

Gene Therapy with Voretigene Neparvovec Improves Vision and Partially Restores Electrophysiological Function in Pre-School Children with Leber Congenital Amaurosis

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection and Overview on Performed Examinations
2.2. Gene Therapy Procedure and Perioperative Treatment
2.3. Efficacy Parameters/Outcome Measures
2.4. Visual Function Questionnaire (NEI-VFQ25)
2.5. Electroretinography (ERG)
2.6. Full-Field Light Sensitivity Testing (FST)
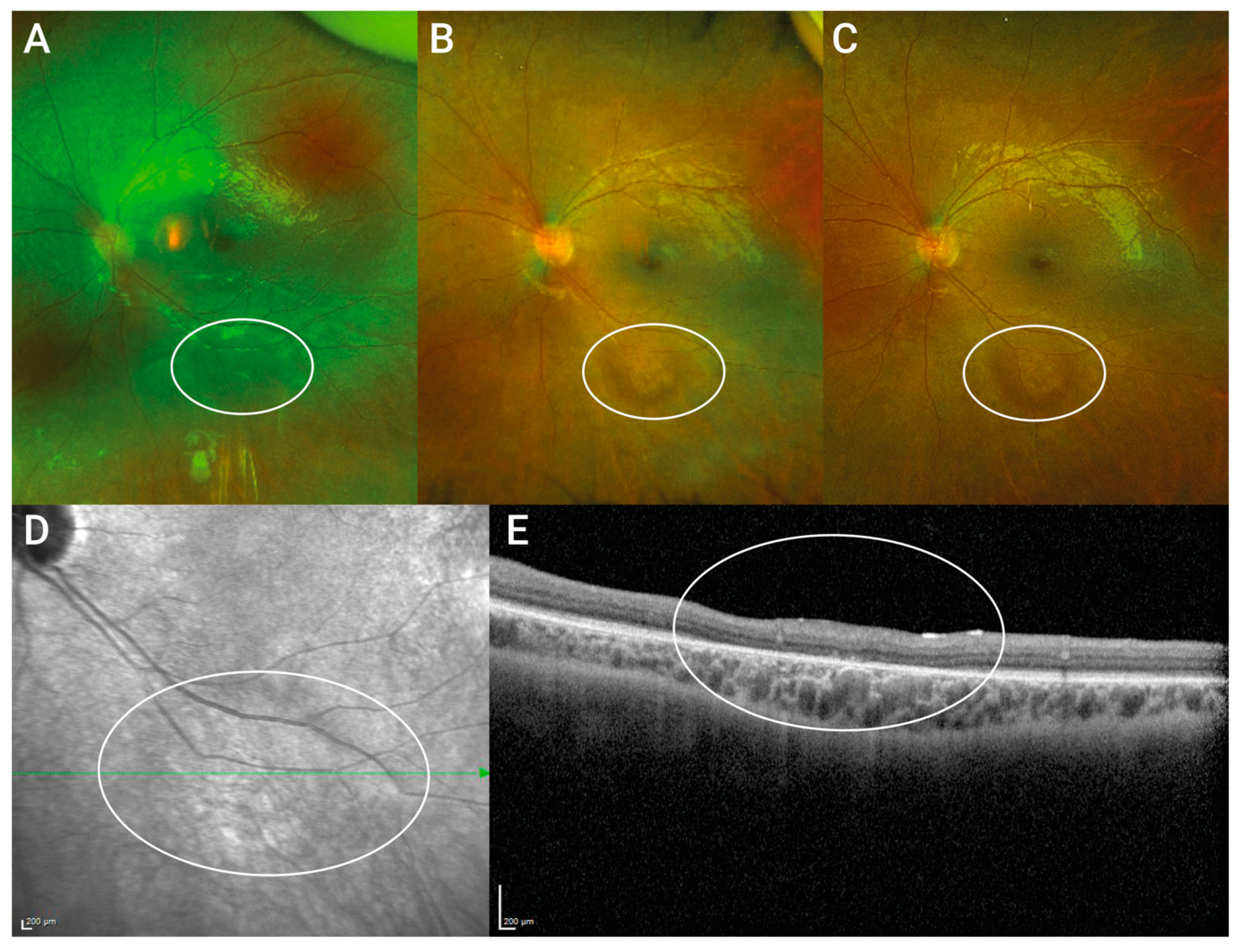
2.7. Optical Coherence Tomography (SD-OCT) and 488 nm Fundus Autofluorescence (FAF)
2.8. Fundus Wide-Angle Imaging
2.9. Statistical Analysis
3. Results
3.1. Patient Demographics and Genetic Background
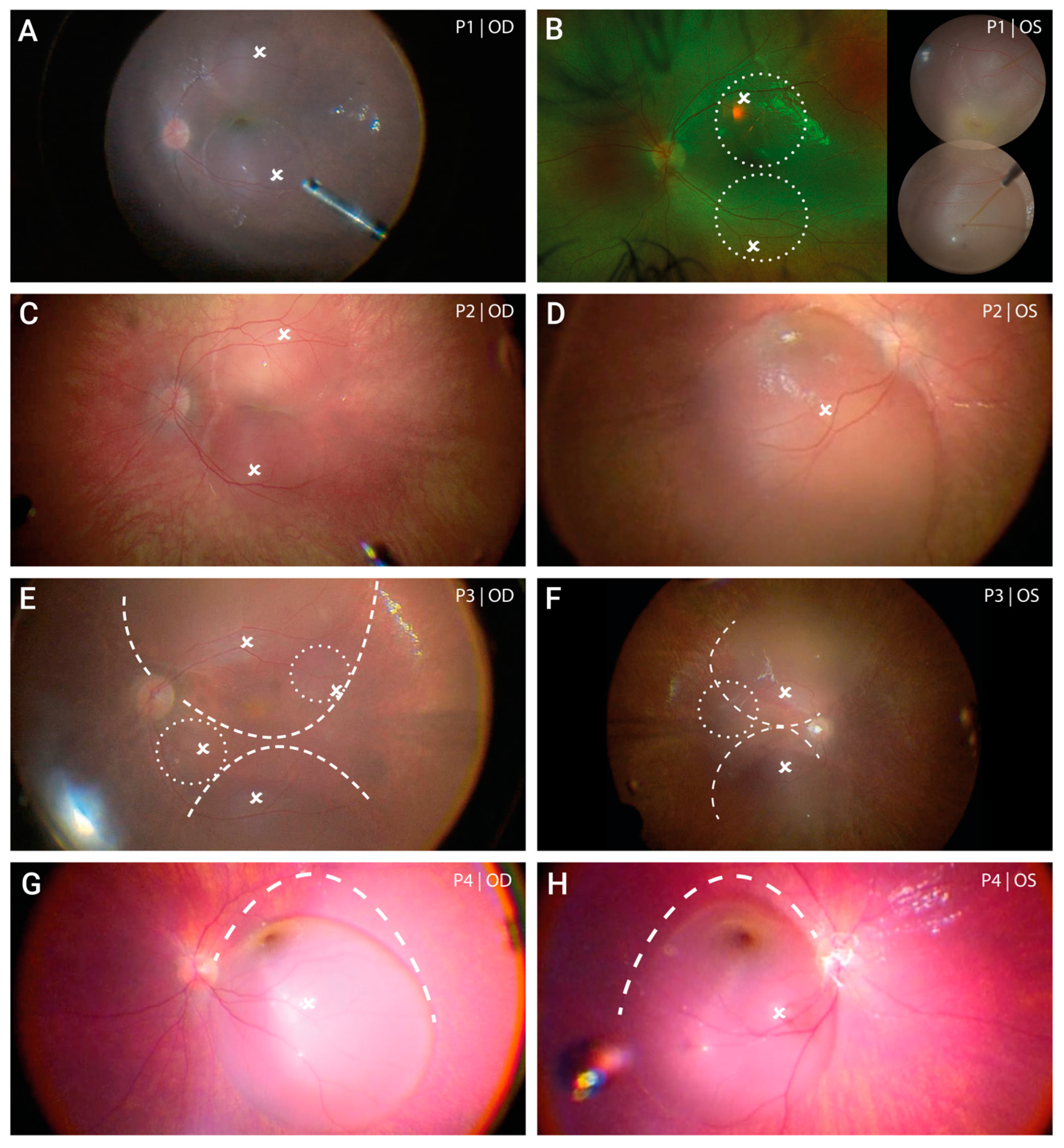
3.2. Baseline Characteristics and Clinical Appearance
3.3. Local Distribution of Voretigene Neparvovec
3.4. Immediate Postoperative Course and Adverse Events
3.5. Efficacy of Voretigene Neparvovec
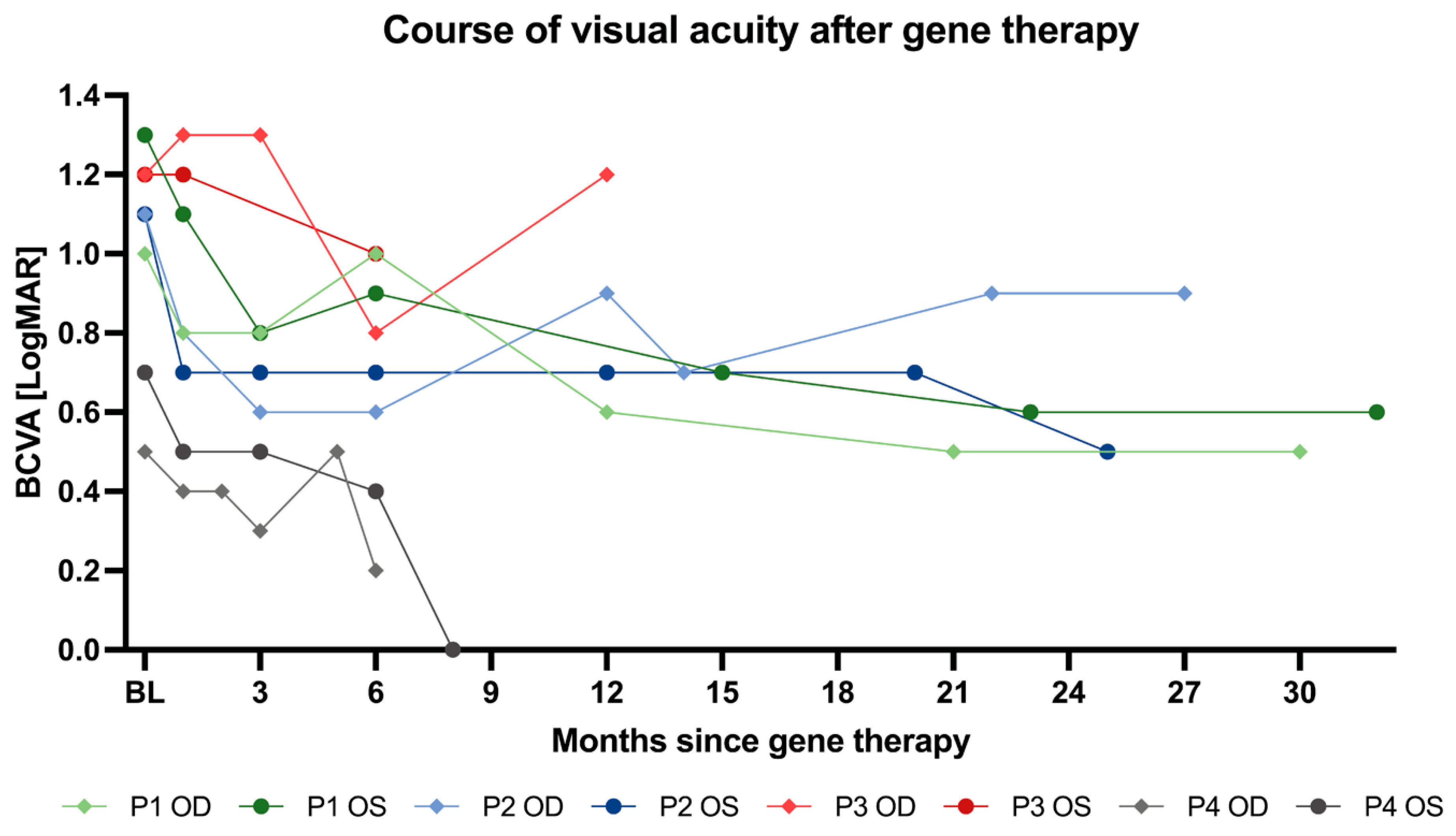
3.5.1. Best Corrected Visual Acuity (BCVA)
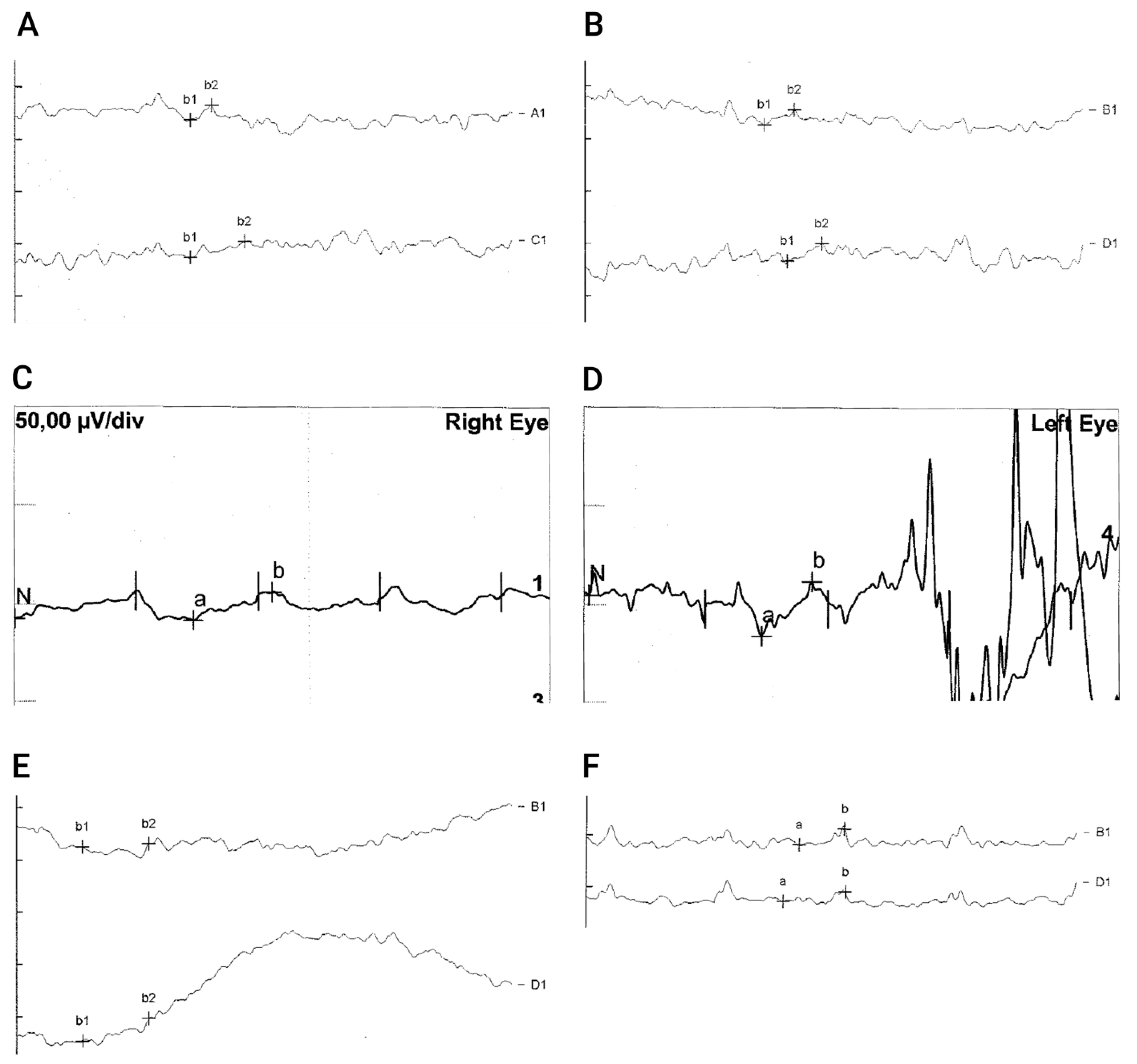
3.5.2. Electroretinography (ERG)
3.5.3. Full-Field Light Sensitivity Testing (FST)
3.5.4. Observation of Vision Guided Behavior/Behavioral Changes
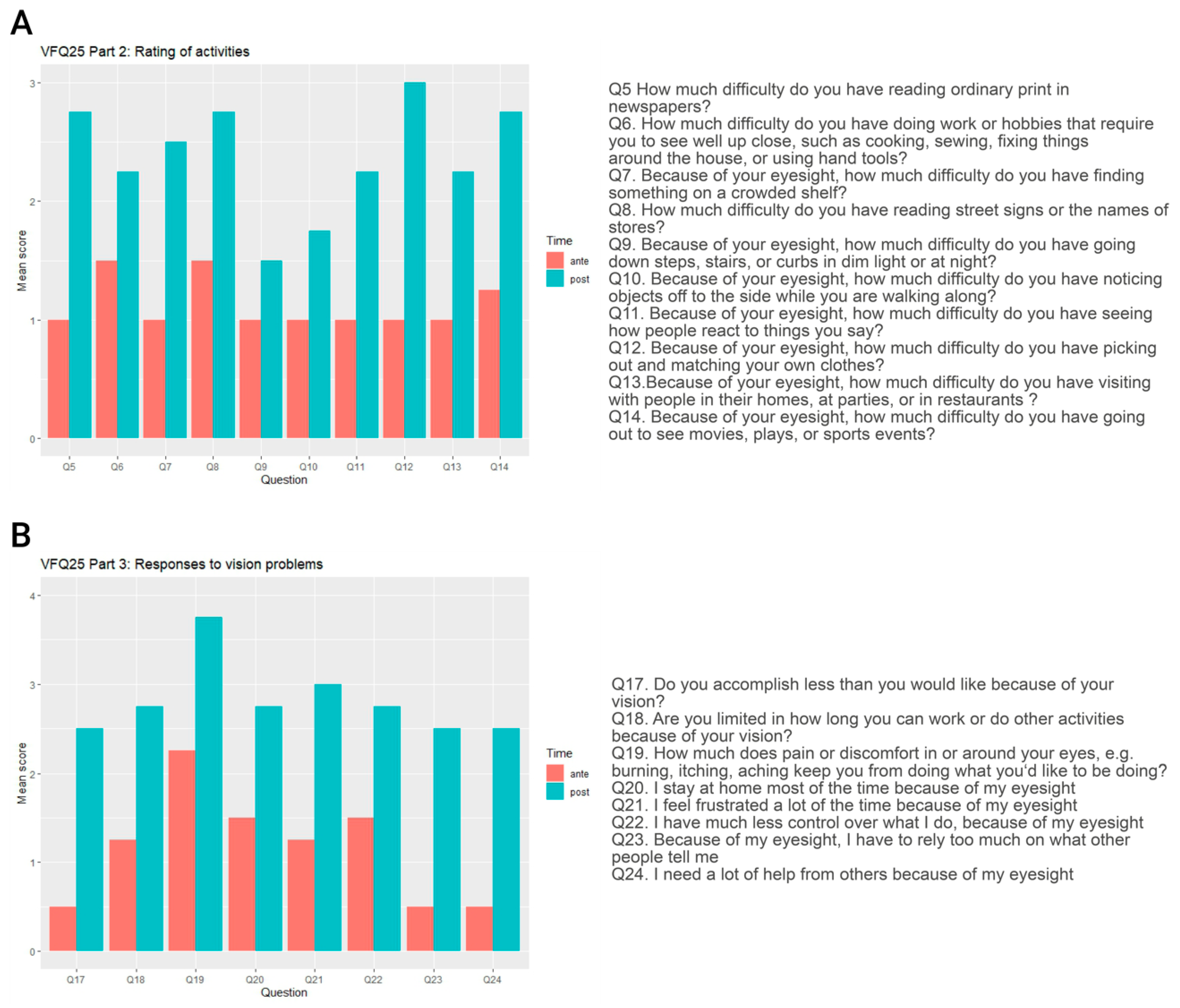
3.5.5. Visual Function Questionnaire
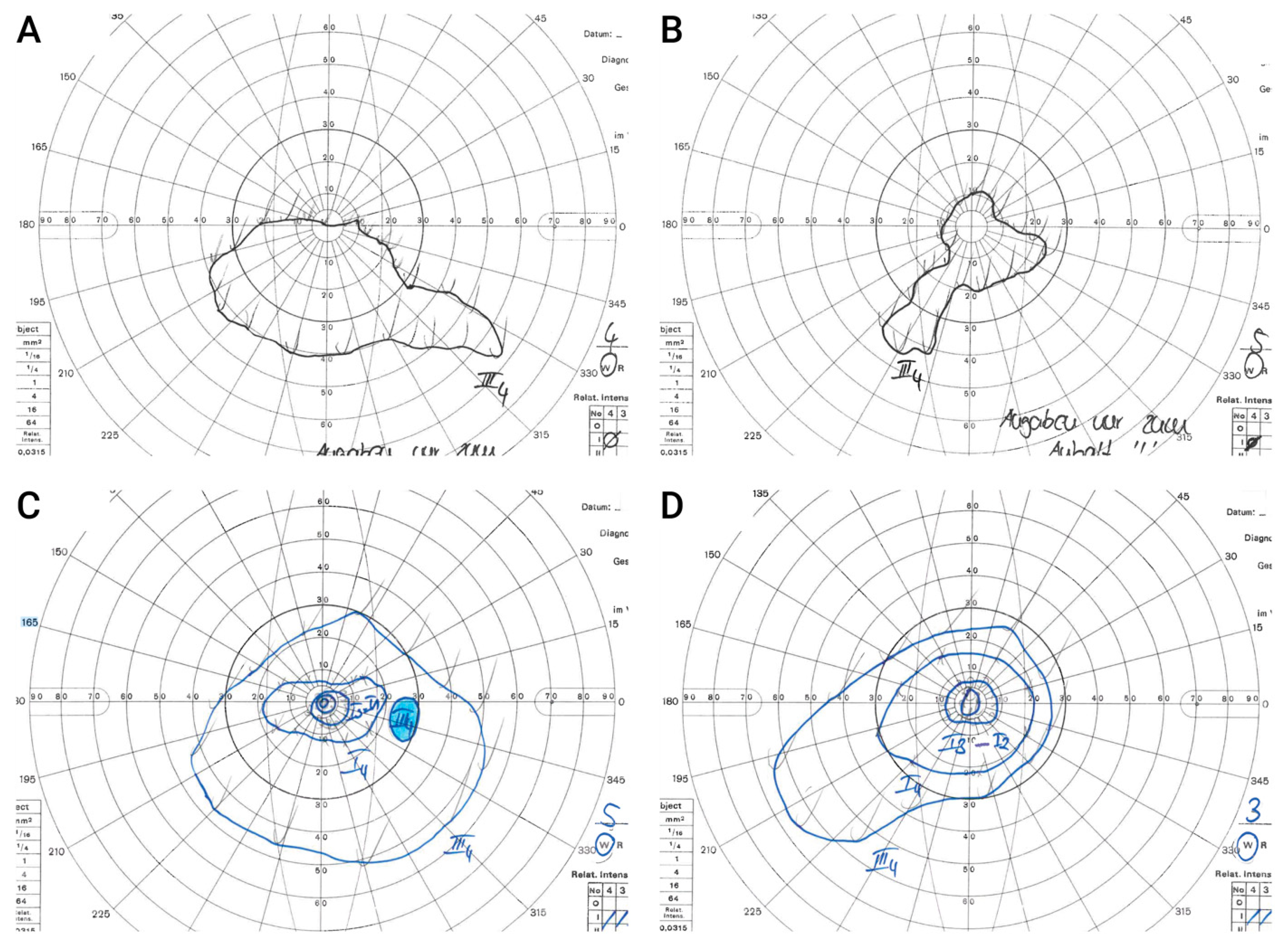
3.5.6. Goldmann Kinetic Perimetry
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aoun, M.; Passerini, I.; Chiurazzi, P.; Karali, M.; De Rienzo, I.; Sartor, G.; Murro, V.; Filimonova, N.; Seri, M.; Banfi, S. Inherited Retinal Diseases Due to RPE65 Variants: From Genetic Diagnostic Management to Therapy. Int. J. Mol. Sci. 2021, 22, 7207. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.C.; Bertelsen, M.; Lorenz, B.; Pennesi, M.E.; Leroy, B.P.; Hamel, C.P.; Pierce, E.; Sallum, J.; Larsen, M.; Stieger, K.; et al. The Natural History of Inherited Retinal Dystrophy Due to Biallelic Mutations in the RPE65 Gene. Am. J. Ophthalmol. 2019, 199, 58–70. [Google Scholar] [CrossRef] [PubMed]
- EMA. Luxturna: EPAR—Product Information 2019. Available online: https://www.ema.europa.eu/en/documents/product-information/luxturna-epar-product-information_en.pdf (accessed on 11 November 2021).
- UFaDA (FDA). Luxturna: Prescribing Information 2017 (Updated 05/2022). Available online: https://www.fda.gov/media/109906/download (accessed on 4 November 2022).
- Michalakis, S.; Gerhardt, M.; Rudolph, G.; Priglinger, S.; Priglinger, C. Gene Therapy for Inherited Retinal Disorders: Update on Clinical Trials. Klin. Monbl. Augenheilkd. 2021, 238, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, B.; Wabbels, B.; Wegscheider, E.; Hamel, C.P.; Drexler, W.; Preising, M.N. Lack of fundus autofluorescence to 488 nanometers from childhood on in patients with early-onset severe retinal dystrophy associated with mutations in RPE65. Ophthalmology 2004, 111, 1585–1594. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Aleman, T.S.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Windsor, E.A.; Schwartz, S.B.; Heon, E.; Stone, E.M. Defining the residual vision in leber congenital amaurosis caused by RPE65 mutations. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2368–2375. [Google Scholar] [CrossRef]
- Mata, N.L.; Moghrabi, W.N.; Lee, J.S.; Bui, T.V.; Radu, R.A.; Horwitz, J.; Travis, G.H. Rpe65 is a retinyl ester binding protein that presents insoluble substrate to the isomerase in retinal pigment epithelial cells. J. Biol. Chem. 2004, 279, 635–643. [Google Scholar] [CrossRef]
- Pierrache, L.H.M.; Ghafaryasl, B.; Khan, M.I.; Yzer, S.; van Genderen, M.M.; Schuil, J.; Boonstra, F.N.; Pott, J.W.R.; de Faber, J.; Tjon-Fo-Sang, M.J.H.; et al. Longitudinal study of RPE65-associated inherited retinal degenerations. Retina 2020, 40, 1812–1828. [Google Scholar] [CrossRef]
- Dejneka, N.S.; Surace, E.M.; Aleman, T.S.; Cideciyan, A.V.; Lyubarsky, A.; Savchenko, A.; Redmond, T.M.; Tang, W.; Wei, Z.; Rex, T.S.; et al. In utero gene therapy rescues vision in a murine model of congenital blindness. Mol. Ther. 2004, 9, 182–188. [Google Scholar] [CrossRef]
- Acland, G.M.; Aguirre, G.D.; Bennett, J.; Aleman, T.S.; Cideciyan, A.V.; Bennicelli, J.; Dejneka, N.S.; Pearce-Kelling, S.E.; Maguire, A.M.; Palczewski, K.; et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol. Ther. 2005, 12, 1072–1082. [Google Scholar] [CrossRef]
- Annear, M.J.; Bartoe, J.T.; Barker, S.E.; Smith, A.J.; Curran, P.G.; Bainbridge, J.W.; Ali, R.R.; Petersen-Jones, S.M. Gene therapy in the second eye of RPE65-deficient dogs improves retinal function. Gene Ther. 2011, 18, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef]
- Testa, F.; Maguire, A.M.; Rossi, S.; Pierce, E.A.; Melillo, P.; Marshall, K.; Banfi, S.; Surace, E.M.; Sun, J.; Acerra, C.; et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology 2013, 120, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Ratnakaram, R.; Heon, E.; Schwartz, S.B.; Roman, A.J.; Peden, M.C.; Aleman, T.S.; Boye, S.L.; Sumaroka, A.; et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: Safety and efficacy in 15 children and adults followed up to 3 years. Arch. Ophthalmol. 2012, 130, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Mangione, C.M.; Lee, P.P.; Gutierrez, P.R.; Spritzer, K.; Berry, S.; Hays, R.D. Development of the 25-item National Eye Institute Visual Function Questionnaire. Arch. Ophthalmol. 2001, 119, 1050–1058. [Google Scholar] [CrossRef]
- Massof, R.W. An interval-scaled scoring algorithm for visual function questionnaires. Optom. Vis. Sci. 2007, 84, E690–E705. [Google Scholar] [CrossRef]
- Ryan, B.; Court, H.; Margrain, T.H. Measuring low vision service outcomes: Rasch analysis of the seven-item National Eye Institute Visual Function Questionnaire. Optom. Vis. Sci. 2008, 85, 112–121. [Google Scholar] [CrossRef]
- Weleber, R.G. The effect of age on human cone and rod ganzfeld electroretinograms. Investig. Ophthalmol. Vis. Sci. 1981, 20, 392–399. [Google Scholar]
- Marmor, M.F.; Brigell, M.G.; McCulloch, D.L.; Westall, C.A.; Bach, M. ISCEV standard for clinical electro-oculography (2010 update). Doc. Ophthalmol. 2011, 122, 1–7. [Google Scholar] [CrossRef]
- Klein, M.; Birch, D.G. Psychophysical assessment of low visual function in patients with retinal degenerative diseases (RDDs) with the Diagnosys full-field stimulus threshold (D-FST). Doc. Ophthalmol. 2009, 119, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Morimura, H.; Fishman, G.A.; Grover, S.A.; Fulton, A.B.; Berson, E.L.; Dryja, T.P. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc. Natl. Acad. Sci. USA 1998, 95, 3088–3093. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, B.; Poliakov, E.; Schambeck, M.; Friedburg, C.; Preising, M.N.; Redmond, T.M. A comprehensive clinical and biochemical functional study of a novel RPE65 hypomorphic mutation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5235–5242. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hu, J.; Jin, R.J.; Aiyar, A.; Jacobson, S.G.; Bok, D.; Jin, M. Temperature-sensitive retinoid isomerase activity of RPE65 mutants associated with Leber Congenital Amaurosis. J. Biochem. 2015, 158, 115–125. [Google Scholar] [CrossRef][Green Version]
- Kondo, H.; Qin, M.; Mizota, A.; Kondo, M.; Hayashi, H.; Hayashi, K.; Oshima, K.; Tahira, T.; Hayashi, K. A homozygosity-based search for mutations in patients with autosomal recessive retinitis pigmentosa, using microsatellite markers. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4433–4439. [Google Scholar] [CrossRef]
- Katagiri, S.; Hosono, K.; Hayashi, T.; Kurata, K.; Mizobuchi, K.; Matsuura, T.; Yoshitake, K.; Iwata, T.; Nakano, T.; Hotta, Y. Early onset flecked retinal dystrophy associated with new compound heterozygous RPE65 variants. Mol. Vis. 2018, 24, 286–296. [Google Scholar]
- Sayer, J.A.; Otto, E.A.; O’Toole, J.F.; Nurnberg, G.; Kennedy, M.A.; Becker, C.; Hennies, H.C.; Helou, J.; Attanasio, M.; Fausett, B.V.; et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006, 38, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.M.; Thompson, D.A.; Srikumari, C.R.; Lorenz, B.; Finckh, U.; Nicoletti, A.; Murthy, K.R.; Rathmann, M.; Kumaramanickavel, G.; Denton, M.J.; et al. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat. Genet. 1997, 17, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; O’Sullivan, J.; Williams, S.G.; Lamb, J.A.; Panda, B.; Sergouniotis, P.I.; Gillespie, R.L.; Daiger, S.P.; et al. Molecular findings from 537 individuals with inherited retinal disease. J. Med. Genet. 2016, 53, 761–767. [Google Scholar] [CrossRef]
- Reichel, F.F.; Seitz, I.; Wozar, F.; Dimopoulos, S.; Jung, R.; Kempf, M.; Kohl, S.; Kortüm, F.C.; Ott, S.; Pohl, L.; et al. Development of retinal atrophy after subretinal gene therapy with voretigene neparvovec. Br. J. Ophthalmol. 2022. [Google Scholar] [CrossRef]
- Gange, W.S.; Sisk, R.A.; Besirli, C.G.; Lee, T.C.; Havunjian, M.; Schwartz, H.; Borchert, M.; Sengillo, J.D.; Mendoza, C.; Berrocal, A.M.; et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmol. Retina 2022, 6, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, C.; Tzekov, R.T.; Zhu, Y.; Li, W. The effect of human gene therapy for RPE65-associated Leber’s congenital amaurosis on visual function: A systematic review and meta-analysis. Orphanet. J. Rare Dis. 2020, 15, 49. [Google Scholar] [CrossRef]
- Maguire, A.M.; High, K.A.; Auricchio, A.; Wright, J.F.; Pierce, E.A.; Testa, F.; Mingozzi, F.; Bennicelli, J.L.; Ying, G.S.; Rossi, S.; et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: A phase 1 dose-escalation trial. Lancet 2009, 374, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Weleber, R.G.; Pennesi, M.E.; Wilson, D.J.; Kaushal, S.; Erker, L.R.; Jensen, L.; McBride, M.T.; Flotte, T.R.; Humphries, M.; Calcedo, R.; et al. Results at 2 Years after Gene Therapy for RPE65-Deficient Leber Congenital Amaurosis and Severe Early-Childhood-Onset Retinal Dystrophy. Ophthalmology 2016, 123, 1606–1620. [Google Scholar] [CrossRef]
- Deng, C.; Zhao, P.Y.; Branham, K.; Schlegel, D.; Fahim, A.T.; Jayasundera, T.K.; Khan, N.; Besirli, C.G. Real-world outcomes of voretigene neparvovec treatment in pediatric patients with RPE65-associated Leber congenital amaurosis. Graefe’s Arch. Clin. Exp. Ophthalmol. 2022, 260, 1543–1550. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Gregori, N.Z.; Sisk, R.A.; Weng, C.Y.; Berrocal, A.M.; Davis, J.L.; Mendoza-Santiesteban, C.E.; Zheng, D.D.; Feuer, W.J.; Lam, B.L. Visual Acuity, Retinal Morphology, and Patients’ Perceptions after Voretigene Neparovec-rzyl Therapy for RPE65-Associated Retinal Disease. Ophthalmol. Retina 2022, 6, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Testa, F.; Melillo, P.; Di Iorio, V.; Iovino, C.; Farinaro, F.; Karali, M.; Banfi, S.; Rossi, S.; Della Corte, M.; Simonelli, F. Visual function and retinal changes after voretigene neparvovec treatment in children with biallelic RPE65-related inherited retinal dystrophy. Sci. Rep. 2022, 12, 17637. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, G.; Lebranchu, P.; Billaud, F.; Adjali, O.; Schmitt, S.; Bézieau, S.; Péréon, Y.; Valabregue, R.; Ivan, C.; Darmon, C.; et al. Safety and Long-Term Efficacy of AAV4 Gene Therapy in Patients with RPE65 Leber Congenital Amaurosis. Mol. Ther. 2018, 26, 256–268. [Google Scholar] [CrossRef]
- Simonelli, F.; Maguire, A.M.; Testa, F.; Pierce, E.A.; Mingozzi, F.; Bennicelli, J.L.; Rossi, S.; Marshall, K.; Banfi, S.; Surace, E.M.; et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol. Ther. 2010, 18, 643–650. [Google Scholar] [CrossRef]
- Khanna, S.; Dell’Osso, L.F. The diagnosis and treatment of infantile nystagmus syndrome (INS). ScientificWorldJournal 2006, 6, 1385–1397. [Google Scholar] [CrossRef]
- Chung, S.T.; LaFrance, M.W.; Bedell, H.E. Influence of motion smear on visual acuity in simulated infantile nystagmus. Optom. Vis. Sci. 2011, 88, 200–207. [Google Scholar] [CrossRef]
- Aguirre, G.K.; Komáromy, A.M.; Cideciyan, A.V.; Brainard, D.H.; Aleman, T.S.; Roman, A.J.; Avants, B.B.; Gee, J.C.; Korczykowski, M.; Hauswirth, W.W.; et al. Canine and human visual cortex intact and responsive despite early retinal blindness from RPE65 mutation. PLoS Med. 2007, 4, e230. [Google Scholar] [CrossRef] [PubMed]
- Kutluer, M.; Huang, L.; Marigo, V. Targeting molecular pathways for the treatment of inherited retinal degeneration. Neural Regen. Res. 2020, 15, 1784–1791. [Google Scholar] [CrossRef]
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2006, 103, 11300–11305. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Strauss, R.W.; Lu, L.; Hafiz, G.; Wolfson, Y.; Shah, S.M.; Sophie, R.; Mir, T.A.; Scholl, H.P. Is There Excess Oxidative Stress and Damage in Eyes of Patients with Retinitis Pigmentosa? Antioxid. Redox. Signal. 2015, 23, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Alibrandi, S.; Scalinci, S.Z.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. Epitranscriptome Analysis of Oxidative Stressed Retinal Epithelial Cells Depicted a Possible RNA Editing Landscape of Retinal Degeneration. Antioxidants 2022, 11, 1967. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa. Antioxidants 2020, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Usui, S.; Zafar, A.B.; Oveson, B.C.; Jo, Y.J.; Lu, L.; Masoudi, S.; Campochiaro, P.A. N-Acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J. Cell Physiol. 2011, 226, 1843–1849. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Age at Time of Treatment, Sex | Mutations in RPE65 | Type of Mutation | Functional Consequence |
|---|---|---|---|
| P1, 4 years male | Exon 11 c.1207.1210dupCTGG | duplication, frameshift | p.(Glu404Alafs*4)→stop codon, non-sense mediated decay of mRNA ** |
| Intron 12 c.1338+1G>A | splicing donor site | →skipping of Exon 12 ** | |
| P2, 3 years male | Exon 2 c.74C>T | missense | p.(Pro25Leu), loss of function |
| Exon 14 c.1543C>T | missense | p.(Arg515Trp), loss of function | |
| + CEP 290 Exon 41 c.5668G>T * | p.(Gly1890 *)→stop codon, non-sense mediated decay of mRNA ** | ||
| P3, 5 years male | c.1207_1210dupCTGG homozygous | duplication, frameshift | p.(Glu404Alafs*4)→stop codon, non-sense mediated decay of mRNA ** |
| P4, 6 years male | Intron 1 c.11+5G>A | splicing donor site | →incomplete splicing of transcript ** |
| Intron 7 c.726-2A>T | splicing acceptor site | →skipping of Exon 8 ** |
| Symptom/Objective | P1 (4y) | P2 (3y) | P3 (5y) | P4 (6y) |
|---|---|---|---|---|
| Lack of eye contact | at birth | at birth | at birth | no |
| Nystagmus (Time of appearance) | few days postnatal | 6 months postnatal | 6 weeks postnatal | not observed |
| Inability of orientation in dim light * | 6 months | from birth | from birth | from birth |
| Discomfort in the dark * | ||||
| OD | 1.0 1.3 - | Fix | 1.2 | 0.5 |
| BCVA OS | Fix | 1.2 | 0.7 | |
| (LogMAR) OU | 1.1 ** | - | - | |
| Refractive error OD OS | +4.00/−0.25/165 +4.00/−0.50/30 | +1.00/−1.50/145° +1.00/−1.50/145° | +4.50/−0.50/180° +4.50/−0.50/5° | +2.25/−1.00/170° +2.25/−1.00/170° |
| Timepoint | BCVA (LogMAR) | Mean Change from Baseline (LogMAR Units) |
|---|---|---|
| Baseline | 1.01 ± 0.27 | |
| Month 1 | 0.85 ± 0.34 | 0.1625 |
| Month 6 | 0.70 ± 0.30 | 0.3125 |
| VFQ25 Subscale | Mean Score Pre-Operative | Mean Score Post-Operative | Mean Change |
|---|---|---|---|
| General vision | 13.75 | 49.375 | +35.625 |
| Near vision/activities | 28.13 | 62.50 | +34.375 |
| Distance vision/activities | 31.25 | 64.58 | +33.33 |
| Visual specific | |||
| -social functioning | 33.33 | 66.66 | +33.33 |
| -role difficulties | 25.00 | 57.81 | +32.81 |
| -dependency | 23.44 | 65.63 | +42.19 |
| Color vision | 25.00 | 75.00 | +50.00 |
| Peripheral vision | 25.00 | 43.75 | +18.75 |
| Overall composite score | 25.61 | 60.66 | +35.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerhardt, M.J.; Priglinger, C.S.; Rudolph, G.; Hufendiek, K.; Framme, C.; Jägle, H.; Salchow, D.J.; Anschütz, A.; Michalakis, S.; Priglinger, S.G. Gene Therapy with Voretigene Neparvovec Improves Vision and Partially Restores Electrophysiological Function in Pre-School Children with Leber Congenital Amaurosis. Biomedicines 2023, 11, 103. https://doi.org/10.3390/biomedicines11010103
Gerhardt MJ, Priglinger CS, Rudolph G, Hufendiek K, Framme C, Jägle H, Salchow DJ, Anschütz A, Michalakis S, Priglinger SG. Gene Therapy with Voretigene Neparvovec Improves Vision and Partially Restores Electrophysiological Function in Pre-School Children with Leber Congenital Amaurosis. Biomedicines. 2023; 11(1):103. https://doi.org/10.3390/biomedicines11010103
Chicago/Turabian StyleGerhardt, Maximilian J., Claudia S. Priglinger, Günther Rudolph, Karsten Hufendiek, Carsten Framme, Herbert Jägle, Daniel J. Salchow, Andreas Anschütz, Stylianos Michalakis, and Siegfried G. Priglinger. 2023. "Gene Therapy with Voretigene Neparvovec Improves Vision and Partially Restores Electrophysiological Function in Pre-School Children with Leber Congenital Amaurosis" Biomedicines 11, no. 1: 103. https://doi.org/10.3390/biomedicines11010103
APA StyleGerhardt, M. J., Priglinger, C. S., Rudolph, G., Hufendiek, K., Framme, C., Jägle, H., Salchow, D. J., Anschütz, A., Michalakis, S., & Priglinger, S. G. (2023). Gene Therapy with Voretigene Neparvovec Improves Vision and Partially Restores Electrophysiological Function in Pre-School Children with Leber Congenital Amaurosis. Biomedicines, 11(1), 103. https://doi.org/10.3390/biomedicines11010103