

Noninvasive DBS-Based Approaches to Assist Clinical Diagnosis and Treatment Monitoring of Gaucher Disease

 ,
, 

Abstract
:1. Introduction
2. Case Presentation
2.1. Clinical Presentation
2.1.1. Family History
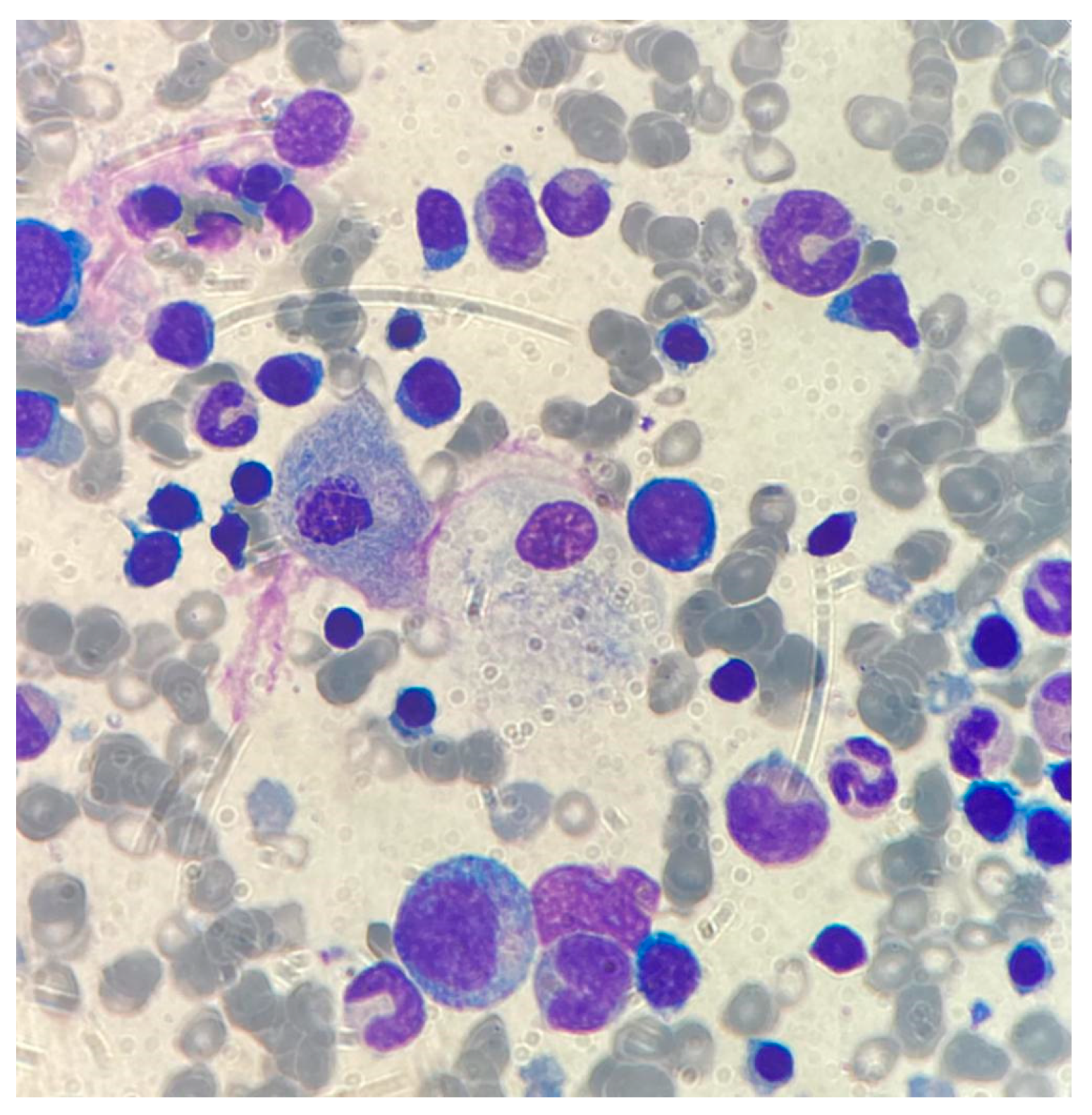
2.1.2. Case 1
2.1.3. Case 2
2.2. Newborn Screening Workflow: First and Second-Level Testing
2.3. Application of NBS Primary Screening and Second-Tier Testing on the Suspected Pediatric Patients and Relatives
2.4. Molecular Confirmation Testing by Sanger Sequencing
3. Diagnosis, Treatment and Follow-Up of the Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nalysnyk, L.; Rotella, P.; Simeone, J.C.; Hamed, A.; Weinreb, N. Gaucher disease epidemiology and natural history: A comprehensive review of the literature. Hematology 2017, 22, 65–73. [Google Scholar] [CrossRef]
- Dionisi-Vici, C.; Rizzo, C.; Burlina, A.B.; Caruso, U.; Sabetta, G.; Uziel, G.; Abeni, D. Inborn errors of metabolism in the Italian pediatric population: A national retrospective survey. J. Pediatr. 2002, 140, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Poorthuis, B.; Wevers, R.; Kleijer, W.; Groener, J.; de Jong, J.; van Weely, S.; Niezen-Koning, K.; van Diggelen, O. The frequency of lysosomal storage diseases in The Netherlands. Hum. Genet. 1999, 105, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E. New perspectives in type 2 Gaucher disease. Adv. Pediatr. 1997, 44, 73–107. [Google Scholar] [PubMed]
- Tylki-Szymańska, A.; Vellodi, A.; El-Beshlawy, A.; Cole, J.A.; Kolodny, E. Neuronopathic Gaucher disease: Demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J. Inherit. Metab. Dis. 2010, 33, 339–346. [Google Scholar] [CrossRef]
- Sidransky, E. Gaucher disease: Complexity in a ‘simple’ disorder. Mol. Genet. Metab. 2004, 83, 6–15. [Google Scholar] [CrossRef]
- Messner, M.C.; Cabot, M.C. Glucosylceramide in humans. Adv. Exp. Med. Biol. 2010, 688, 156–164. [Google Scholar] [CrossRef]
- Svennerholm, L.; Håkansson, G.; Månsson, J.E.; Nilsson, O. Chemical differentiation of the Gaucher subtypes. Prog. Clin. Biol. Res. 1982, 95, 231–252. [Google Scholar]
- Cox, T.M. Gaucher disease: Understanding the molecular pathogenesis of sphingolipidoses. J. Inherit. Metab. Dis. 2001, 24 (Suppl. S2), 106–121. [Google Scholar] [CrossRef]
- Moran, M.T.; Schofield, J.P.; Hayman, A.R.; Shi, G.P.; Young, E.; Cox, T.M. Pathologic gene expression in Gaucher disease: Up-regulation of cysteine proteinases including osteoclastic cathepsin K. Blood 2000, 96, 1969–1978. [Google Scholar] [CrossRef]
- Reed, M.; Baker, R.J.; Mehta, A.B.; Hughes, D.A. Enhanced differentiation of osteoclasts from mononuclear precursors in patients with Gaucher disease. Blood Cells Mol. Dis. 2013, 51, 185–194. [Google Scholar] [CrossRef]
- Berger, J.; Lecourt, S.; Vanneaux, V.; Rapatel, C.; Boisgard, S.; Caillaud, C.; Boiret-Dupré, N.; Chomienne, C.; Marolleau, J.-P.; Larghero, J.; et al. Glucocerebrosidase deficiency dramatically impairs human bone marrow haematopoiesis in an in vitro model of Gaucher disease. Br. J. Haematol. 2010, 150, 93–101. [Google Scholar] [CrossRef]
- Gelb, M.H.; Lukacs, Z.; Ranieri, E.; Schielen, P.C.J.I. Newborn Screening for Lysosomal Storage Disorders: Methodologies for Measurement of Enzymatic Activities in Dried Blood Spots. Int. J. Neonatal Screen. 2019, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Pession, A.; Di Rocco, M.; Venturelli, F.; Tappino, B.; Morello, W.; Santoro, N.; Giordano, P.; Filippini, B.; Rinieri, S.; Russo, G.; et al. GAU-PED study for early diagnosis of Gaucher disease in children with splenomegaly and cytopenia. Orphanet J. Rare Dis. 2023, 18, 151. [Google Scholar] [CrossRef] [PubMed]
- Cormand, B.; Montfort, M.; Chabás, A.; Vilageliu, L.; Grinberg, D. Genetic fine localization of the beta-glucocerebrosidase (GBA) and prosaposin (PSAP) genes: Implications for Gaucher disease. Hum. Genet. 1997, 100, 75–79. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Sawada, T.; Kido, J.; Sugawara, K.; Yoshida, S.; Matsumoto, S.; Shimazu, T.; Matsushita, Y.; Inoue, T.; Hirose, S.; Endo, F.; et al. Newborn screening for Gaucher disease in Japan. Mol. Genet. Metab. Rep. 2022, 31, 100850. [Google Scholar] [CrossRef]
- Orvisky, E.; Park, J.K.; E LaMarca, M.; I Ginns, E.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: Correlation with phenotype and genotype. Mol. Genet. Metab. 2002, 76, 262–270. [Google Scholar] [CrossRef]
- Gary, S.E.; Ryan, E.; Steward, A.M.; Sidransky, E. Recent advances in the diagnosis and management of Gaucher disease. Expert. Rev. Endocrinol. Metab. 2018, 13, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Dardis, A.; Michelakakis, H.; Rozenfeld, P.; Fumic, K.; Wagner, J.; Pavan, E.; Fuller, M.; Revel-Vilk, S.; Hughes, D.; Cox, T.; et al. Patient centered guidelines for the laboratory diagnosis of Gaucher disease type 1. Pract. Guidel. Orphanet J. Rare Dis. 2022, 17, 442. [Google Scholar] [CrossRef] [PubMed]
- Polo, G.; Burlina, A.P.; Ranieri, E.; Colucci, F.; Rubert, L.; Pascarella, A.; Duro, G.; Tummolo, A.; Padoan, A.; Plebani, M.; et al. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: A comparative study. Clin. Chem. Lab. Med. 2019, 57, 1863–1874. [Google Scholar] [CrossRef]
- Hurvitz, N.; Dinur, T.; Becker-Cohen, M.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Abramov, A.; Zimran, A.; et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. Int. J. Mol. Sci. 2019, 20, 3033. [Google Scholar] [CrossRef]
- Cozma, C.; Cullufi, P.; Kramp, G.; Hovakimyan, M.; Velmishi, V.; Gjikopulli, A.; Tomori, S.; Fischer, S.; Oppermann, S.; Grittner, U.; et al. Treatment Efficiency in Gaucher Patients Can Reliably Be Monitored by Quantification of Lyso-Gb1 Concentrations in Dried Blood Spots. Int. J. Mol. Sci. 2020, 21, 4577. [Google Scholar] [CrossRef] [PubMed]
- Anurathapan, U.; Tim-Aroon, T.; Zhang, W.; Sanpote, W.; Wongrungsri, S.; Khunin, N.; Chutipongtanate, S.; Chirdkiatgumchai, V.; Ngiwsara, L.; Jaovisidha, S.; et al. Comprehensive and long-term outcomes of enzyme replacement therapy followed by stem cell transplantation in children with Gaucher disease type 1 and 3. Pediatr. Blood Cancer 2023, 70, e30149. [Google Scholar] [CrossRef] [PubMed]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef]
- Castillon, G.; Chang, S.-C.; Moride, Y. Global Incidence and Prevalence of Gaucher Disease: A Targeted Literature Review. J. Clin. Med. 2022, 12, 85. [Google Scholar] [CrossRef]
- Bouayadi, O.; Lyagoubi, A.; Aarab, A.; Lamrabat, S.; Berhili, A.; Bensalah, M.; Seddik, R. Gaucher Disease: An Underdiagnosed Pathology in the Eastern Moroccan Population. EJIFCC 2019, 30, 82–87. [Google Scholar]
- Pastores, G.M.; Hughes, D.A. Gaucher Disease; National Institute of Health: Bethesda, MD, USA, 1993.
- Zimran, A.; Sorge, J.; Gross, E.; Kubitz, M.; West, C.; Beutler, E. Prediction of severity of Gaucher’s disease by identification of mutations at DNA level. Lancet 1989, 2, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Malini, E.; Grossi, S.; Deganuto, M.; Rosano, C.; Parini, R.; Dominisini, S.; Cariati, R.; Zampieri, S.; Bembi, B.; Filocamo, M.; et al. Functional analysis of 11 novel GBA alleles. Eur. J. Hum. Genet. 2014, 22, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Goker-Alpan, O.; Kishnani, P.S.; Longo, N.; Burrow, T.A.; Bernat, J.A.; Gupta, P.; Henderson, N.; Pedro, H.; Prada, C.E.; et al. The diagnosis and management of Gaucher disease in pediatric patients: Where do we go from here? Mol. Genet. Metab. 2022, 136, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, U.; Mengel, E.; Berrah, A.; AlSayed, M.; Broomfield, A.; Donald, A.; El Dein, H.M.S.; Freisens, S.; Hwu, W.-L.; Peterschmitt, M.J.; et al. Throwing a spotlight on under-recognized manifestations of Gaucher disease: Pulmonary involvement, lymphadenopathy and Gaucheroma. Mol. Genet. Metab. 2021, 133, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K.; Vellodi, A.; McHugh, K. Mesenteric mass in a young girl—An unusual site for Gaucher’s disease. Pediatr. Radiol. 2002, 32, 674–676. [Google Scholar] [CrossRef]
- Bossù, G.; Pedretti, L.; Bertolini, L.; Esposito, S. Pediatric Gaucher Disease Presenting with Massive Splenomegaly and Hepatic Gaucheroma. Children 2023, 10, 869. [Google Scholar] [CrossRef]
- Oto, Y.; Inoue, T.; Nagai, S.; Tanaka, S.; Itabashi, H.; Shiraisihi, M.; Nitta, A.; Murakami, N.; Ida, H.; Matsubara, T. Successful treatment of Gaucher disease type 1 by enzyme replacement therapy over a 10-year duration in a Japanese pediatric patient: A case report. Exp. Ther. Med. 2021, 21, 246. [Google Scholar] [CrossRef]
- Dinur, T.; Istaiti, M.; Frydman, D.; Becker-Cohen, M.; Szer, J.; Zimran, A.; Revel-Vilk, S. Patient reported outcome measures in a large cohort of patients with type 1 Gaucher disease. Orphanet J. Rare Dis. 2020, 15, 284. [Google Scholar] [CrossRef]
- Weinreb, N.J.; Camelo, J.S., Jr.; Charrow, J.; McClain, M.R.; Mistry, P.; Belmatoug, N.; International Collaborative Gaucher Group (ICGG) Gaucher Registry (NCT00358943) Investigators. Gaucher disease type 1 patients from the ICGG Gaucher Registry sustain initial clinical improvements during twenty years of imiglucerase treatment. Mol. Genet. Metab. 2021, 132, 100–111. [Google Scholar] [CrossRef]
- Deegan, P.; Fernandez-Sasso, D.; Giraldo, P.; Lau, H.; Panahloo, Z.; Zimran, A. Treatment patterns from 647 patients with Gaucher disease: An analysis from the Gaucher Outcome Survey. Blood Cells Mol. Dis. 2018, 68, 218–225. [Google Scholar] [CrossRef]
- Safety and Efficacy of Eliglustat with or without Imiglucerase in Pediatric Patients with Gaucher Disease (GD) Type 1 and Type 3 (ELIKIDS)—Full Text View. Available online: ClinicalTrials.gov (accessed on 14 September 2023).
- Dinur, T.; Bauer, P.; Beetz, C.; Kramp, G.; Cozma, C.; Iurașcu, M.-I.; Becker-Cohen, M.; Istaiti, M.; Rolfs, A.; Zimran, A.; et al. Gaucher Disease Diagnosis Using Lyso-Gb1 on Dry Blood Spot Samples: Time to Change the Paradigm? Int. J. Mol. Sci. 2022, 23, 1627. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Features at Diagnosis | Case 1 | Case 2 |
|---|---|---|
| Gender/Age | M/22 months | F/41 months |
| Duration of symptoms | 2 months | - |
| Presenting sign/symptom | Abdominal distension, asthenia | Abdominal distension |
| WBC/PMN/uL | 3200/1200 | 3100/1400 |
| Hb gr/dl | 6.0 | 10.0 |
| Platelets/uL | 80,000 | 98,000 |
| Abdominal circumference | 58 cm | 51 cm |
| Liver size (US longitudinal diameter) | 13 cm | 11 cm |
| Spleen size (US, longitudinal diameter) | 16 cm | 15 cm |
| Bone deformities or lytic lesions | No | No |
| Somato-sensorial potential | Normal | Normal |
| Performance status (Lansky scale) at diagnosis | 40% | 50% |
| Age at ERT start | 22 months | 41 months |
| Current age | 29 months | 46 months |
| Current Performance status (Lansky scale) | 80% | 70% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, C.; Ferrante, R.; Valentinuzzi, S.; Zucchelli, M.; Buccolini, C.; Di Rado, S.; Trotta, D.; Stuppia, L.; Federici, L.; Aricò, M. Noninvasive DBS-Based Approaches to Assist Clinical Diagnosis and Treatment Monitoring of Gaucher Disease. Biomedicines 2023, 11, 2672. https://doi.org/10.3390/biomedicines11102672
Rossi C, Ferrante R, Valentinuzzi S, Zucchelli M, Buccolini C, Di Rado S, Trotta D, Stuppia L, Federici L, Aricò M. Noninvasive DBS-Based Approaches to Assist Clinical Diagnosis and Treatment Monitoring of Gaucher Disease. Biomedicines. 2023; 11(10):2672. https://doi.org/10.3390/biomedicines11102672
Chicago/Turabian StyleRossi, Claudia, Rossella Ferrante, Silvia Valentinuzzi, Mirco Zucchelli, Carlotta Buccolini, Sara Di Rado, Daniela Trotta, Liborio Stuppia, Luca Federici, and Maurizio Aricò. 2023. "Noninvasive DBS-Based Approaches to Assist Clinical Diagnosis and Treatment Monitoring of Gaucher Disease" Biomedicines 11, no. 10: 2672. https://doi.org/10.3390/biomedicines11102672
APA StyleRossi, C., Ferrante, R., Valentinuzzi, S., Zucchelli, M., Buccolini, C., Di Rado, S., Trotta, D., Stuppia, L., Federici, L., & Aricò, M. (2023). Noninvasive DBS-Based Approaches to Assist Clinical Diagnosis and Treatment Monitoring of Gaucher Disease. Biomedicines, 11(10), 2672. https://doi.org/10.3390/biomedicines11102672