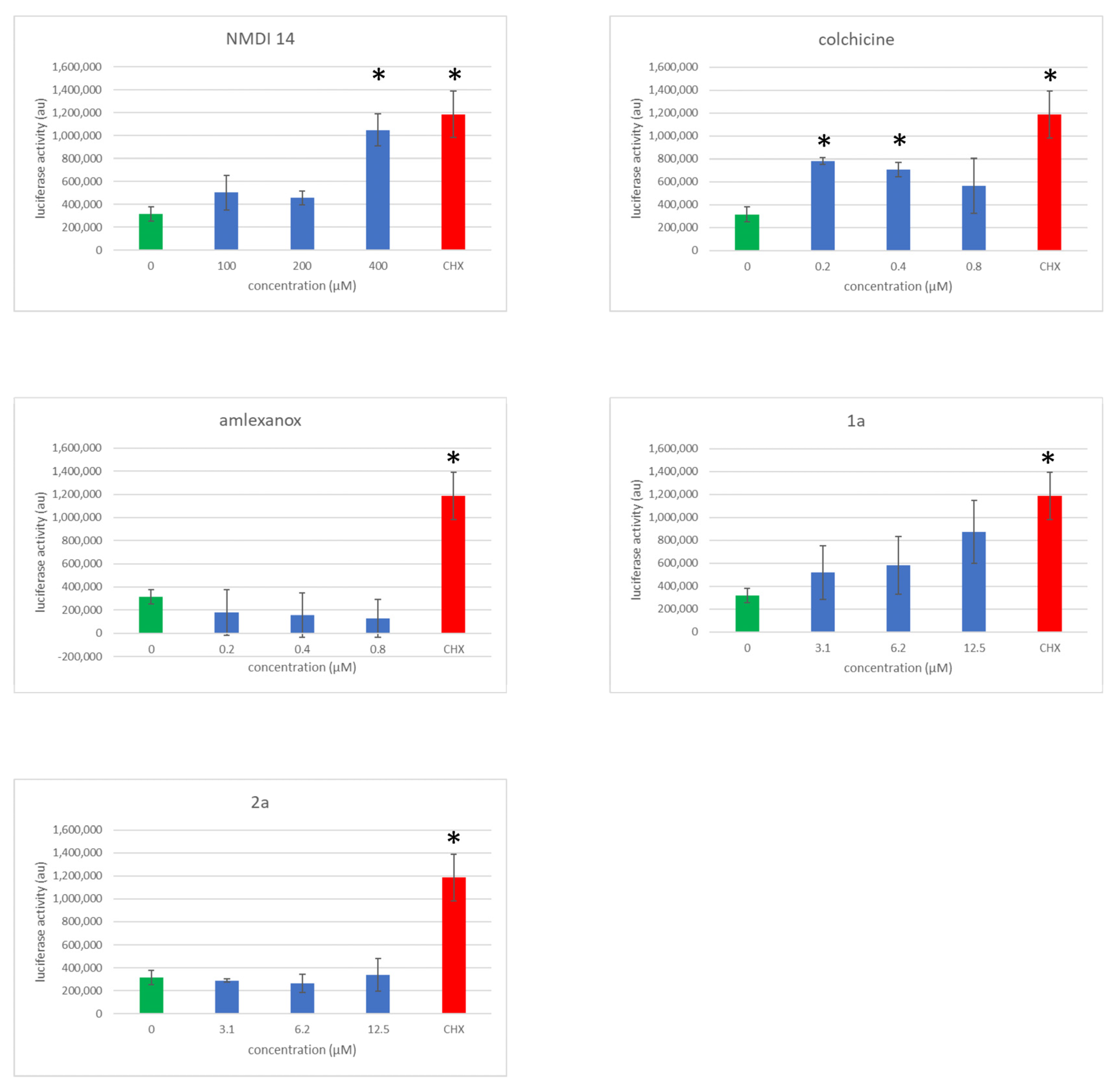
3.5. Evaluation of NMD Inhibitor Molecule Cytotoxicity
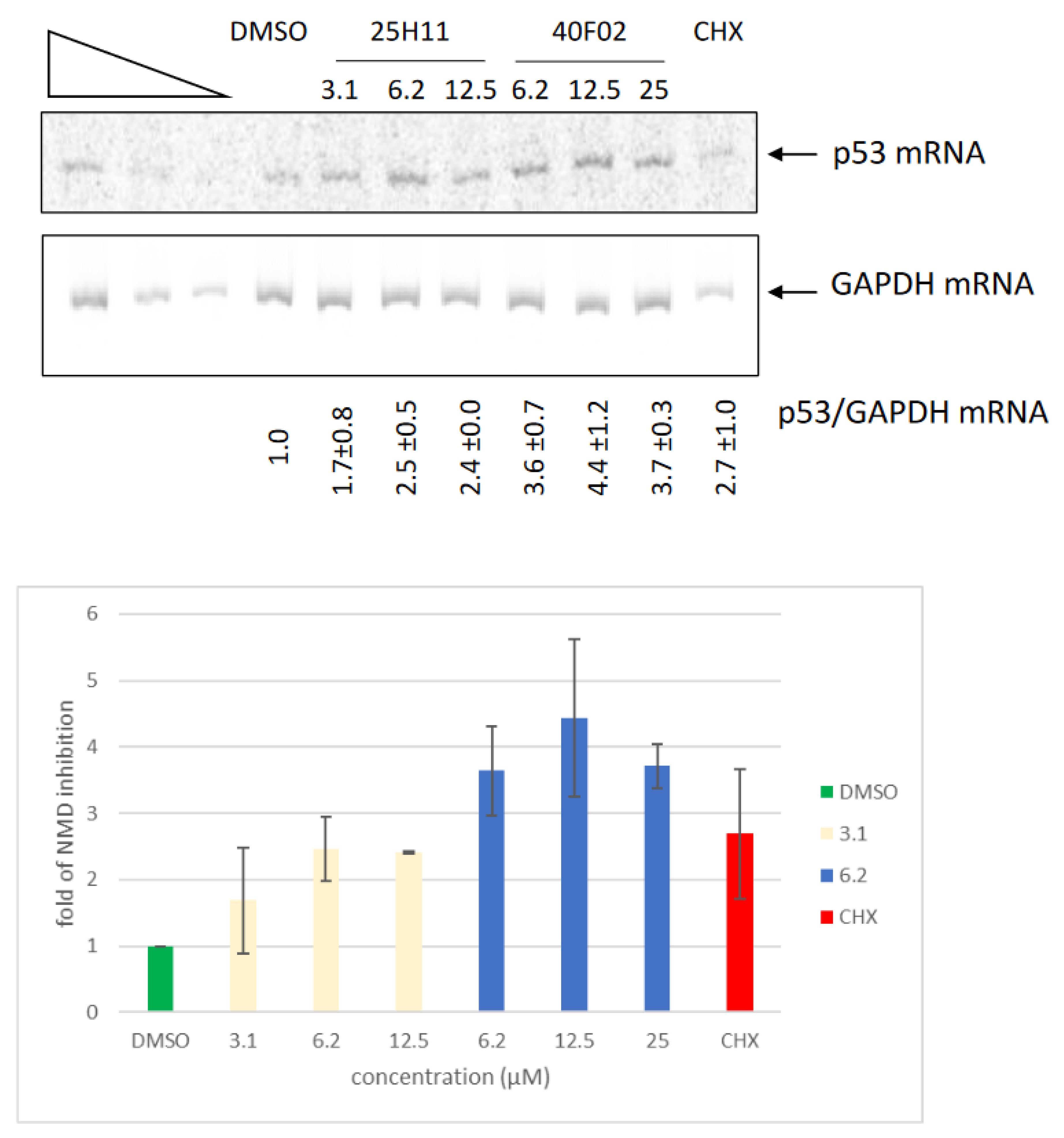
3.6. Inhibition of Endogenous NMD by Molecules 1a and 2a
4. Conclusions
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008, 29, 1037–1047. [Google Scholar] [CrossRef]
- Yi, Z.; Sanjeev, M.; Singh, G. The Branched Nature of the Nonsense-Mediated mRNA Decay Pathway. Trends Genet. 2021, 37, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, G.; Fernandes, R.; Garcia-Moreno, J.F.; Romao, L. Nonsense-mediated RNA decay and its bipolar function in cancer. Mol. Cancer 2021, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Mailliot, J.; Schaffitzel, C. Nonsense-Mediated mRNA Decay Factor Functions in Human Health and Disease. Biomedicines 2023, 11, 722. [Google Scholar] [CrossRef]
- Popp, M.W.; Maquat, L.E. Nonsense-mediated mRNA Decay and Cancer. Curr. Opin. Genet. Dev. 2018, 48, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, F. Nonsense-Mediated mRNA Decay, a Finely Regulated Mechanism. Biomedicines 2022, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 2017, 50, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, F.; Bagchi, D.; Le Hir, H.; Croquette, V. Human Upf1 is a highly processive RNA helicase and translocase with RNP remodelling activities. Nat. Commun. 2015, 6, 7581. [Google Scholar] [CrossRef]
- Kanaan, J.; Raj, S.; Decourty, L.; Saveanu, C.; Croquette, V.; Le Hir, H. UPF1-like helicase grip on nucleic acids dictates processivity. Nat. Commun. 2018, 9, 3752. [Google Scholar] [CrossRef] [PubMed]
- Franks, T.M.; Singh, G.; Lykke-Andersen, J. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense- mediated mRNA decay. Cell 2010, 143, 938–950. [Google Scholar] [CrossRef]
- Kashima, I.; Yamashita, A.; Izumi, N.; Kataoka, N.; Morishita, R.; Hoshino, S.; Ohno, M.; Dreyfuss, G.; Ohno, S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006, 20, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.; Leroy, C.; Salome-Desnoulez, S.; Werkmeister, E.; Kong, R.; Mongy, M.; Le Hir, H.; Lejeune, F. A role for AKT1 in nonsense-mediated mRNA decay. Nucleic Acids Res. 2021, 49, 11022–11037. [Google Scholar] [CrossRef]
- Cho, H.; Abshire, E.T.; Popp, M.W.; Proschel, C.; Schwartz, J.L.; Yeo, G.W.; Maquat, L.E. AKT constitutes a signal-promoted alternative exon-junction complex that regulates nonsense-mediated mRNA decay. Mol. Cell 2022, 82, 2779–2796.e2710. [Google Scholar] [CrossRef] [PubMed]
- Martins-Dias, P.; Romao, L. Nonsense suppression therapies in human genetic diseases. Cell. Mol. Life Sci. 2021, 78, 4677–4701. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, F. Triple effect of nonsense-mediated mRNA decay inhibition on cancer. Single Cell Biol. 2016, 5, 136. [Google Scholar] [CrossRef]
- Karam, R.; Wengrod, J.; Gardner, L.B.; Wilkinson, M.F. Regulation of nonsense-mediated mRNA decay: Implications for physiology and disease. Biochim. Biophys. Acta 2013, 1829, 624–633. [Google Scholar] [CrossRef]
- Zanello, G.; Garrido-Estepa, M.; Crespo, A.; O’Connor, D.; Nabbout, R.; Waters, C.; Hall, A.; Taglialatela, M.; Chan, C.H.; Pearce, D.A.; et al. Targeting shared molecular etiologies to accelerate drug development for rare diseases. EMBO Mol. Med. 2023, 15, e17159. [Google Scholar] [CrossRef]
- Popp, M.W.; Maquat, L.E. Attenuation of nonsense-mediated mRNA decay facilitates the response to chemotherapeutics. Nat. Commun. 2015, 6, 6632. [Google Scholar] [CrossRef]
- Jia, J.; Furlan, A.; Gonzalez-Hilarion, S.; Leroy, C.; Gruenert, D.C.; Tulasne, D.; Lejeune, F. Caspases shutdown nonsense-mediated mRNA decay during apoptosis. Cell Death Differ. 2015, 22, 1754–1763. [Google Scholar] [CrossRef]
- Durand, S.; Cougot, N.; Mahuteau-Betzer, F.; Nguyen, C.H.; Grierson, D.S.; Bertrand, E.; Tazi, J.; Lejeune, F. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J. Cell Biol. 2007, 178, 1145–1160. [Google Scholar] [CrossRef]
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Deprez, B.; Lejeune, F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Werkmeister, E.; Gonzalez-Hilarion, S.; Leroy, C.; Gruenert, D.C.; Lafont, F.; Tulasne, D.; Lejeune, F. Premature termination codon readthrough in human cells occurs in novel cytoplasmic foci and requires UPF proteins. J. Cell Sci. 2017, 130, 3009–3022. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, J.; Shu, M.D.; Steitz, J.A. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 2000, 103, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Grand, L.; Pouleriguen, T.; Queneau, Y.; da Silva, P.; Rahbe, Y.; Poessel, J.L.; Moebs-Sanchez, S. Synthesis of new dicinnamoyl 4-deoxy quinic acid and methyl ester derivatives and evaluation of the toxicity against the pea aphid Acyrthosiphon pisum. Org. Biomol. Chem. 2016, 14, 2487–2497. [Google Scholar] [CrossRef]
- Li, X.; Sivignon, C.; da Silva, P.; Rahbé, Y.; Queneau, Y.; Moebs-Sanchez, S. Design and synthesis of 3, 5- hetero diesters of 4-deoxy quinic acid and their aphicidal activity against Acyrthosiphon pisum. Tetrahedron 2021, 83, 131982–131992. [Google Scholar] [CrossRef]
- Benhabiles, H.; Gonzalez-Hilarion, S.; Amand, S.; Bailly, C.; Prevotat, A.; Reix, P.; Hubert, D.; Adriaenssens, E.; Rebuffat, S.; Tulasne, D.; et al. Optimized approach for the identification of highly efficient correctors of nonsense mutations in human diseases. PLoS ONE 2017, 12, e0187930. [Google Scholar] [CrossRef] [PubMed]
- Correa-Cerro, L.S.; Wassif, C.A.; Waye, J.S.; Krakowiak, P.A.; Cozma, D.; Dobson, N.R.; Levin, S.W.; Anadiotis, G.; Steiner, R.D.; Krajewska-Walasek, M.; et al. DHCR7 nonsense mutations and characterisation of mRNA nonsense mediated decay in Smith-Lemli-Opitz syndrome. J. Med. Genet. 2005, 42, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Trzaska, C.; Amand, S.; Bailly, C.; Leroy, C.; Marchand, V.; Duvernois-Berthet, E.; Saliou, J.M.; Benhabiles, H.; Werkmeister, E.; Chassat, T.; et al. 2,6-Diaminopurine as a highly potent corrector of UGA nonsense mutations. Nat. Commun. 2020, 11, 1509. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Grigoryan, A.; Wang, D.; Wang, J.; Breda, L.; Rivella, S.; Cardozo, T.; Gardner, L.B. Identification and characterization of small molecules that inhibit nonsense mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 2014, 74, 3104–3113. [Google Scholar] [CrossRef]
- Cozens, A.L.; Yezzi, M.J.; Chin, L.; Simon, E.M.; Finkbeiner, W.E.; Wagner, J.A.; Gruenert, D.C. Characterization of immortal cystic fibrosis tracheobronchial gland epithelial cells. Proc. Natl. Acad. Sci. USA 1992, 89, 5171–5175. [Google Scholar] [CrossRef]
- Sureau, A.; Gattoni, R.; Dooghe, Y.; Stevenin, J.; Soret, J. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 2001, 20, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.T.; Sharifi, N.A.; Meyers, J.L.; Martinez-Murillo, F.; Dietz, H.C. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 2004, 36, 1073–1078. [Google Scholar] [CrossRef]
- Palma, M.; Lejeune, F. Deciphering the molecular mechanism of stop codon readthrough. Biol. Rev. Camb. Philos. Soc. 2021, 96, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Kolonias, D.; Giangrande, P.H.; Gilboa, E. Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature 2010, 465, 227–230. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrard, J.; Ratajczak, F.; Elsens, J.; Leroy, C.; Kong, R.; Geoffroy, L.; Comte, A.; Fournet, G.; Joseph, B.; Li, X.; et al. Identifying Potent Nonsense-Mediated mRNA Decay Inhibitors with a Novel Screening System. Biomedicines 2023, 11, 2801. https://doi.org/10.3390/biomedicines11102801
Carrard J, Ratajczak F, Elsens J, Leroy C, Kong R, Geoffroy L, Comte A, Fournet G, Joseph B, Li X, et al. Identifying Potent Nonsense-Mediated mRNA Decay Inhibitors with a Novel Screening System. Biomedicines. 2023; 11(10):2801. https://doi.org/10.3390/biomedicines11102801
Chicago/Turabian StyleCarrard, Julie, Fiona Ratajczak, Joséphine Elsens, Catherine Leroy, Rebekah Kong, Lucie Geoffroy, Arnaud Comte, Guy Fournet, Benoît Joseph, Xiubin Li, and et al. 2023. "Identifying Potent Nonsense-Mediated mRNA Decay Inhibitors with a Novel Screening System" Biomedicines 11, no. 10: 2801. https://doi.org/10.3390/biomedicines11102801
APA StyleCarrard, J., Ratajczak, F., Elsens, J., Leroy, C., Kong, R., Geoffroy, L., Comte, A., Fournet, G., Joseph, B., Li, X., Moebs-Sanchez, S., & Lejeune, F. (2023). Identifying Potent Nonsense-Mediated mRNA Decay Inhibitors with a Novel Screening System. Biomedicines, 11(10), 2801. https://doi.org/10.3390/biomedicines11102801