
Identification of a Novel Missense Mutation of POLR3A Gene in a Cohort of Sicilian Patients with Leukodystrophy

 , ,
, ,  ,
,  , ,
, ,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Preparation
2.2. Genotypic Identification by NGS Sequencing
2.3. Sanger Sequencing
3. Results
3.1. Study Subjects
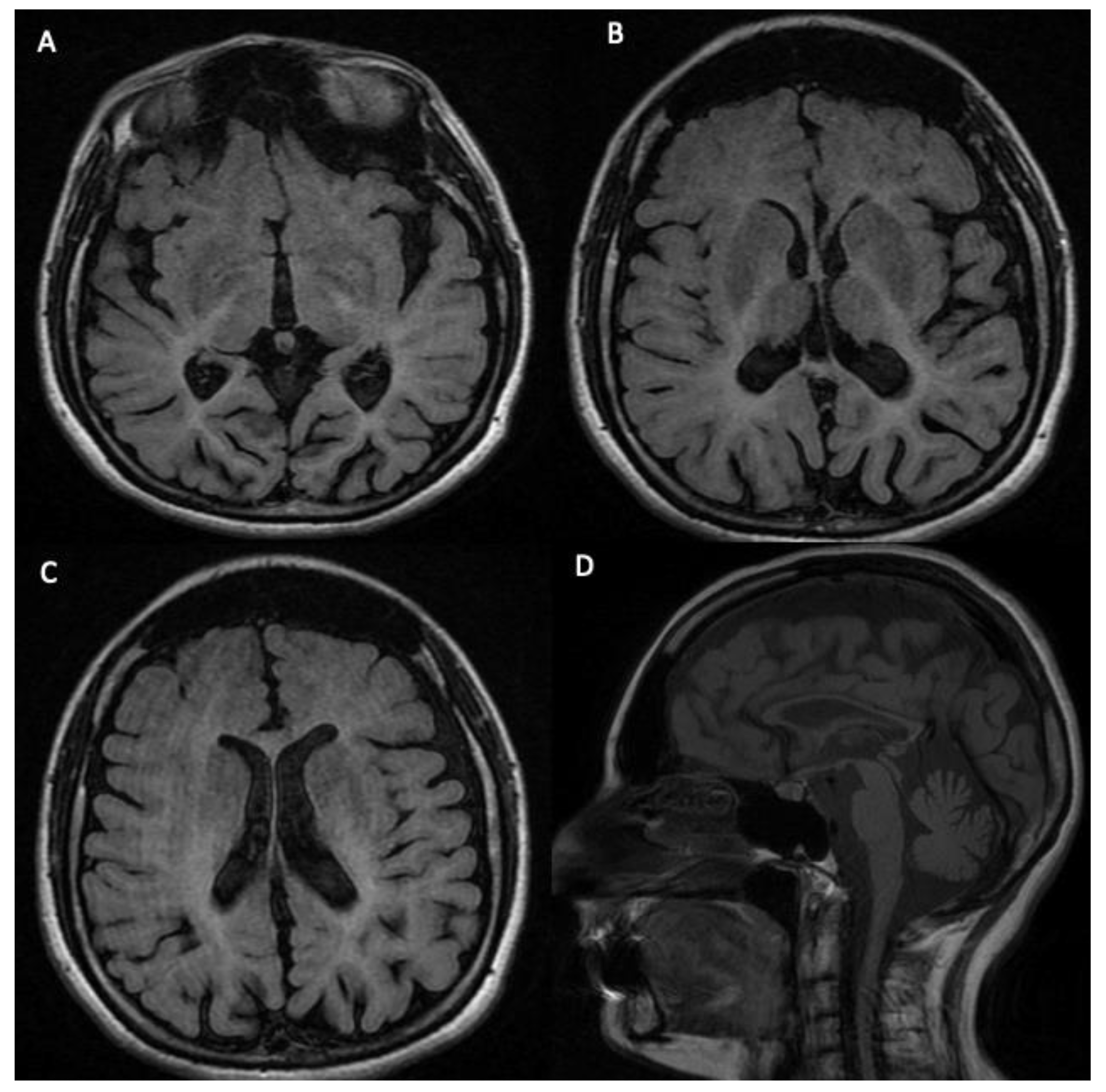
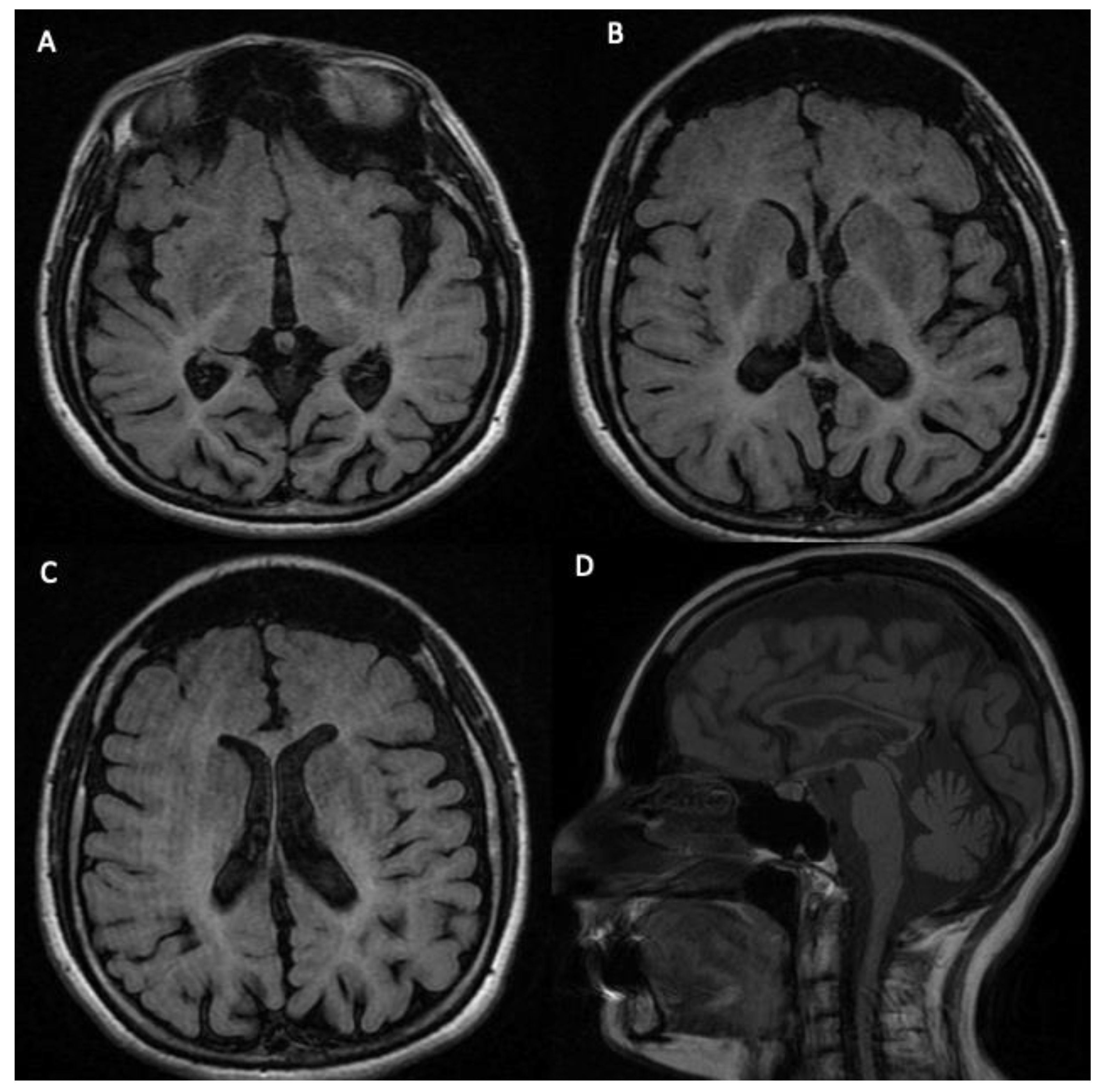
3.2. Phenotypic Features of the Analyzed Subject
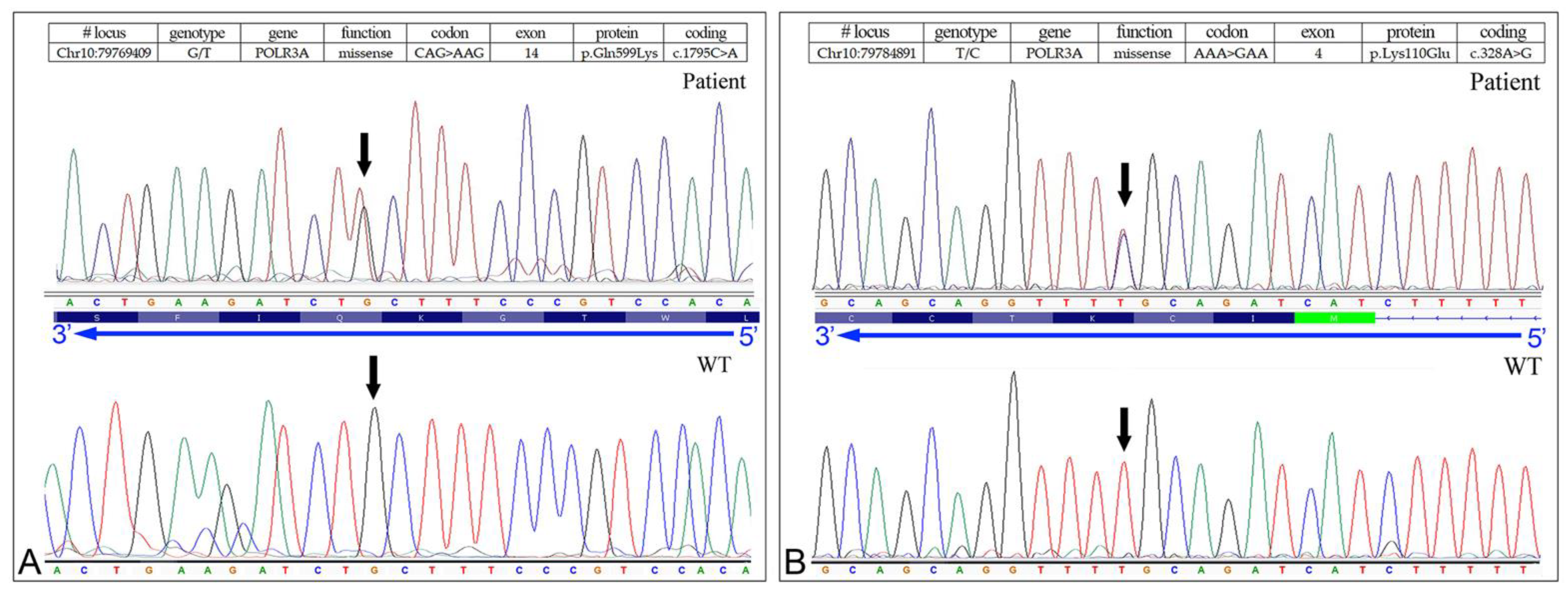
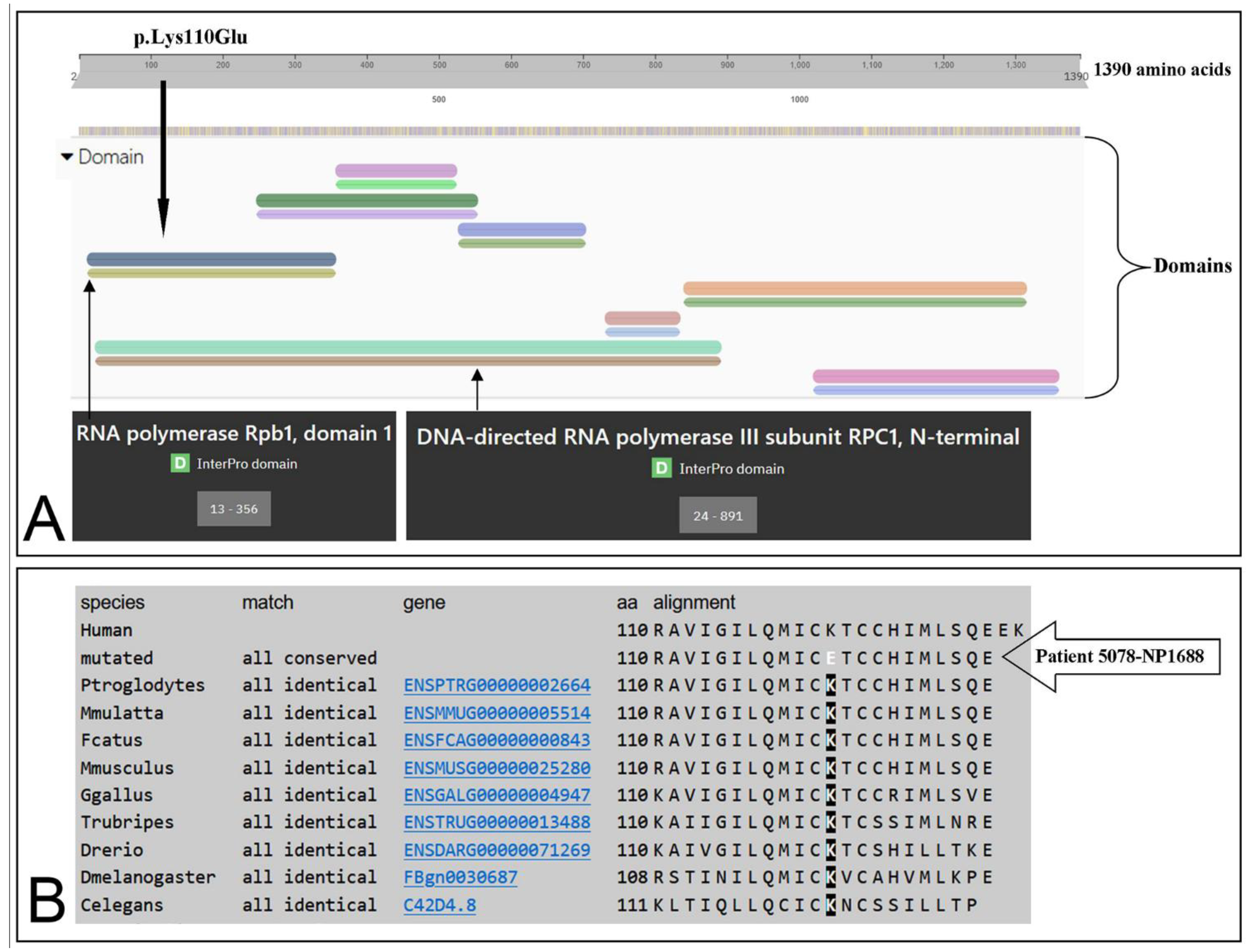
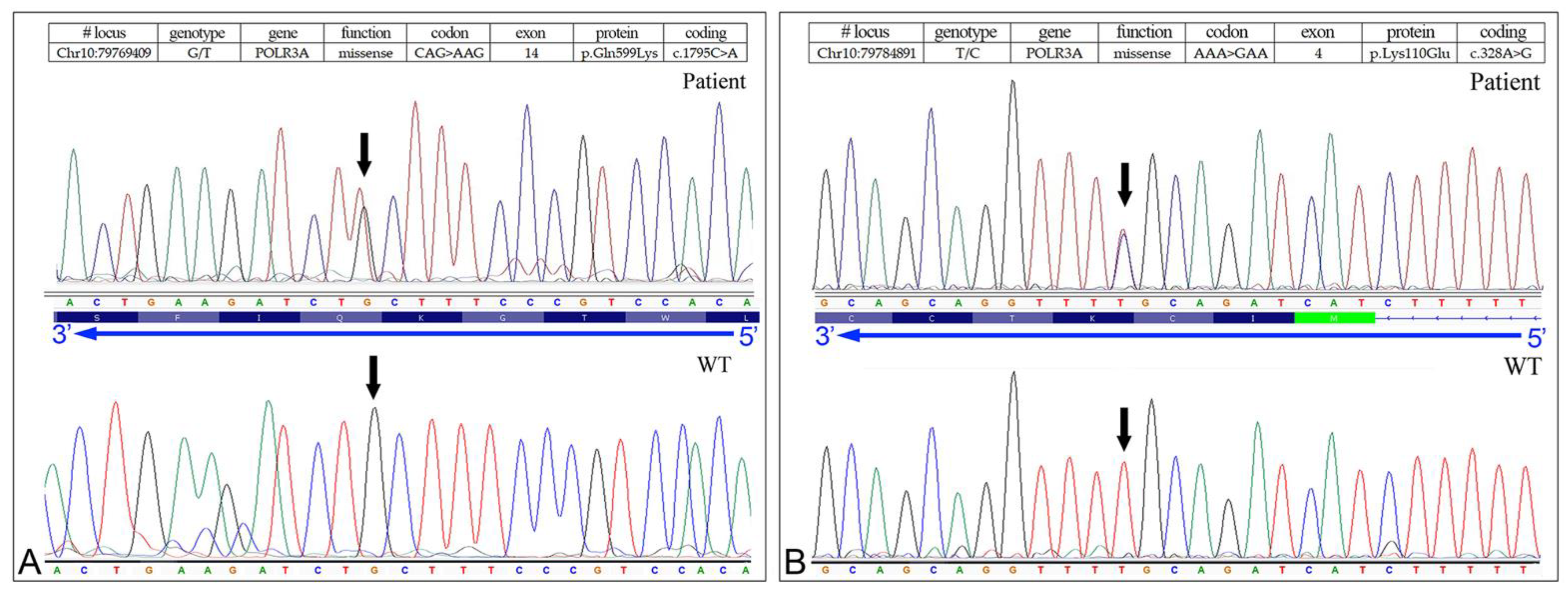
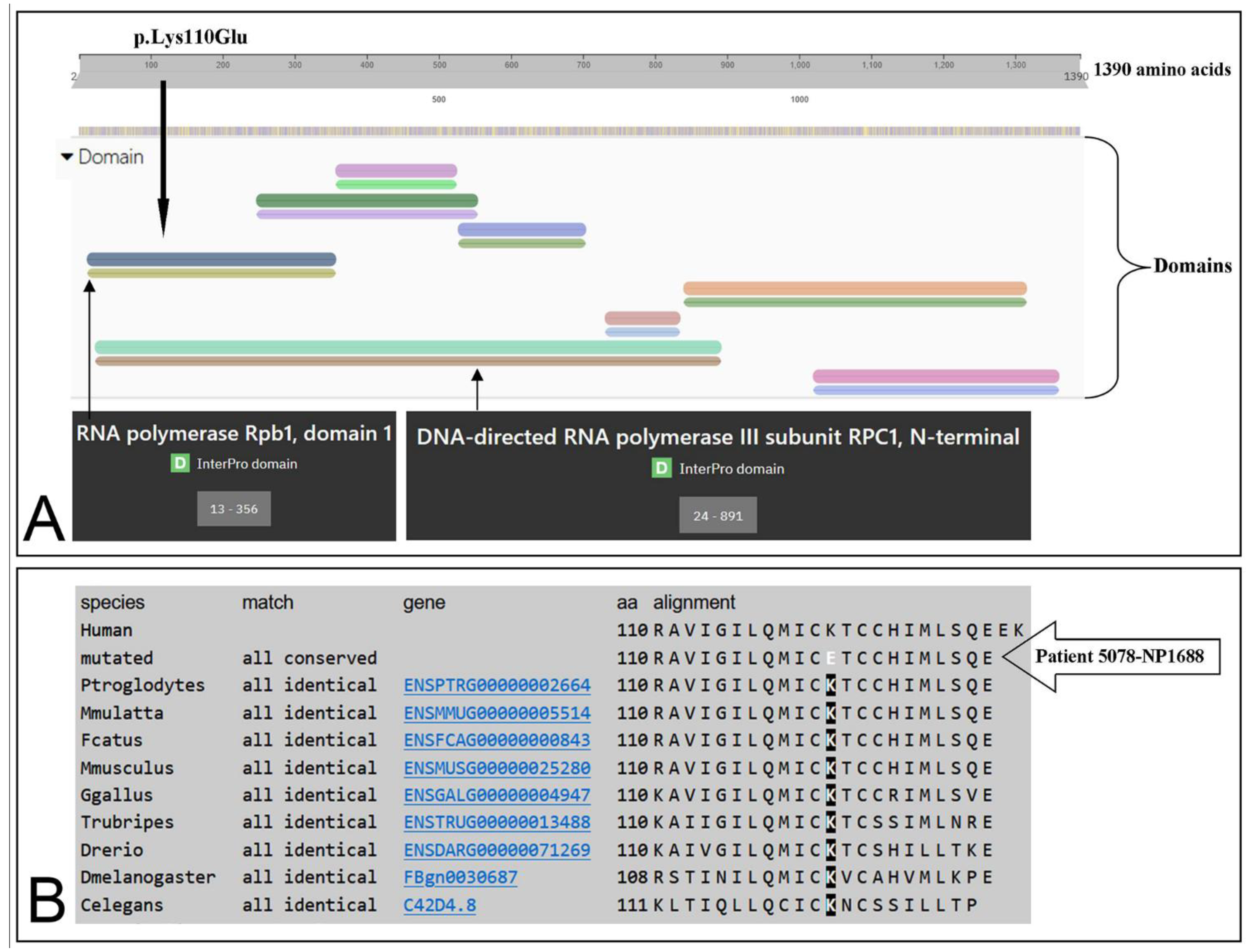
3.3. Identification of the Missense Mutation in the POLR3A (NM_007055.3) Gene
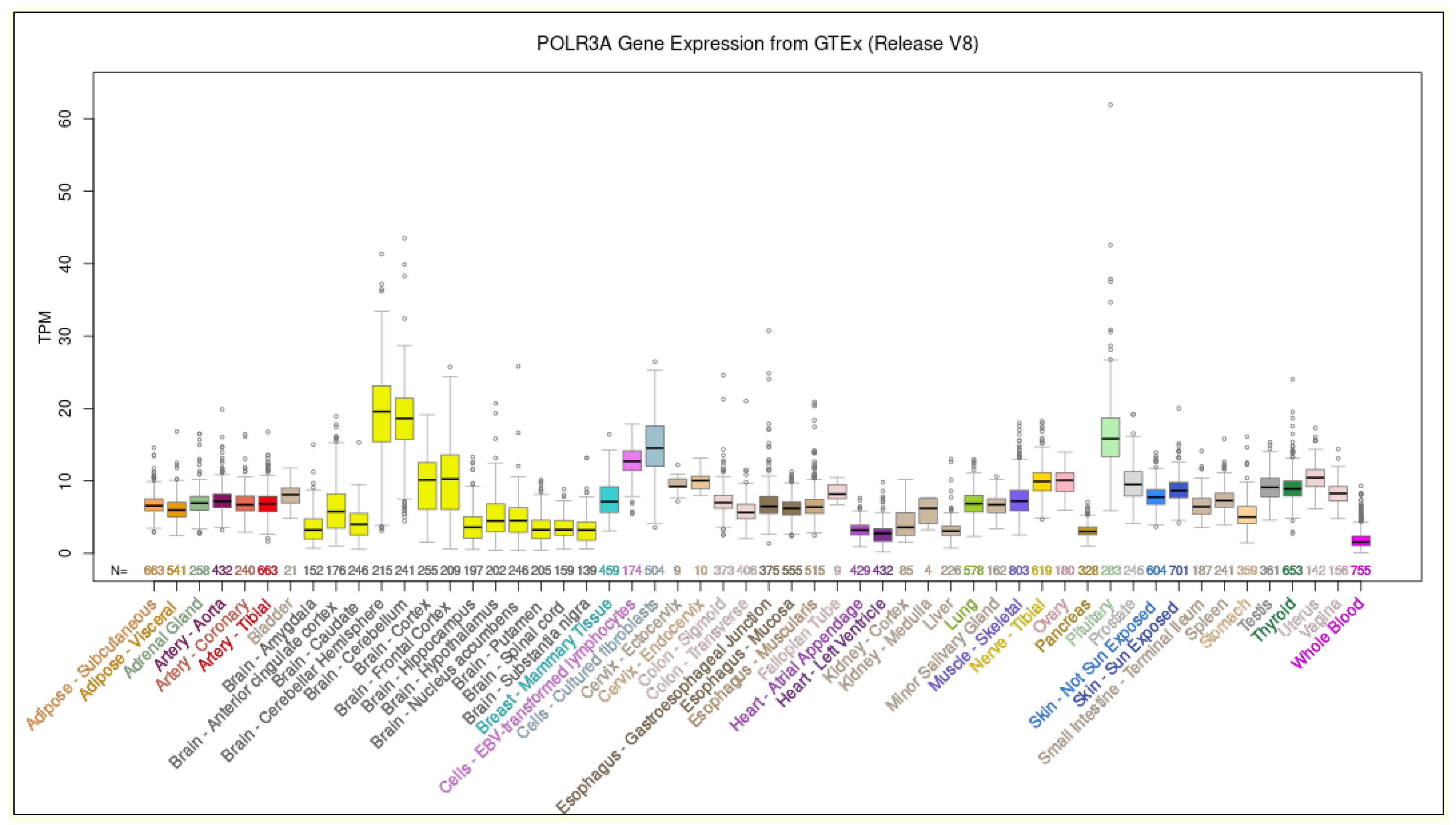
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bernard, G.; Chouery, E.; Putorti, M.L.; Tétreault, M.; Takanohashi, A.; Carosso, G.; Clément, I.; Boespflug-Tanguy, O.; Rodriguez, D.; Delague, V.; et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011, 89, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Choquet, K.; Forget, D.; Meloche, E.; Dicaire, M.-J.; Bernard, G.; Vanderver, A.; Schiffmann, R.; Fabian, M.-R.; Teichmann, M.; Coulombe, B.; et al. Leukodystrophy-associated POLR3A mutations down-regulate the RNA polymerase III transcript and important regulatory RNA BC200. J. Biol. Chem. 2019, 294, 7445–7459. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.; Vanderver, A. POLR3-Related Leukodystrophy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Takanashi, J.; Osaka, I.; Saitsu, H.; Sasaki, H.; Mori, M.; Shibayama, H.; Tanaka, M.; Nomura, Y.; Terao, Y.; Inoue, K.; et al. Different patterns of cerebellar abnormality and hypomyelination between POLR3A and POLR3B mutations. Brain Dev. 2014, 36, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Azmanov, D.N.; Siira, S.; Chamova, T.; Kaprelyan, A.; Guergueltcheva, V.; Shearwood, A.J.; Liu, G.; Morar, B.; Rackham, O.; Bynevelt, M.; et al. Transcriptome-wide effects of a POLR3A gene mutation in patients with an unusual phenotype of striatal involvement. Hum. Mol. Genet. 2016, 25, 4302–4314. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Calì, F.; Chiavetta, V.; Ruggeri, G.; Piccione, M.; Selicorni, A.; Palazzo, D.; Bonsignore, M.; Cereda, A.; Elia, M.; Failla, P.; et al. Mutation spectrum of NF1 gene in Italianpatients with neurofibromatosis type 1 usingIonTorrent PGM™ platform. Eur. J. Med. Genet. 2017, 60, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Vanderver, A.; van Spaendonk, R.M.L.; Schiffmann, R.; Brais, B.; Bugiani, M.; Sistermans, E.; Catsman-Berrevoets, C.; Kros, J.M.; Pinto, P.S.; et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014, 83, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.S.; de Paiva, A.R.B.; Zhang, W.J.; Bugiardini, E.; Freua, F.; Lucato, L.T.; Macedo-Souza, L.I.; Lakshmanan, R.; Kinsella, J.A.; Merwick, A.; et al. Clinical and genetic characterization of leukoencephalopathies in adults. Brain 2017, 140, 1204–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewari, V.V.; Mehta, R.; Sreedhar, C.M.; Tewar, K.; Mohammad, A.; Gupta, N.; Gulati, S.; Kabra, M. A novel homozygous mutation in POLR3A gene causing 4H syndrome: A case report. BMC Pediatr. 2018, 18, 126. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Age | Gender | Brain Magnetic Resonance Imaging (MRI) | NGS Results |
|---|---|---|---|---|
| 4041-NP1139 | 31 | F | Diffuse hypomyelination of the subcortical and deep white matter, enlarged high convexity CSF spaces, ponto-cerebellar hypoplasia, and thin corpus callosum. | Causative mutations |
| 5078-NP1688 | 23 | M | Slight peri- and supra-ventricular white matter hyperintensities and cortical atrophy. | Causative mutations |
| 3277-CS130A | 40 | F | Diffuse hyperintensities in the corona radiata and centra semiovale. | Negative |
| 3809-VR103A | 26 | F | Slight and diffuse signal alteration in the subcortical and deep white matter, and thin corpus callosum. | Negative |
| 5303-VR813A | 24 | F | Large and irregular hyperintensities in the subcortical and perventricular with matter on T2-weighed and FLAIR sequences. | Negative |
| Sample | Gene | Function | Genotype | Codon | Protein | Coding | HGMD a | ACMG b | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| 4041-NP1139 | POLR3A | Missense | C/T | AGT | p.Gly1240Ser | c.3718G>A | Yes | L.P. | [10,11] |
| POLR3A | Missense | G/T | AAG | p.Gln599Lys | c.1795C>A | Yes | L.P. | [10] | |
| 5078-NP1688 | POLR3A | Missense | G/T | AAG | p.Gln599Lys | c.1795C>A | Yes | L.P. | [10] |
| POLR3A | Missense | T/C | GAA | p.Lys110Glu | c.328A>G | - | L.P. | (c) |
| In Silico Predictive Tool | Prediction/Score/PHRED-Scaled |
|---|---|
| CADD_pred | 27.2 (>30 highly pathogenic; >20 pathogenic) |
| DANN_score | 0.999 (range from 0 to 1) * |
| FATHMM_pred | Tolerated |
| GERP++_RS | 5.67 (range from −12.3 to 6.17) * |
| LRT_pred | Deleterious |
| M-CAP_pred | Damaging |
| MetaSVM_pred | Tolerated |
| MetaLR_pred | Tolerated |
| MutationAssessor_pred | Medium |
| MutationTaster_pred | Damaging |
| PROVEAN_pred | Deleterious |
| Polyphen2_HDIV_pred | Probably damaging |
| SIFT_pred | Deleterious |
| SiPhy_29way_logOdds | 15.915 (range from 0 to 37.9718) * |
| Fathmm-MKL_coding_pred | Damaging |
| ACMG criteria | Likely Pathogenic ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musumeci, A.; Calì, F.; Scuderi, C.; Vinci, M.; Vitello, G.A.; Musumeci, S.A.; Chiavetta, V.; Federico, C.; Amore, G.; Saccone, S.; et al. Identification of a Novel Missense Mutation of POLR3A Gene in a Cohort of Sicilian Patients with Leukodystrophy. Biomedicines 2022, 10, 2276. https://doi.org/10.3390/biomedicines10092276
Musumeci A, Calì F, Scuderi C, Vinci M, Vitello GA, Musumeci SA, Chiavetta V, Federico C, Amore G, Saccone S, et al. Identification of a Novel Missense Mutation of POLR3A Gene in a Cohort of Sicilian Patients with Leukodystrophy. Biomedicines. 2022; 10(9):2276. https://doi.org/10.3390/biomedicines10092276
Chicago/Turabian StyleMusumeci, Antonino, Francesco Calì, Carmela Scuderi, Mirella Vinci, Girolamo Aurelio Vitello, Sebastiano Antonino Musumeci, Valeria Chiavetta, Concetta Federico, Greta Amore, Salvatore Saccone, and et al. 2022. "Identification of a Novel Missense Mutation of POLR3A Gene in a Cohort of Sicilian Patients with Leukodystrophy" Biomedicines 10, no. 9: 2276. https://doi.org/10.3390/biomedicines10092276
APA StyleMusumeci, A., Calì, F., Scuderi, C., Vinci, M., Vitello, G. A., Musumeci, S. A., Chiavetta, V., Federico, C., Amore, G., Saccone, S., Di Rosa, G., & Nicotera, A. G. (2022). Identification of a Novel Missense Mutation of POLR3A Gene in a Cohort of Sicilian Patients with Leukodystrophy. Biomedicines, 10(9), 2276. https://doi.org/10.3390/biomedicines10092276