

Apabetalone, a Clinical-Stage, Selective BET Inhibitor, Opposes DUX4 Target Gene Expression in Primary Human FSHD Muscle Cells
 ,
,
Abstract
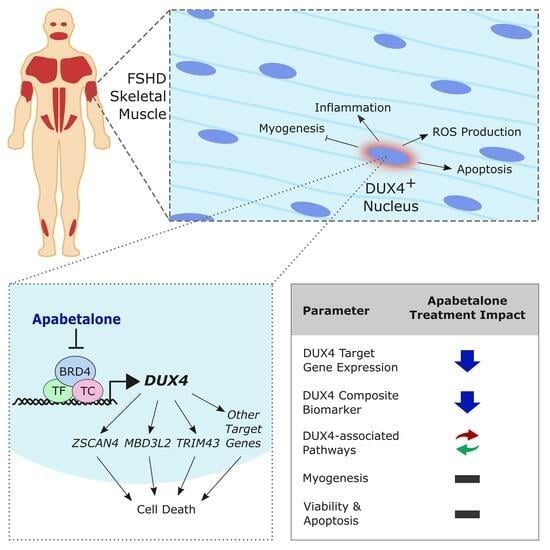
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. mRNA Expression Quantification by Real-Time PCR
2.4. Immunofluorescence
2.5. Cell Viability and Apoptosis Assays
2.6. RNA-Sequencing
2.7. Composite Biomarker Calculation
2.8. Pathway Analysis
2.9. Statistical Analysis
3. Results
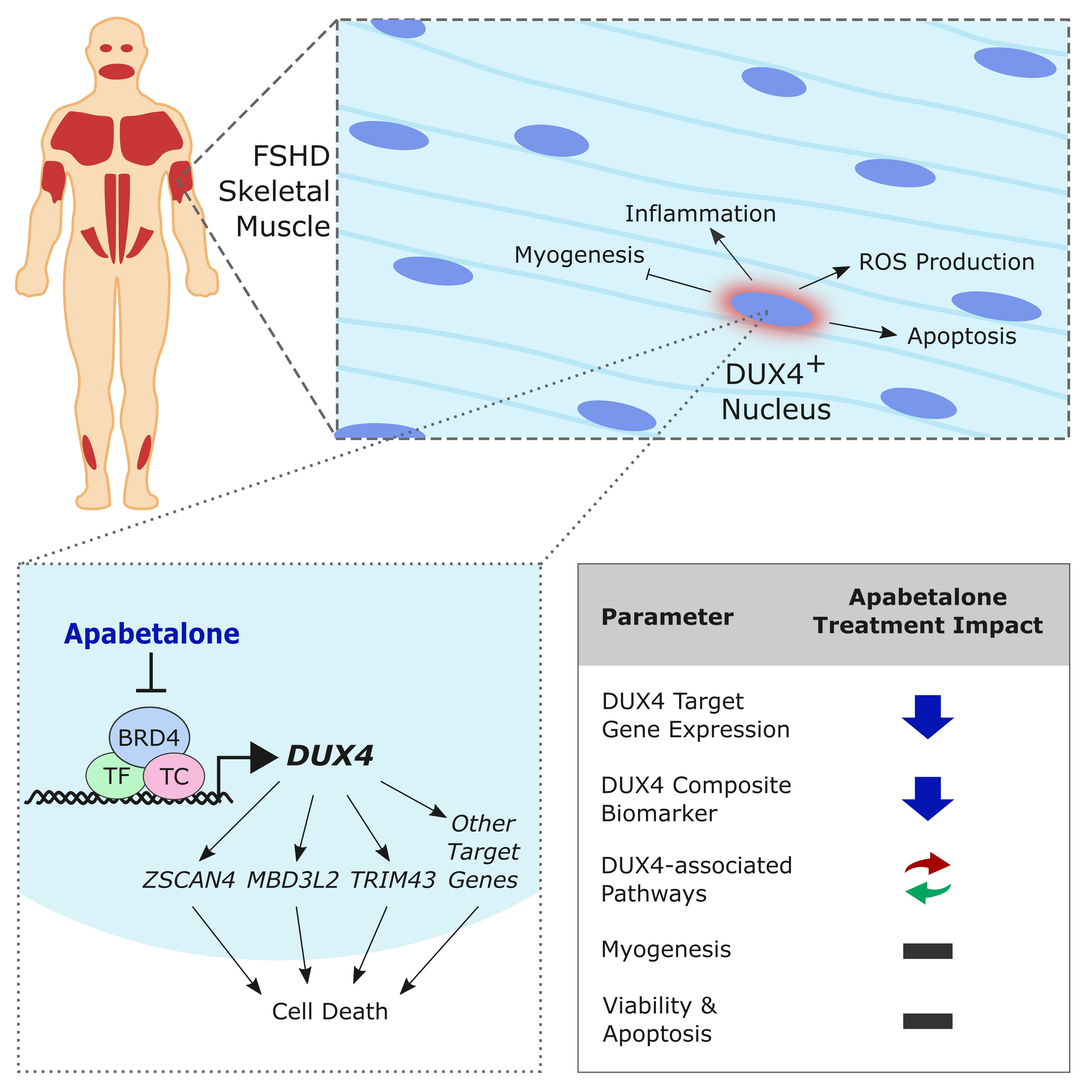
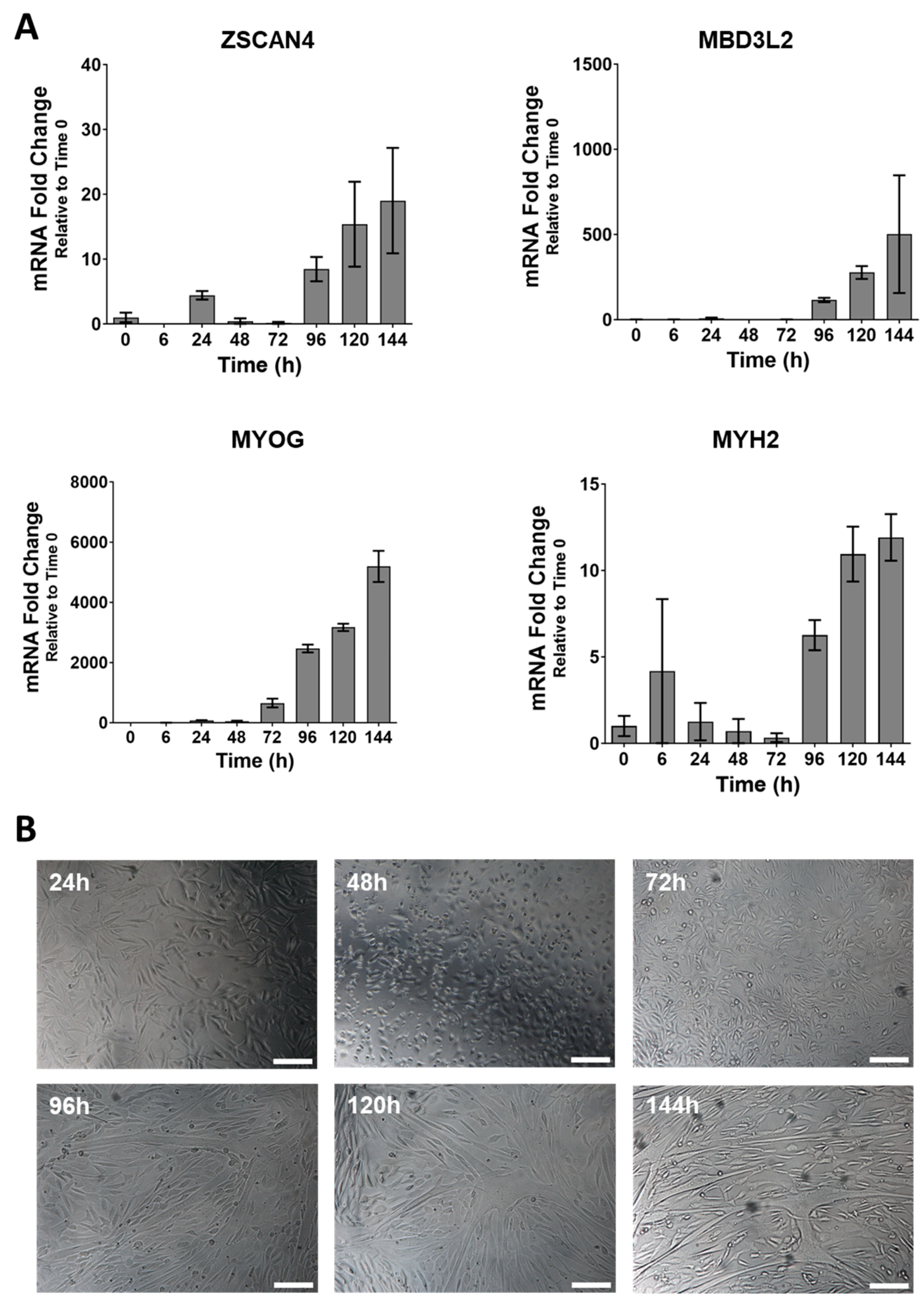
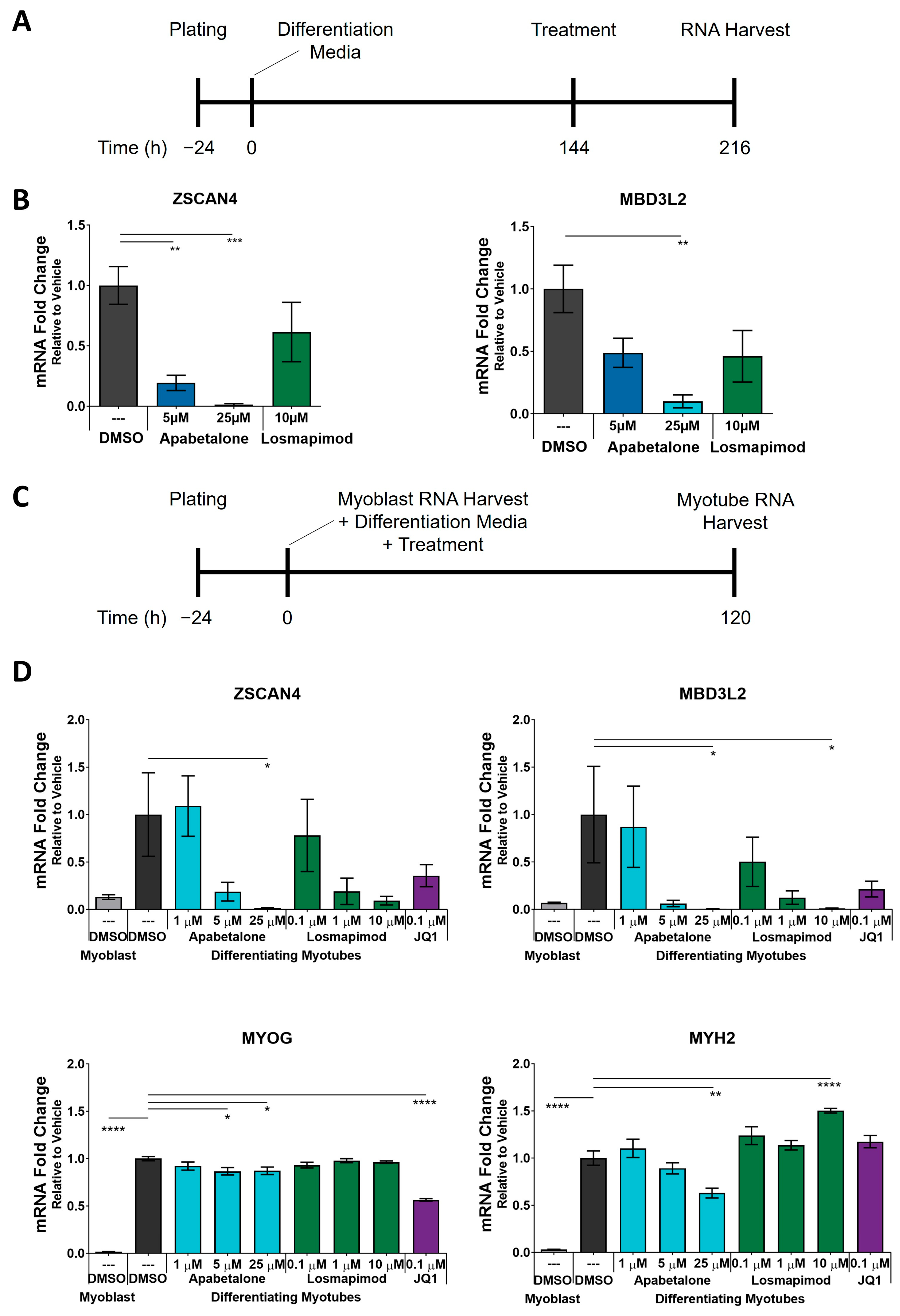
3.1. DUX4 Target Gene Expression Increases during Differentiation of Primary FSHD Muscle Cells and Is Countered by Apabetalone Treatment
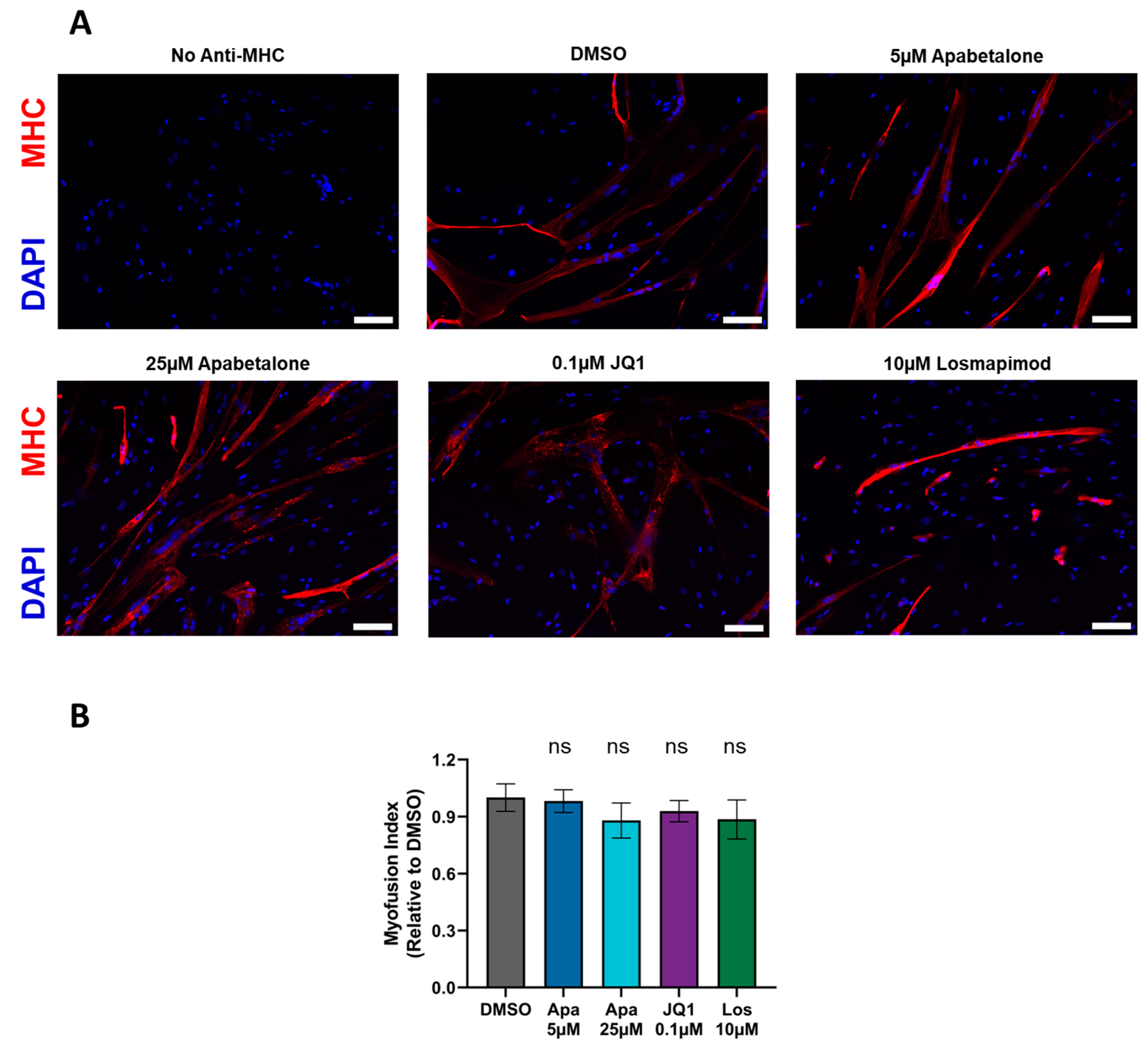
3.2. Myotube Fusion Is Unaffected by Apabetalone, JQ1, or Losmapimod Treatment
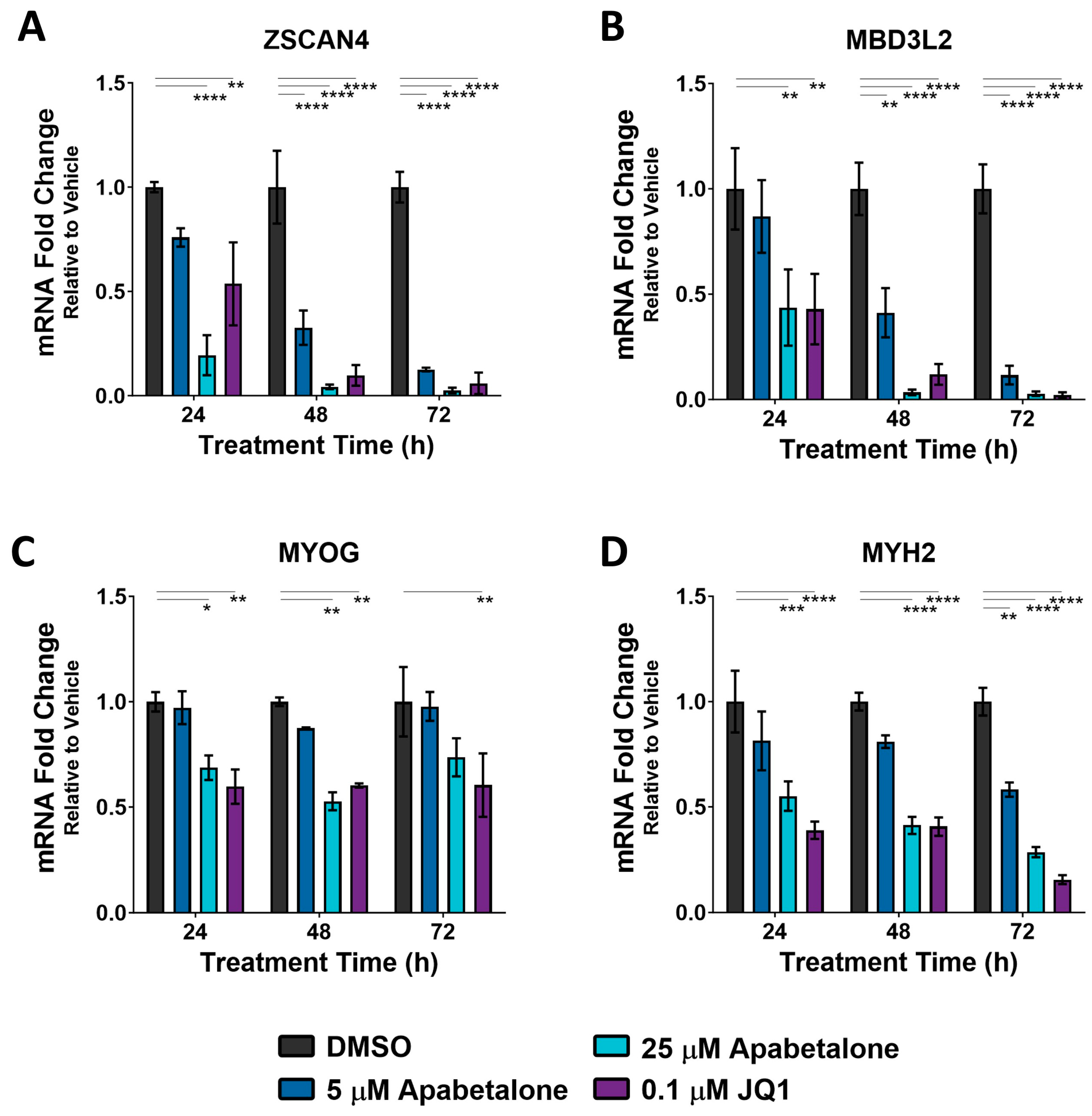
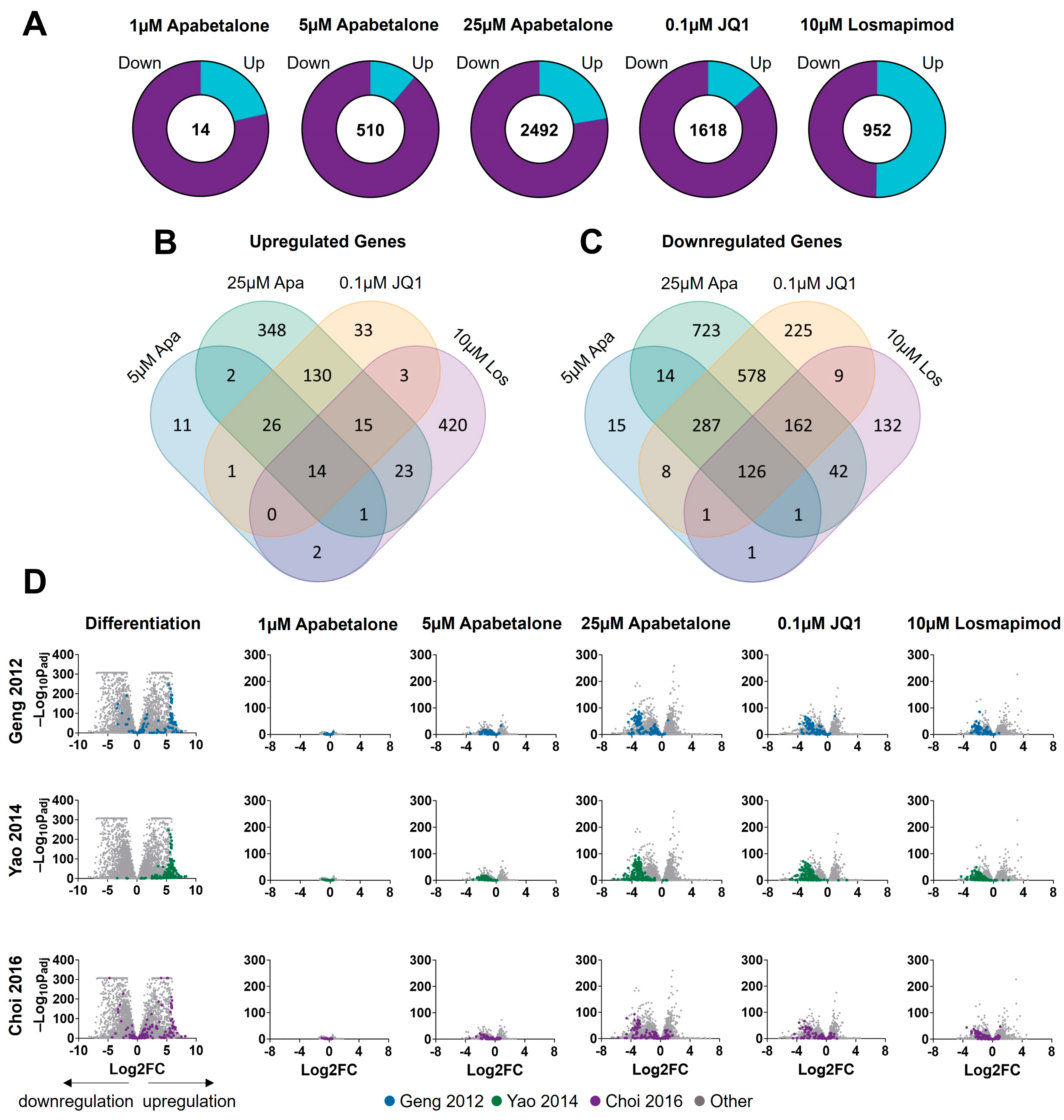
3.3. Apabetalone-Mediated DUX4 Target Gene Inhibition Compares Favorably to That of Losmapimod
3.4. Apabetalone Benefits Individual and Composite Markers of FSHD
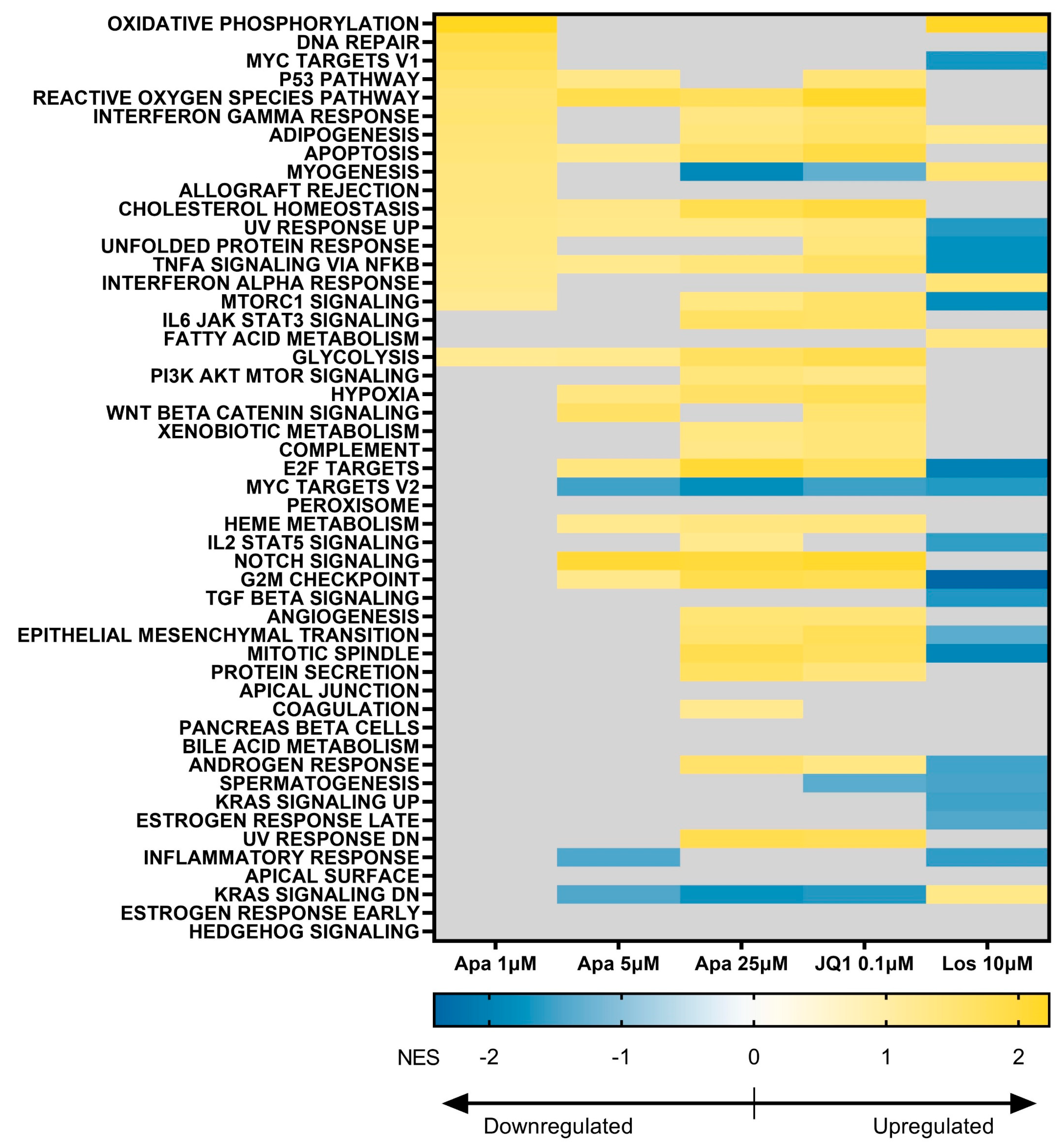
3.5. BET Inhibition and p38 Inhibition Have Different Impacts on DUX4-linked Pathways
3.6. Cell Viability and Apoptosis in pHSMCs Are Not Negatively Impacted with Apabetalone Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orrell, R.W. Facioscapulohumeral dystrophy and scapuloperoneal syndromes. Handb. Clin. Neurol. 2011, 101, 167–180. [Google Scholar] [CrossRef]
- Deenen, J.C.; Arnts, H.; van der Maarel, S.M.; Padberg, G.W.; Verschuuren, J.J.; Bakker, E.; Weinreich, S.S.; Verbeek, A.L.; van Engelen, B.G. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 2014, 83, 1056–1059. [Google Scholar] [CrossRef]
- Lemmers, R.J.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camano, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef]
- Deidda, G.; Cacurri, S.; Grisanti, P.; Vigneti, E.; Piazzo, N.; Felicetti, L. Physical mapping evidence for a duplicated region on chromosome 10qter showing high homology with the facioscapulohumeral muscular dystrophy locus on chromosome 4qter. Eur. J. Hum. Genet. 1995, 3, 155–167. [Google Scholar] [CrossRef]
- Wijmenga, C.; Sandkuijl, L.A.; Moerer, P.; van der Boorn, N.; Bodrug, S.E.; Ray, P.N.; Brouwer, O.F.; Murray, J.C.; van Ommen, G.J.; Padberg, G.W.; et al. Genetic linkage map of facioscapulohumeral muscular dystrophy and five polymorphic loci on chromosome 4q35-qter. Am. J. Hum. Genet. 1992, 51, 411–415. [Google Scholar]
- Lemmers, R.J.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, M.L.; Lemmers, R.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 activates germline genes, retroelements, and immune mediators: Implications for facioscapulohumeral dystrophy. Dev. Cell 2012, 22, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.M.; Petek, L.M.; Miller, D.G. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 2015, 24, 5901–5914. [Google Scholar] [CrossRef]
- Lek, A.; Zhang, Y.; Woodman, K.G.; Huang, S.; DeSimone, A.M.; Cohen, J.; Ho, V.; Conner, J.; Mead, L.; Kodani, A.; et al. Applying genome-wide CRISPR-Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci. Transl. Med. 2020, 12, eaay0271. [Google Scholar] [CrossRef]
- Mariot, V.; Joubert, R.; Le Gall, L.; Sidlauskaite, E.; Hourde, C.; Duddy, W.; Voit, T.; Bencze, M.; Dumonceaux, J. RIPK3-mediated cell death is involved in DUX4-mediated toxicity in facioscapulohumeral dystrophy. J. Cachexia Sarcopenia Muscle 2021, 12, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.E.; Dyle, M.C.; Albanese, R.; Matheny, T.; Sudheendran, K.; Cortazar, M.A.; Forman, T.; Fu, R.; Gillen, A.E.; Caruthers, M.H.; et al. Compromised nonsense-mediated RNA decay results in truncated RNA-binding protein production upon DUX4 expression. Cell Rep. 2023, 42, 112642. [Google Scholar] [CrossRef] [PubMed]
- Block, G.J.; Narayanan, D.; Amell, A.M.; Petek, L.M.; Davidson, K.C.; Bird, T.D.; Tawil, R.; Moon, R.T.; Miller, D.G. Wnt/beta-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum. Mol. Genet. 2013, 22, 4661–4672. [Google Scholar] [CrossRef]
- Choi, S.H.; Gearhart, M.D.; Cui, Z.; Bosnakovski, D.; Kim, M.; Schennum, N.; Kyba, M. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016, 44, 5161–5173. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; Gearhart, M.D.; Toso, E.A.; Recht, O.O.; Cucak, A.; Jain, A.K.; Barton, M.C.; Kyba, M. p53-independent DUX4 pathology in cell and animal models of facioscapulohumeral muscular dystrophy. Dis. Model Mech. 2017, 10, 1211–1216. [Google Scholar] [CrossRef]
- Bosnakovski, D.; Chan, S.S.K.; Recht, O.O.; Hartweck, L.M.; Gustafson, C.J.; Athman, L.L.; Lowe, D.A.; Kyba, M. Muscle pathology from stochastic low level DUX4 expression in an FSHD mouse model. Nat. Commun. 2017, 8, 550. [Google Scholar] [CrossRef] [PubMed]
- Haynes, P.; Kernan, K.; Zhou, S.L.; Miller, D.G. Expression patterns of FSHD-causing DUX4 and myogenic transcription factors PAX3 and PAX7 are spatially distinct in differentiating human stem cell cultures. Skelet. Muscle 2017, 7, 13. [Google Scholar] [CrossRef]
- Wallace, L.M.; Garwick, S.E.; Mei, W.; Belayew, A.; Coppee, F.; Ladner, K.J.; Guttridge, D.; Yang, J.; Harper, S.Q. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann. Neurol 2011, 69, 540–552. [Google Scholar] [CrossRef]
- Banerji, C.R.S.; Panamarova, M.; Hebaishi, H.; White, R.B.; Relaix, F.; Severini, S.; Zammit, P.S. PAX7 target genes are globally repressed in facioscapulohumeral muscular dystrophy skeletal muscle. Nat. Commun. 2017, 8, 2152. [Google Scholar] [CrossRef]
- Snider, L.; Geng, L.N.; Lemmers, R.J.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181. [Google Scholar] [CrossRef]
- Block, G.J.; Petek, L.M.; Narayanan, D.; Amell, A.M.; Moore, J.M.; Rabaia, N.A.; Tyler, A.; van der Maarel, S.M.; Tawil, R.; Filippova, G.N.; et al. Asymmetric bidirectional transcription from the FSHD-causing D4Z4 array modulates DUX4 production. PLoS ONE 2012, 7, e35532. [Google Scholar] [CrossRef]
- Tassin, A.; Laoudj-Chenivesse, D.; Vanderplanck, C.; Barro, M.; Charron, S.; Ansseau, E.; Chen, Y.W.; Mercier, J.; Coppee, F.; Belayew, A. DUX4 expression in FSHD muscle cells: How could such a rare protein cause a myopathy? J. Cell Mol. Med. 2013, 17, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Shadle, S.C.; Zhong, J.W.; Campbell, A.E.; Conerly, M.L.; Jagannathan, S.; Wong, C.J.; Morello, T.D.; van der Maarel, S.M.; Tapscott, S.J. DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy. PLoS Genet. 2017, 13, e1006658. [Google Scholar] [CrossRef] [PubMed]
- Banerji, C.R.S.; Zammit, P.S. Pathomechanisms and biomarkers in facioscapulohumeral muscular dystrophy: Roles of DUX4 and PAX7. EMBO Mol. Med. 2021, 13, e13695. [Google Scholar] [CrossRef]
- Zammit, P.S.; Relaix, F.; Nagata, Y.; Ruiz, A.P.; Collins, C.A.; Partridge, T.A.; Beauchamp, J.R. Pax7 and myogenic progression in skeletal muscle satellite cells. J. Cell Sci. 2006, 119, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Maltzahn, J.V.; Jones, A.E.; Parks, R.J.; Rudnicki, M.A. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. USA 2013, 110, 16474–16479. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; Xu, Z.; Gang, E.J.; Galindo, C.L.; Liu, M.; Simsek, T.; Garner, H.R.; Agha-Mohammadi, S.; Tassin, A.; Coppee, F.; et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. Embo. J. 2008, 27, 2766–2779. [Google Scholar] [CrossRef]
- Oliva, J.; Galasinski, S.; Richey, A.; Campbell, A.E.; Meyers, M.J.; Modi, N.; Zhong, J.W.; Tawil, R.; Tapscott, S.J.; Sverdrup, F.M. Clinically Advanced p38 Inhibitors Suppress DUX4 Expression in Cellular and Animal Models of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol Exp. Ther. 2019, 370, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Rojas, L.A.; Valentine, E.; Accorsi, A.; Maglio, J.; Shen, N.; Robertson, A.; Kazmirski, S.; Rahl, P.; Tawil, R.; Cadavid, D.; et al. p38α Regulates Expression of DUX4 in a Model of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol. Exp. Ther. 2020, 374, 489–498. [Google Scholar] [CrossRef]
- Campbell, A.E.; Oliva, J.; Yates, M.P.; Zhong, J.W.; Shadle, S.C.; Snider, L.; Singh, N.; Tai, S.; Hiramuki, Y.; Tawil, R.; et al. BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet. Muscle 2017, 7, 16. [Google Scholar] [CrossRef]
- Kulikowski, E.; Rakai, B.D.; Wong, N.C.W. Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med. Res. Rev. 2021, 41, 223–245. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, L.M.; Kharenko, O.A.; Stotz, S.C.; Rakai, B.D.; Sarsons, C.D.; Gilham, D.; Wasiak, S.; Fu, L.; Sweeney, M.; Johansson, J.O.; et al. Breaking boundaries: Pan BETi disrupt 3D chromatin structure, BD2-selective BETi are strictly epigenetic transcriptional regulators. Biomed. Pharmacother. 2022, 152, 113230. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, T.I.; Virbasius, C.M.; Zhu, L.J.; Green, M.R.; Jones, P.L. Identification of Epigenetic Regulators of DUX4-fl for Targeted Therapy of Facioscapulohumeral Muscular Dystrophy. Mol. Ther. 2018, 26, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef]
- Ray, K.K.; Nicholls, S.J.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Effect of Apabetalone Added to Standard Therapy on Major Adverse Cardiovascular Events in Patients With Recent Acute Coronary Syndrome and Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2020, 323, 1565–1573. [Google Scholar] [CrossRef]
- Provencher, S.; Potus, F.; Blais-Lecours, P.; Bernard, S.; Martineau, S.; Breuils-Bonnet, S.; Weatherald, J.; Sweeney, M.; Kulikowski, E.; Boucherat, O.; et al. BET Protein Inhibition for Pulmonary Arterial Hypertension: A Pilot Clinical Trial. Am. J. Respir. Crit. Care Med. 2022, 205, 1357–1360. [Google Scholar] [CrossRef]
- Gilham, D.; Smith, A.L.; Fu, L.; Moore, D.Y.; Muralidharan, A.; Reid, S.P.M.; Stotz, S.C.; Johansson, J.O.; Sweeney, M.; Wong, N.C.W.; et al. Bromodomain and Extraterminal Protein Inhibitor, Apabetalone (RVX-208), Reduces ACE2 Expression and Attenuates SARS-CoV-2 Infection In Vitro. Biomedicines 2021, 9, 437. [Google Scholar] [CrossRef]
- Abramson, J.S.; Blum, K.A.; Flinn, I.W.; Gutierrez, M.; Goy, A.; Maris, M.; Cooper, M.; O’Meara, M.; Borger, D.; Mertz, J.; et al. BET Inhibitor CPI-0610 Is Well Tolerated and Induces Responses in Diffuse Large B-Cell Lymphoma and Follicular Lymphoma: Preliminary Analysis of an Ongoing Phase 1 Study. Blood 2015, 126, 1491. [Google Scholar] [CrossRef]
- Cruz, J.M.; Hupper, N.; Wilson, L.S.; Concannon, J.B.; Wang, Y.; Oberhauser, B.; Patora-Komisarska, K.; Zhang, Y.; Glass, D.J.; Trendelenburg, A.U.; et al. Protein kinase A activation inhibits DUX4 gene expression in myotubes from patients with facioscapulohumeral muscular dystrophy. J. Biol. Chem. 2018, 293, 11837–11849. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yao, Z.; Snider, L.; Balog, J.; Lemmers, R.J.; Van Der Maarel, S.M.; Tawil, R.; Tapscott, S.J. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum. Mol. Genet. 2014, 23, 5342–5352. [Google Scholar] [CrossRef] [PubMed]
- Banerji, C.R.S.; Zammit, P.S. PAX7 target gene repression is a superior FSHD biomarker than DUX4 target gene activation, associating with pathological severity and identifying FSHD at the single-cell level. Hum. Mol. Genet. 2019, 28, 2224–2236. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef]
- Rodriguez Cruz, P.M.; Cossins, J.; Beeson, D.; Vincent, A. The Neuromuscular Junction in Health and Disease: Molecular Mechanisms Governing Synaptic Formation and Homeostasis. Front. Mol. Neurosci. 2020, 13, 610964. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, P.; Bou Saada, Y.; Dib, C.; Ansseau, E.; Barat, A.; Hamade, A.; Dessen, P.; Robert, T.; Lazar, V.; Louzada, R.A.N.; et al. DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free Radic. Biol. Med. 2016, 99, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.J.; Han, L.H.; Cong, R.S.; Liang, J. Caspase family proteases and apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Haarhaus, M.; Ray, K.K.; Nicholls, S.J.; Schwartz, G.G.; Kulikowski, E.; Johansson, J.O.; Sweeney, M.; Halliday, C.; Lebioda, K.; Wong, N.; et al. Apabetalone lowers serum alkaline phosphatase and improves cardiovascular risk in patients with cardiovascular disease. Atherosclerosis 2019, 290, 59–65. [Google Scholar] [CrossRef]
- Jagannathan, S.; Shadle, S.C.; Resnick, R.; Snider, L.; Tawil, R.N.; van der Maarel, S.M.; Bradley, R.K.; Tapscott, S.J. Model systems of DUX4 expression recapitulate the transcriptional profile of FSHD cells. Hum. Mol. Genet. 2016, 25, 4419–4431. [Google Scholar] [CrossRef]
- Tyler, D.S.; Vappiani, J.; Caneque, T.; Lam, E.Y.N.; Ward, A.; Gilan, O.; Chan, Y.C.; Hienzsch, A.; Rutkowska, A.; Werner, T.; et al. Click chemistry enables preclinical evaluation of targeted epigenetic therapies. Science 2017, 356, 1397–1401. [Google Scholar] [CrossRef]
- Roberts, T.C.; Etxaniz, U.; Dall’Agnese, A.; Wu, S.-Y.; Chiang, C.-M.; Brennan, P.E.; Wood, M.J.A.; Puri, P.L. BRD3 and BRD4 BET Bromodomain Proteins Differentially Regulate Skeletal Myogenesis. Sci. Rep. 2017, 7, 6153. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene of Interest | Donor 1 | Donor 2 | |||
|---|---|---|---|---|---|
| Apabetalone IC50 (µM) | JQ1 IC50 (µM) | Apabetalone IC50 (µM) | JQ1 IC50 (µM) | ||
| DUX4 Targets | ZSCAN4 | 1.2 | 0.014 | 0.69 | 0.011 |
| MBD3L2 | 0.59 | 0.015 | nd | nd | |
| Differentiation | MYOG | 42 | 0.28 | >50 | 0.47 |
| MYH2 | 10 | 0.033 | >50 | 0.12 | |
| PAX7 | 33 | >3.0 | nd | nd | |
| Apabetalone | JQ1 | Losmapimod | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 µM | 5 µM | 25 µM | 0.1 µM | 10 µM | ||||||
| NES | FDR | NES | FDR | NES | FDR | NES | FDR | NES | FDR | |
| DUX4 Upregulated Pathways (Rickard 2015 [9]) | ||||||||||
| Spliceosome | ns | ns | ns | ns | −1.96 | 0.02 | ||||
| Basal Transcription Factors | ns | ns | ns | ns | −1.86 | 0.04 | ||||
| DUX4 Downregulated Pathways (Rickard 2015 [9]) | ||||||||||
| Focal Adhesion | ns | ns | ns | ns | −1.36 | 0.23 | ||||
| Apoptosis | ns | ns | ns | 1.31 | 0.21 | ns | ||||
| Lysosome | 1.73 | 0.02 | 1.92 | 0.02 | 2.62 | <0.001 | 2.46 | <0.001 | ns | |
| Glutathione Metabolism | 1.36 | 0.23 | ns | 1.68 | 0.04 | 1.62 | 0.09 | ns | ||
| Gap Junction | ns | ns | ns | ns | −1.49 | 0.23 | ||||
| Vascular Smooth Muscle Contraction | ns | ns | ns | ns | −1.36 | 0.24 | ||||
| Other Glycan Degradation | ns | ns | 1.84 | 0.02 | 1.57 | 0.08 | ns | |||
| p53 Signaling Pathway | ns | ns | 1.42 | 0.15 | ns | |||||
| GnRH Signaling Pathway | ns | ns | 1.26 | 0.23 | 1.26 | 0.24 | ns | |||
| DUX4 Bidirectionally Disrupted Pathways (Rickard 2015 [9]) | ||||||||||
| Endocytosis | ns | ns | 1.30 | 0.23 | ns | ns | ||||
| Adherens Junction | ns | ns | ns | ns | −1.55 | 0.17 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarsons, C.D.; Gilham, D.; Tsujikawa, L.M.; Wasiak, S.; Fu, L.; Rakai, B.D.; Stotz, S.C.; Carestia, A.; Sweeney, M.; Kulikowski, E. Apabetalone, a Clinical-Stage, Selective BET Inhibitor, Opposes DUX4 Target Gene Expression in Primary Human FSHD Muscle Cells. Biomedicines 2023, 11, 2683. https://doi.org/10.3390/biomedicines11102683
Sarsons CD, Gilham D, Tsujikawa LM, Wasiak S, Fu L, Rakai BD, Stotz SC, Carestia A, Sweeney M, Kulikowski E. Apabetalone, a Clinical-Stage, Selective BET Inhibitor, Opposes DUX4 Target Gene Expression in Primary Human FSHD Muscle Cells. Biomedicines. 2023; 11(10):2683. https://doi.org/10.3390/biomedicines11102683
Chicago/Turabian StyleSarsons, Christopher D., Dean Gilham, Laura M. Tsujikawa, Sylwia Wasiak, Li Fu, Brooke D. Rakai, Stephanie C. Stotz, Agostina Carestia, Michael Sweeney, and Ewelina Kulikowski. 2023. "Apabetalone, a Clinical-Stage, Selective BET Inhibitor, Opposes DUX4 Target Gene Expression in Primary Human FSHD Muscle Cells" Biomedicines 11, no. 10: 2683. https://doi.org/10.3390/biomedicines11102683
APA StyleSarsons, C. D., Gilham, D., Tsujikawa, L. M., Wasiak, S., Fu, L., Rakai, B. D., Stotz, S. C., Carestia, A., Sweeney, M., & Kulikowski, E. (2023). Apabetalone, a Clinical-Stage, Selective BET Inhibitor, Opposes DUX4 Target Gene Expression in Primary Human FSHD Muscle Cells. Biomedicines, 11(10), 2683. https://doi.org/10.3390/biomedicines11102683